

Abstract
Heme oxygenase (HO) and cytochrome P450 (P450)-derived epoxyeicosatrienoic acids (EETs) participate in vascular protection, and recent studies suggest these two systems are functionally linked. We examined the consequences of HO deficiency on P450-derived EETs with regard to body weight, adiposity, insulin resistance, blood pressure, and vascular function in HO-2-null mice. The HO-2-null mice were obese, displayed insulin resistance, and had high blood pressure. HO-2 deficiency was associated with decreases in cyp2c expression, EET levels, HO-1 expression, and HO activity and with an increase in superoxide production and an impairment in the relaxing response to acetylcholine. In addition, HO-2-null mice exhibited increases in serum levels of tumor necrosis factor (TNF)-α and macrophage chemoattractant protein (MCP)-1 and a decrease in serum adiponectin levels. Treatment of HO-2-null mice with a dual-activity EET agonist/soluble epoxide hydrolase inhibitor increased renal and vascular EET levels and HO-1 expression, lowered blood pressure, prevented body weight gain, increased insulin sensitivity, reduced subcutaneous and visceral fat, and decreased serum TNF-α and MCP-1, while increasing adiponectin and restoring the relaxing responses to acetylcholine. The decrease in cyp2c expression and EETs levels in HO-2-null mice underscores the importance of the HO system in the regulation of epoxygenase levels and suggests that protection against obesity-induced cardiovascular complications requires interplay between these two systems. A deficiency in one of these protective systems may contribute to the adverse manifestations associated with the clinical progression of the metabolic syndrome.
The heme oxygenase (HO) isoforms HO-1 and HO-2, responsible for the degradation of heme, have diverse physiological functions. HO-2 (constitutive) contributes to basal physiological functions, including renal channel activity, transport, and vascular tone (Abraham and Kappas, 2008 and references therein). HO-1 (inducible) is considered to have a role in regulation of blood pressure based on rapid induction by shear stress, starvation, statins, and its ability to provide renal cytoprotection against oxidative injury (for review, see Abraham and Kappas, 2008). CO and bilirubin, the products of heme degradation, have the capacity to modulate renal function and to attenuate vasoconstrictor action. HO-1 and HO-2 gene expression is known to increase cellular antioxidant and anti-inflammatory properties. This is associated with an increase in adiponectin and signaling mechanisms, resulting in the stimulation of NO bioavailability (Kruger et al., 2006; L'Abbate et al., 2007; Abraham and Kappas, 2008). HO-1-derived CO regulates vascular tone in part by decreasing cytochrome P450 (P450)-derived vasoconstrictor (Kaide et al., 2001) and cyclooxygenase-2 activity (Abraham and Kappas, 2008). Sacerdoti et al. (2006, 2007) describe the interaction of HO-1 gene expression with the P450 epoxygenase metabolites epoxyeicosatrienoic acids (EETs) and show in vascular endothelial cells that an increase in EET levels was associated with an increase in HO activity.
Epoxidation of arachidonic acid to EETs is catalyzed by a large number of P450 isoforms (Zeldin, 2001). These isoforms demonstrate tissue-specific expression and exhibit relative regioselectivity and stereospecificity. Members of the CYP2C and CYP2J families are the predominant epoxygenases in liver, kidney, brain, and blood vessels of rodents and humans (Zeldin, 2001; Campbell and Falck, 2007). In mice, the major arachidonate CYP2C epoxygenases are CYP2c29, CYP2c37, CYP2c38, and CYP2c44 (Tsao et al., 2001; DeLozier et al., 2004; Nakagawa et al., 2006). Upon formation, EETs are subjected to rapid hydrolysis by epoxide hydrolases to their respective dihydroxyeicosatrienoic acids (DHETs) as well as to esterification, primarily to glycerophospholipids (Karara et al., 1991). The pathophysiological significance of EETs and DHETs stems from numerous studies describing potent stereospecific biological effects, including vasodilation, stimulation of ion transport, inhibition of inflammatory response, and stimulation of epithelial cell growth (Imig, 2005; Spector and Norris, 2007). Studies showing that induction of CYP2C23 in rats (Muller et al., 2004) or inhibition of the soluble epoxide hydrolase (sEH) (Imig, 2005) provides protection against angiotensin II-induced renal injury and that mice lacking sEH have lower blood pressure than wild-type control mice (Sinal et al., 2000) further enforce a role for EETs as important modulators of the cardiovascular system. We previously identified in mouse and rat a significant role for HO-2 in regulating renal homeostasis (Goodman et al., 2006; Abraham and Kappas, 2008). HO-2 deletion leads to chronic inflammatory conditions and aggravates diabetes-mediated renal injury (Goodman et al., 2006). Mice lacking HO-2 display increased iron accumulation and marked oxidative stress (Dennery et al., 1998; Goodman et al., 2006). Selective inhibition of HO-2 by small interfering RNA activates caspases and increases reactive oxygen species (ROS) (Abraham and Kappas, 2008), whereas ROS decreases EET levels (Larsen et al., 2008). Therefore, the distinctive renal and vascular expression of the HO-2 protein suggests that HO-2 gene expression plays a crucial role in renal function and vascular tone, possibly via the regulation, at least in part, of epoxygenase activity and EET levels. Hence, the present study was designed to investigate the consequences of HO-2 deletion on P450 epoxygenase and to examine whether EETs play a role in the manifestations of the metabolic syndrome in HO-2-null mice.
Materials and Methods
Materials.
The parent EET analog 11-(nonyloxy)undec-8(Z)-enoic acid (NUDA) acts as a potent vasorelaxant in mesenteric and renal arteries (Dimitropoulou et al., 2007; Imig et al., 2008). For the in vivo studies described here, NUDA was conjugated to l-aspartic acid to give (S)-2-(11-(nonyloxy)undec-8(Z)-enamido)succinic acid (NUDSA) to minimize β-oxidation and to improve its solubility in aqueous milieu.
Animal Experimentation.
All animal experiments followed an institutionally approved protocol in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The HO-2-null mice are direct descendants of the HO-2 mutants. These well characterized HO-2-null mice have a C57BL/6 × 129/Sv genetic background that was used on age- and gender-matched controls. Eight-month-old HO-2(−/−)-null and B6/129SF2/J (WT) mice were used for the studies. Mice were divided into three groups (10 mice/group): WT, HO-2(−/−), and HO-2(−/−) + NUDSA. The dual-activity EET agonist/sEH inhibitor NUDSA was injected intraperitoneally for 2 weeks daily at a dose of 1.5 mg/100 g of body weight/day. Food intake did not change in the mice in any treatment group (7.3 ± 0.4 g/day), and they had access to water ab libitum. Glucose monitoring was performed using an automated analyzer (Life Scan Inc., Milpitas, CA). Blood pressure was measured by the tail-cuff method before and every 7 days after NUDSA administration. Body weights of HO-2(−/−) and WT mice at the beginning of the experiment were 28 ± 2 and 20 ± 2 g, respectively. On the last day before sacrifice, mice were housed in metabolic cages for urine collection. Total urine did not change in the mice in any treatment group (1 ± 0.2 ml/day). At the time of sacrifice, the body weight of all mice was measured. The subcutaneous and visceral fat in the abdomen; mesenteric fat; and fat around the liver, kidney, spleen, and heart were dissected free, pooled for each mouse, and weighed. Blood samples were collected in K3EDTA tubes at sacrifice, and the plasma was separated. Aortas were dissected and processed for functional studies and kidneys were harvested, drained of blood, and flash-frozen in liquid nitrogen. Specimens were maintained at −80°C until needed. In some experiments, renal interlobar arteries were dissected and processed for measurements of EETs and DHETs.
Assessment of Vascular Reactivity.
The aorta was removed, cleaned of fat and loose connective tissue, placed in ice-cold Krebs-bicarbonate solution, and sectioned into 3-mm-long rings. Two rings per aorta were mounted on stainless steel hooks and suspended in 5-ml tissue baths. The bath was filled with Krebs' solution at 37°C, pH 7.4, with 95% O2, 5% CO2 as a gas phase. The rings were incubated in individually thermostated (37°C) DMT myograph baths (DMT, Atlanta, GA), with a passive tension of 0.2 g for 1 h. Force was recorded from force displacement transducers via the PowerLab system (ADInstruments Ltd., Chalgrove, Oxfordshire, UK), running Chart 5 software. The Krebs' buffer solution in the tissue bath was replaced every 15 min, and the tension was readjusted each time. At the end of the equilibration period, the maximal force generated by addition of a depolarizing solution of 60 mM KCl was determined. Vasorelaxation responses of phenylephrine-constricted arteries to cumulative increments in the acetylcholine (10−9–10−3 M) were examined in the presence of indomethacin (10 μM).
Measurement of EETs and DHETs.
Renal cortical tissues were homogenized in 66% methanol containing a 500-pg mixture of internal standards [prostaglandin E2-d4, 8(9)-EET-d11, 11(12)-EET-d8, 12-hydroxyeicosatetraenoic acid-d8, 20-hydroxyeicosatetraenoic acid-d6, and 11,12-DHET-d11]. EETs and DHETs were extracted using solid phase C18-ODS AccuBond II 500-mg cartridges (Agilent Technologies, Santa Clara, CA). In brief, each sample was centrifuged for 15 min at 4°C. Then, the supernatant was collected, diluted with 15 ml of water, and acidified to pH 4.0 with HCl. Cartridges were primed first with 20 ml of methanol followed by 20 ml of water. Samples were loaded, washed with 20 ml of water and 8 ml of hexane, and eluted with 8 ml of methanol. The collected methanol fraction was dried under nitrogen and resupended in 200 μl of methanol and stored at −80°C until analysis by liquid chromatography-tandem mass spectrometry. Renal interlobar arteries (four to six segments per tube) were incubated in 1 ml of oxygenated Krebs' buffer containing 1 mM NADPH at 37°C for 1 h. Thereafter, internal standards were added to each sample after acidification to pH 4.0 with glacial acetic acid. Eicosanoids were extracted with twice the volume of ethyl acetate (three times), dried under nitrogen, and stored at −80°C for further analysis. Identification and quantification of EETs and DHETs was performed with a Q-trap 3200 linear ion trap quadruple liquid chromatography-tandem mass spectrometry system equipped with a Turbo V ion source operated in negative electrospray mode (Applied Biosystems, Foster City, CA). Extracted samples were suspended in 10 μl of methanol and injected into the high-performance liquid chromatography via an Agilent 1200 standard series autosampler equipped with a thermostat set at 4°C (Agilent Technologies). The high-performance liquid chromatographic component consisted of an Agilent 1100 series binary gradient pump equipped with an Eclipse plus C18 column (50 × 4.6 mm; 1.8 mm) (Agilent Technologies). The column was eluted at a flow rate of 0.5 ml/min with 100% mobile phase A [methanol/water/acetic acid (60:40:0.01, v/v/v)] from 0 to 2 min and a gradient increasing to 100% B (100% methanol) at 13 min. Multiple reaction monitoring was used with a dwell time of 25 or 50 ms for each compound, with the following source parameters: ion spray voltage, −4500 V; curtain gas, 40 U; ion source gas flow rate 1, 65 U; ion source gas flow 2, 50 U; and temperature, 600°C. Synthetic standards were used to obtain standard curves (5–500 pg) for each compound. These standard curves were used and extrapolated to calculate the final EET and DHET concentrations, which were presented as picograms per milligram of protein.
Western Blot Analysis.
Frozen tissues were pulverized under liquid nitrogen and placed in a homogenization buffer, and homogenates were centrifuged at 27,000g for 10 min at 4°C. The supernatant was isolated, and protein levels were determined by immunoblotting with antibodies against HO-1 and HO-2 (AKELA Pharma, Inc., Montreal, QC, Canada), AMPK, pAMPK, and adiponectin antibodies were obtained from Cell Signaling Technology Inc. (Danvers, MA), and eNOS and peNOS were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies against the mouse cyp2c44 and the rat CYP2C11, which cross-reacts with the mouse cyp2c29, 2c37, and 2c38, were gifts from Dr. Jorge Capdevila (Vanderbilt University, Nashville, TN). Antibodies were prepared in the following dilutions: HO-1, HO-2, eNOS, peNOS, AMPK, pAMPK, and adiponectin, 1:1000 dilution; and CYP2C11 and cyp2C44, 1:5000 dilution. Immunoblotting was performed as described previously (Abraham and Kappas, 2008).
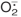 Production.
Production.
Arterial segments were placed in scintillation vials (two segments per vial) containing 1 ml of Krebs-HEPES buffer, pH 7.4, and lucigenin (5 μM) for 30 min at 37°C. Lucigenin chemiluminescence was measured in a liquid scintillation counter (LS6000TA; Beckman Instruments, Fullerton, CA), and superoxide production was quantified as described previously (Peterson et al., 2009).
Blood Glucose, Insulin Tolerance, and Cytokines/Chemokines Measurements.
After a 12-h fast, mice were injected intraperitoneally with either insulin (2.0 U/kg) or glucose (2.0g/kg body weight) to determine insulin sensitivity (insulin tolerance test) and glucose intolerance. Blood samples were taken at various time points (0–120 min) as described previously (Li et al., 2008). The levels of MCP-1 and TNF-α and the high-molecular-weight form of adiponectin were determined using an enzyme-linked immunosorbent assay (Thermo Fisher LSR, Rockford, IL).
Measurement of HO-Derived Bilirubin Formation.
HO activity was assayed in homogenates of renal cortex or vessels tissue. In brief, 300 μg of protein was used for bilirubin measurement in the presence and absence of heme and a NADPH-generating system (Kruger et al., 2006).
Statistical Analyses.
Statistical significance between experimental groups was determined by the Fisher method of analysis of multiple comparisons (p < 0.05). For comparison between treatment groups, the null hypothesis was tested by a single-factor analysis of variance for multiple groups or unpaired t test for two groups. Vascular responses, generated from the concentration-response data derived from each vessel, were fitted separately to a logistic function by nonlinear regression. Maximal asymptote of the curve and the concentration of agonist producing 50% of the maximal response (EC50) were calculated using commercially available software (Prism 2.01; GraphPad Software Inc., San Diego, CA). Vascular concentration-response data were analyzed by a two-way analysis of variance followed by a Duncan multiple range test. Data are presented as mean ± S.E.
Results
Effect of HO-2 Deletion on Body Weight and Fat Content.
As seen in Fig. 1, A and B, HO-2 deletion increased body weight compared with age-matched controls. The increases in body weight gain were manifested by increases of 30 to 40% (p < 0.01) in subcutaneous and visceral fat and the ratio of visceral to subcutaneous fat. Subcutaneous fat in HO-2 mice was 1.48 ± 0.09 g compared with 1.16 ± 0.11 g in WT mice (p < 0.05) (Fig. 1C); visceral fat was 1.87 ± 0.07 versus 1.32 ± 0.13 g in HO-2-null and WT mice, respectively (p < 0.05) (Fig. 1C). The ratio of visceral to subcutaneous fat was higher in HO-2-null mice [1.07 ± 0.10 versus 1.4 ± 0.12 in WT and HO-2(−/−) mice, respectively, p < 0.01] (Fig. 1D), suggesting that the increases in visceral fat may be a major player in HO-2 adiposity.
Fig. 1.

A, representative photographs showing WT and HO-2(−/−) mice at 14 weeks. Top, body appearance and subcutaneous tissue appearance. Bottom, dissected subcutaneous and visceral fat. B, body weight in grams of WT and HO-2(−/−) mice at 14 weeks. Values are means ± S.E., n = 4; *, p < 0.01 versus WT mice. C, weight in grams of subcutaneous and visceral fat of WT and HO-2(−/−) mice at 14 weeks. Values are means ± S.E., n = 4; *, p < 0.05 versus WT mice. D, ratio of subcutaneous and visceral fat of WT and HO-2(−/−) mice at 14 weeks, n = 4; *, p < 0.01 versus WT mice.
Effect of HO-2 Deletion on Blood Pressure, Blood Glucose, and Insulin Sensitivity.
Systolic blood pressure was significantly higher in HO-2-null mice than in WT mice (110.9 ± 1.5 versus 90.6 ± 2.9 mm Hg; p < 00.01) as was diastolic blood pressure (79.1 ± 2.3 versus 67.4 ± 2.8 mm Hg; p < 00.01) (Fig. 2A).
Fig. 2.

A, blood pressure of HO-2(−/−) mice. Values are means ± S.E., n = 4; *, p < 0.01 versus WT systolic blood pressure; **, p < 0.01 versus WT diastolic blood pressure. B, blood glucose of WT and HO-2(−/−) mice. Values are means ± S.E., n = 4; *, p < 0.01 versus WT mice. C, insulin sensitivity of WT and HO-2(−/−) mice. Intraperitoneal insulin tolerance test was performed as described under Materials and Methods. Values are means ± S.E., n = 4; *, p < 0.01 versus WT mice; #, p < 0.01 versus 0 min.
Plasma glucose levels in HO-2-null mice were higher than those in WT mice. Blood glucose levels in HO-2-null mice were 255 ± 6.6 mg/dl compared with 127 ± 7.6 mg/dl in WT mice (p < 0.01) (Fig. 2B). Insulin administration to WT mice produced a rapid decrease in glucose (Fig. 2C). In HO-2-null mice, the drop in blood glucose was significant (p < 0.01), but blood glucose remained at higher levels compared with WT mice for the duration of the experiment; at 90 min, blood glucose for HO-2-null mice was 134 ± 3.8 mg/dl compared with 44.5 ± 4.9 mg/dl for WT mice (Fig. 2C), suggesting increased insulin resistance in HO-2-null mice. The increases in body weight and fat content coupled with hypertension and insulin resistance are key indicators of the metabolic syndrome in these mice.
Effect of HO-2 Deletion on Superoxide Production, EETs Levels, and Relaxation to Acetylcholine.
The HO-2-null mice have been shown to experience oxidative stress (Basuroy et al., 2006; Goodman et al., 2006). Indeed, compared with WT mice, HO-2 mice exhibited elevated levels of in renal and vascular tissues. In renal tissues, superoxide levels amounted to 3.57 ± 0.19 × 104 cpm compared with 1.51 ± 0.35 × 104 cpm in WT mice (p < 0.01) (Fig. 3A). Similar results were obtained in aortic tissues; superoxide production was approximately 3-fold higher in HO-2-null aortas than in WT aortas (Fig. 3A).
Fig. 3.

A, levels of production in WT and HO-2(−/−) mouse kidney. Values are means ± S.E., n = 4; *, p < 0.05 versus WT mice. B, levels of EETs and EETs + DHETs in kidney of WT and HO-2(−/−) mice. Values are means ± S.E., n = 4; *, p < 0.01 versus WT mice. C, levels of EETs and EETs + DHETs in renal interlobar arteries of WT and HO-2(−/−) mice. Values are means ± S.E., n = 4; *, p < 0.05 versus WT mice. D, relaxation-response curves to acetylcholine (Ach) in aortic rings from WT and HO-2(−/−) mice. Values are means ± S.E., n = 4; *, p < 0.05 versus WT mice.
The increased production of superoxide was associated with a marked decrease in the levels of all four EET regioisomers, including 14,15-EET, 11,12-EET, 8,9-EET, and 5,6-EET, in renal cortical and vascular tissues. As shown in Fig. 3B, levels of EETs in renal cortical tissues were four times lower in HO-2(−/−) mice compared with WT mice. Likewise, levels of EETs in renal vascular tissues (interlobar arteries) of HO-2-null mice amounted to 0.44 ± 0.15 ng/mg compared with 1.78 ± 0.42 ng/mg in WT mice (p < 0.05) (Fig. 3C). The sum of EETs and DHETs, which is a determinant of epoxygenase activity, in HO-2-null mice was 4.6- and 2.3-fold lower than that measured in WT mice in renal and vascular tissues, respectively (Fig. 3, B and C).
Vascular function measured as the relaxation to acetylcholine was significantly impaired in the HO-2-null mice. As seen in Fig. 3D, the relaxing response of HO-2-null aortas to acetylcholine was impaired; at 10−4 M acetylcholine, the percentage of relaxation was 45.3 ± 9.9 in HO-2 versus 65.2 ± 1.9 in WT mice (p < 0.05); and at 10−5 M, relaxation was 65.7 ± 6.18 versus 90.2 ± 6.15 in WT aortas, suggesting that endothelial function is impaired in the HO-2-null mice.
Effect of EET Agonist on HO-2-Null Mice Body Weight, Fat Content, Blood Glucose, Blood Pressure, and Endothelial Function.
As seen in Fig. 4A, treatment with the EET agonist NUDSA visibly reduced weight gain in HO-2-null mice. Body weights of HO-2-null mice after 2 weeks of treatment were decreased from 33 ± 0.6 to 22.6 ± 0.3 g (p < 0.05) (Fig. 4B). This was associated with a marked decrease in subcutaneous fat from 1.45 ± 0.11 to 0.23 ± 0.027 g (p < 0.01) (Fig. 4C) and in visceral fat from 1.82 ± 0.05 to 0.23 ± 0.28 g (p < 0.01) (Fig. 4D) after treatment.
Fig. 4.

Effect of NUDSA on body weight and fat content in HO-2(−/−) mice. A, representative photographs showing mouse from each group after 2 weeks of treatment. Top, body appearance. Middle, subcutaneous tissue appearance. Bottom, dissected subcutaneous and visceral fat. B, body weight. Values are means ± S.E., n = 6; *, p < 0.05 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice. C, weight of subcutaneous fat. Values are means ± S.E., n = 6; *, p < 0.05 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice. D, weight of visceral fat. Values are means ± S.E., n = 6; *, p < 0.01 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice.
Administration of NUDSA to HO-2(−/−) mice significantly decreased blood glucose and blood pressure and increased insulin sensitivity. As seen in Fig. 5A, blood glucose of HO-2-null mice, which was 280.6 ± 9.6 mg/dl, decreased by 40% to 172.7 ± 5.5 mg/dl (p < 0.01), but it remained higher compared with that in age-matched WT mice. Administration of NUDSA also increased insulin sensitivity in HO-2(−/−) mice (Fig. 5B). Insulin administration to the NUDSA-treated HO-2(−/−) mice resulted in rapid decrease in glucose compared with untreated HO-2(−/−) mice receiving vehicle alone, suggesting improved insulin sensitivity in the EET-agonist treated HO-2(−/−) mice (Fig. 5B). Figure 5C depicts the effect of NUDSA on blood pressure in the HO-2-null mice. The increase in systolic and diastolic blood pressure in untreated HO-2-null mice was attenuated by treatment with NUDSA. In fact blood pressure was normalized to pressure measured for the WT mice decreasing systolic pressure from 119 ± 2 to 101 ± 3 mm Hg (p < 0.01) and diastolic from 80 ± 2 to 75 ± 2 mm Hg (p < 0.01) (Fig. 5C).
Fig. 5.

Effect of NUDSA on blood glucose, blood pressure, and endothelial function in HO-2(−/−) mice. A, blood glucose. Values are means ± S.E.; n = 6; *, p < 0.01 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice. B, insulin sensitivity of untreated HO-2(−/−) mice (from Fig. 2C) and HO-2(−/−) mice treated with NUDSA (*, p < 0.05 versus untreated mice). C, blood pressure. Values are means ± S.E., n = 6; *, p < 0.01 versus WT systolic blood pressure; **, p < 0.01 versus WT diastolic blood pressure; #, p < 0.01 versus HO-2(−/−) systolic blood pressure. D, relaxation-response curves to acetylcholine (Ach). Values are means ± S.E., n = 6; #, p < 0.05 versus HO-2(−/−) mice. E, EC50 value of acetylcholine. Values are means ± SE, n = 6; *, p < 0.01 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice.
As indicated above (Fig. 3D), HO-2-null mice displayed endothelial dysfunction. Thus, concentration-dependent relaxations to acetylcholine were significantly (p < 0.05) impaired in HO-2-null aortas compared with WT mice, with EC50 (log M) values of −3.98 ± 0.12 and −4.85 ± 0.12, respectively. Treatment with NUDSA restored relaxation to acetylcholine in the HO-2-null aortas, reaching an EC50 (log M) value of −4.853 ± 0.23, which was not different from that of WT mice (Fig. 5, D and E).
Effect of EET-Agonist on HO-1 Expression, Superoxide Production, and P450 Epoxygenase Levels.
It has been shown that HO-2 deletion impairs HO-1 inducibility leading to a decrease in HO activity and increase in oxidative stress (Seta et al., 2006). Indeed, Western blot analysis showed a significant decrease (p < 0.01) in the ratio of HO-1 to actin in the renal cortex in HO-2(−/−) mice compared with WT mice (Fig. 6A). HO activity in the renal cortex of HO-2(−/−) and WT mice was measured by the bilirubin formation and was found to be significantly decreased (p < 0.05). HO activity in WT mice was 0.65 ± 0.12 nmol bilirubin formed ng/h compared with 0.39 ± 0.14 nmol bilirubin formed ng/h. NUDSA treatment increased bilirubin formation to 0.79 ± 0.16 nmol bilirubin formed ng/h. A daily injection of NUDSA for 2 weeks resulted in a significant 2-fold increase in HO-1 protein levels in HO-2(−/−) mice (Fig. 6A). The levels of in renal cortex obtained from untreated HO-2(−/−) were 3.65 ± 0.19 × 104 cpm/mg compared with 1.85 ± 0.35 × 104 cpm/mg (p < 0.05) in WT mice (Fig. 6B). Treatment of HO-2(−/−) mice with NUDSA significantly decreased levels in the renal cortex to 1.49 ± 0.33 × 104cpm/mg protein, which was not different from that measured in WT mice (Fig. 6B).
Fig. 6.

Effect of NUDSA on HO-1 and CYP2c protein levels and superoxide production in aortas HO-2(−/−) mice. A, Western blot and densitometry analysis of renal HO-1 protein. Values are means ± S.E., n = 3; *, p < 0.01 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice. B, superoxide production. Values are means ± S.E., n = 4; *, p < 0.05 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice. C, representative Western blot and densitometry analysis of cyp2c44 protein in aorta. Values are means ± S.E., n = 3; *, p < 0.01 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice. D, representative Western blot and densitometry analysis of CYP2C11-immunoreactive proteins (iCYP2C11, i.e., 2c29, 2c38, and 2c37) in aorta. Values are means ± S.E., n = 3; *, p < 0.01 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice.
It is interesting to note that the expression of arachidonate cyp2c epoxygenases in mice was differentially regulated by HO-2 deletion. As seen in Fig. 6, C and D, aortic CYP2C11 but not cyp2c44 immunoreactivity was markedly reduced in HO-2-null mice. Antibodies against the rat CYP2C11 cross-react with the mouse cyp2c29, 2c37, and 2c38. cyp2c29 and cyp2c37 are expressed primarily in lung, aorta, and white blood cells as well as in the kidney (DeLozier et al., 2004). Treatment with NUDSA increased (p < 0.01) the protein levels of cyp2c44 and CYP2C11-immunoreactive proteins (cyp2c2, 2c3, and 2c38) by 3- and 2-fold, respectively. Similar results were obtained in the kidney (data not shown).
The changes in CYP2C11 immunoreactive protein levels paralleled changes in the levels of renal and vascular EETs and the estimated epoxygenase activity. In WT mice, renal EETs were composed of 14,15-, 11,12-, 8,9-, and 5,6-EETs at a ratio of 4:2:2:1, whereas in the aorta, this ratio was 1:0.25:4:3 (Fig. 7, A and D). As seen in Fig. 7, A–C, renal EETs levels as well as total epoxygenase activity (EETS + DHETs) were 10 and 25% of that measured in the WT mice. The reduction was comparable for all four EET regioisomers (Fig. 7, A and D). Treatment with NUDSA increased EETs levels by 5.8-fold and epoxygenase activity by 2.9-fold. The ratio of EETs to DHETs also increased after treatment with NUDSA, suggesting that in addition to increasing epoxygenase activity, it also decreased the activity of sEH. Similar results were obtained in renal vascular tissues (Fig. 7, D and E). NUDSA increased EETs levels and epoxygenase activity (EETs + DHETs) in renal interlobar arteries by 2.5- and 2.3-fold, respectively, and the ratio of EETs to DHETs by 7.7-fold (p < 0.05).
Fig. 7.

Effect of NUDSA on renal and vascular EETs/DHETs in HO-2(−/−) mice. A, EETs levels in the kidney. Values are means ± S.E., n = 4; *, p < 0.01 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice. B, levels of EETs + DHETs in the kidney. Values are means ± S.E., n = 4; *, p < 0.01 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice. C, EET-to-DHET ratio in kidney after EET treatment. Values are means ± S.E., n = 4; *, p < 0.05 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice. D, levels of EETs in renal interlobar arteries. Values are means ± S.E., n = 4; *, p < 0.05 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice. E, levels of EETs + DHETs in renal interlobar arteries. Values are means ± S.E., n = 4; *, p < 0.05 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice. F, EET-to-DHET ratio in renal interlobar arteries. Values are means ± S.E., n = 4; *, p < 0.01 versus WT; #, p < 0.05 versus HO-2(−/−) mice.
Effect of EET-Agonist on Adiponectin, pAMPK, and peNOS Levels.
Western blot analysis clearly demonstrated that HO-2 deletion is associated with significant decreases in the vascular expression of adiponectin, pAMPK, and peNOS compared with those in age-matched WT mice (Fig. 8, A–C). Treatment with NUDSA increased these levels by 2- to 2.5-fold to levels significantly higher than those measured in WT mice. Similar results were obtained with renal tissue (data not shown).
Fig. 8.

Effect of NUDSA on expression levels of vascular adiponectin, pAMPK, and peNOS and serum cytokines. A, Western blot and densitometry analysis of adiponectin in aortic tissues. Values are means ± S.E., n = 3; *, p < 0.01 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice. B, Western blot and densitometry analysis pAMPK and AMPK proteins. Values are means ± S.E., n = 3; *, p < 0.01 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice. C, Western blot and densitometry analysis peNOS and eNOS proteins. Values are means ± S.E., n = 3; *, p < 0.01 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice. D, serum adiponectin levels. Values are means ± S.E., n = 6; *, p < 0.01 versus WT mice; #, p < 0.01 versus HO-2(−/−) mice. E, serum TNF-α levels. Values are means ± S.E., n = 6; *, p < 0.01 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice. F, serum MCP-1 levels. Values are means ± S.E., n = 6; *, p < 0.01 versus WT mice; #, p < 0.05 versus HO-2(−/−) mice.
Levels of plasma adiponectin in HO-2(−/−) mice were significantly lower than those in age-matched WT mice (6.21 ± 0.65 versus 12.61 ± 1.22 μg/ml; p < 0.01) (Fig. 8D). Treatment with NUDSA increased plasma adiponectin in HO-2(−/−) mice to 10.87 ± 1.18 μg/ml. The HO-2-null mice also displayed increasing levels of inflammatory molecules, including TNF-α and MCP-1 compared with age-matched WT mice. As seen in Fig. 8E, plasma TNF-α levels were 5-fold higher in HO-2-null mice. Likewise, levels of MCP-1 were 60% higher (p < 0.05) than those in WT mice (Fig. 8F). Treatment of HO-2-null mice with NUDSA decreased the levels of both TNF-α and MCP-1 to levels not significantly different from that in WT mice.
Discussion
The present study clearly shows that HO-2 deletion leads to manifestations of the metabolic syndrome, including obesity, hypertension, and insulin resistance. These changes in the HO-2-null mouse were associated with impaired vascular function, increased inflammatory signals, and alterations in cytoprotective circuits, including HO-1, epoxygenase-derived EETs, and adiponectin. It is important to note that treatment of these mice with an EET agonist ameliorated the apparent metabolic syndrome phenotype of the HO-2-null mouse by preventing the decline in the HO-1-adiponectin axis that plays a critical role in preventing adiposity-mediated vascular and renal damage. This novel finding suggests that EETs are an integral component of this axis and are critical for the regulation of adipocyte stem cell-mediated release of adiponectin and of cardiovascular function.
The first cytoprotective circuit that was altered by HO-2 deletion was the HO system. In the present study we show that HO-2 deletion leads to reduced HO-1 expression and a decrease in HO activity. Consequently, this leads to a reduction in the antioxidant and anti-inflammatory capacity bestowed by the HO-derived metabolites biliverdin and CO (Abraham and Kappas, 2008). Whereas HO-1 has been recognized as the major cytoprotective moiety of the HO system in response to injury, primarily because of its rapid inducibility and its distinctive responsiveness to stress, recent studies using HO-2-null mice suggest that expression of HO-2 is also critical for HO-derived cytoprotection. HO-2 being constitutively expressed can be critical in cytoprotection, particularly in tissues where it is highly expressed such as the brain and the cornea (Chang et al., 2003; Seta et al., 2006; Herder et al., 2007). HO-2 deficiency enhanced diabetes-induced renal dysfunction and morphologic injury, and HO-1 up-regulation in the HO-2-null mouse rescued and prevented the morphologic damage (Goodman et al., 2006). HO-2 is critical for HO-1 expression and that the subsequent failure to up-regulate the HO system may contribute to unresolved inflammation and the development of chronic inflammatory conditions (Seta et al., 2006). HO-1 induction or overexpression abrogates the injurious consequences of obesity and hypertension, whereas inhibition of HO activity exacerbates these conditions (Abraham and Kappas, 2008). Therefore, it was not surprising that HO-2-null mice exhibited similar characteristics as ob mice in which the HO-1 suppression displays metabolic syndrome (Li et al., 2008; Peterson et al., 2009). Hence, the diminished activity of the HO system may contribute to increased oxidative stress and inflammatory conditions and consequently to derangements in the regulation of adipogenesis, and increased blood pressure.
The second cytoprotective system is the epoxygenase-EET pathway. We show here that the HO-2-null mice exhibited decreased activity of the arachidonic acid metabolic pathway that yields EETs. The inability of HO-2-null mice to sustain normal levels of EET may be the result of the increased levels of ROS and sEH coupled with the decreased expression of P450-epoxygenases. The expression levels of CYP2C11-immunoreactive proteins were significantly decreased in renal tissue of HO-2-null mice. EETs have been shown to contribute to antihypertensive mechanisms through their natriuretic and vasodilatory actions (Imig, 2005). The decrease in levels of EETs could also be construed as a major detrimental factor of the impaired vascular function in HO-2-null mice, an important component of blood pressure regulation. EETs play an important role in the regulation of ion transport and vascular tone and consequently in the control of blood pressure (Roman, 2002; Imig, 2005). EETs inhibit sodium transport and relax blood vessels through their EDHF property (Zeldin, 2001; Campbell and Falck, 2007).
EET agonist administration was associated with an increase in EETs and adiponectin levels. Because the EET agonist NUDSA increased both EET levels and HO-1 expression, it is not clear which, EETs or HO-1, is responsible for the increase of adiponectin levels and the decrease in inflammatory cytokines TNF and MCP-1. Both EETs and HO-1 exhibit anti-inflammatory properties (Spiecker and Liao, 2005; Abraham and Kappas, 2008). Increased adiponectin levels improve renal function (Sharma et al., 2008), decrease blood pressure, and attenuate diabetes-mediated vascular dysfunction (for review, see Wang and Scherer, 2008). Increased levels of HO-1-adiponectin are associated with an increase in pAMPK, peNOS, EPC homing, and NO bioavailability (L'Abbate et al., 2007; Sambuceti et al., 2009). These perturbations occur in sequence, commencing with increased levels of HO-1 expression after EET-agonist treatment, suggesting that increased expression of HO-1 is the trigger for the phenotype changes and resistance to oxidative stress.
Another important finding of the present study is the presence of excessive amounts of superoxide in kidney and aorta isolated from HO-2-null mice, suggesting that HO-2 deletion increased ROS levels, probably contributing to the decrease in EET levels (Larsen et al., 2008). The associated increase in HO-1 adiponectin and decrease in superoxide may therefore contribute to the activation of the epoxygenase, thereby forming a three-way cytoprotective circuit that includes EET, HO-1, and adiponectin. We propose that the HO-2 deletion and the resultant decrease in HO activity contribute to the increase in adiposity and vascular dysfunction. In fact, a direct link has been proposed between obesity-induced hypertension and related alterations in renal function as a result of increased sodium retention (Hall, 2003).
The present study is the first to show that deletion of HO-2 decreases cyp2c epoxygenases and increases sEH activity, deduced from the decreased EET-to-DHET ratio. Administration of an EET agonist/sEH inhibitor increased cyp2c epoxygenases and EET levels, probably by a mechanism that not only includes inhibition of sEH but also induction of cyp2c via activation of peroxisome proliferator-activated receptor-α (Ng et al., 2007). Interactions between HO-1 and EETs have been reported recently (Sacerdoti et al., 2006).
Furthermore, the observed perturbations in the P450 epoxygenase-derived EETs suggest that the increase in sEH accounts for the rapid decrease in EETs and conversion to the inactive metabolite DHET. P450 epoxygenase-derived eicosanoids have a relatively short half-life, and their biological effects are often limited to cells in proximity to their site of biosynthesis. Therefore, deletion of HO-2 may cause a local decrease in the concentration of these metabolites in a specific renal region, which accounts for the increase in blood pressure. However, the control of blood pressure requires the complex interaction of multiple regulatory systems. In addition to the renin-angiotensin aldosterone system and endothelial-derived relaxing factor (e.g., eNOS and prostacyclin), the involvement of P450-derived epoxygenases in blood pressure regulation have been reported previously (Zeldin, 2001; Roman, 2002; Capdevila, 2007). Deletion of the cyp4a10 gene, which leads to a marked decrease in renal EETs, results in increased blood pressure (Nakagawa et al., 2006). Increase in blood pressure was also observed in cyp2j5 null mice (Athirakul et al., 2008). In contrast, disruption of the sEH gene leads to a decrease in blood pressure (Sinal et al., 2000), although a recent study by (Luria et al., 2007) reported no change in blood pressure in the sEH null mice.
EET-agonist treatment and the associated increases in EET-adiponectin levels were associated with an increase in pAMPK signaling. pAMPK and pAKT levels appear to move in tandem with perturbations in the levels of HO-1 and adiponectin. Adiponectin is critical for endothelial cell survival and function through the activation of eNOS, via cross-talk between pAKT and pAMPK (Ouchi et al., 2004; Wang and Scherer, 2008). Activation of AMPK is important in cellular energy homeostasis through the stimulation of glucose transport, the switching off of energy consumption by decreasing lipogenesis, increasing fatty acid oxidation and ATP levels (Tomas et al., 2002; Laderoute et al., 2006). An increase in AMPK-serine/threonine kinase protein kinase B signaling is considered an important metabolic response to attenuate ROS-mediated endothelial dysfunction, because pAMPK and pAKT use eNOS as a substrate and enhance the levels of peNOS, suggesting that the HO-1-mediated prevention of vascular dysfunction and -increased vascular response to acetylcholine may be related to increases in adiponectin-pAMPK signaling pathways.
In conclusion, our studies reveal a novel role for the HO system in regulating obesity and adiponectin levels and demonstrate the interdependence of three protective circuits, namely, HO, EETs, and adiponectin, in the prevention of hypertension, obesity, and insulin resistance, the major manifestations of the metabolic syndrome. The present data are of considerable significance from a clinical and basic science perspective, clearly defining the existence of an EET-HO-1-adiponectin regulatory axis that can be manipulated by EET agonists to increase vascular resistance to dysfunction in obesity and diabetes and thus ameliorate the adverse manifestations seen with the clinical progression of the metabolic syndrome.
Acknowledgments
We thank Jennifer Brown for outstanding editorial assistance in the preparation of the manuscript.
This work was supported in part by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK068134]; the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL55601, HL34300]; the National Institutes of Health National Institute of General Medical Sciences [Grant GM31278]; and The Robert A. Welch Foundation.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.109.157545
- HO
- heme oxygenase
- P450
- cytochrome P450
- EET
- epoxyeicosatrienoic acid
- DHET
- dihydroxyeicosatrienoic acid
- sEH
- soluble epoxide hydrolase
- ROS
- reactive oxygen species
- NUDA
- 11-(nonyloxy)undec-8(Z)-enoic acid
- NUDSA
- 11-(nonyloxy)undec-8(Z)-enoic acid
- WT
- wild type
- d
- deuterium
- AMPK
- AMP-activated protein kinase
- pAMPK
- phosphorylated AMP-activated protein kinase
- pAKT
- phosphorylated serine/threonine kinase protein kinase B
- eNOS
- endothelial nitric-oxide synthase
- peNOS
- phosphorylated nitric-oxide synthase
- MCP
- macrophage chemoattractant protein
- TNF
- tumor necrosis factor.
References
- Abraham NG, Kappas A. (2008) Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev 60:79–127 [DOI] [PubMed] [Google Scholar]
- Athirakul K, Bradbury JA, Graves JP, DeGraff LM, Ma J, Zhao Y, Couse JF, Quigley R, Harder DR, Zhao X, et al. ( 2008) Increased blood pressure in mice lacking cytochrome P450 2J5. FASEB J 22:4096–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuroy S, Bhattacharya S, Tcheranova D, Qu Y, Regan RF, Leffler CW, Parfenova H. (2006) HO-2 provides endogenous protection against oxidative stress and apoptosis caused by TNF-alpha in cerebral vascular endothelial cells. Am J Physiol Cell Physiol 291:C897–C908 [DOI] [PubMed] [Google Scholar]
- Campbell WB, Falck JR. (2007) Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension 49:590–596 [DOI] [PubMed] [Google Scholar]
- Capdevila JH. (2007) Regulation of ion transport and blood pressure by cytochrome P450 monooxygenases. Curr Opin Nephrol Hypertens 16:465–470 [DOI] [PubMed] [Google Scholar]
- Chang EF, Wong RJ, Vreman HJ, Igarashi T, Galo E, Sharp FR, Stevenson DK, Noble-Haeusslein LJ. (2003) Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J Neurosci 23:3689–3696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLozier TC, Tsao CC, Coulter SJ, Foley J, Bradbury JA, Zeldin DC, Goldstein JA. (2004) CYP2C44, a new murine CYP2C that metabolizes arachidonic acid to unique stereospecific products. J Pharmacol Exp Ther 310:845–854 [DOI] [PubMed] [Google Scholar]
- Dennery PA, Spitz DR, Yang G, Tatarov A, Lee CS, Shegog ML, Poss KD. (1998) Oxygen toxicity and iron accumulation in the lungs of mice lacking heme oxygenase-2. J Clin Invest 101:1001–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitropoulou C, West L, Field MB, White RE, Reddy LM, Falck JR, Imig JD. (2007) Protein phosphatase 2A and Ca2+-activated K+ channels contribute to 11,12-epoxyeicosatrienoic acid analog mediated mesenteric arterial relaxation. Prostaglandins Other Lipid Mediat 83:50–61 [DOI] [PubMed] [Google Scholar]
- Goodman AI, Chander PN, Rezzani R, Schwartzman ML, Regan RF, Rodella L, Turkseven S, Lianos EA, Dennery PA, Abraham NG. (2006) Heme oxygenase-2 deficiency contributes to diabetes-mediated increase in superoxide anion and renal dysfunction. J Am Soc Nephrol 17:1073–1081 [DOI] [PubMed] [Google Scholar]
- Hall JE. (2003) The kidney, hypertension, and obesity. Hypertension 41:625–633 [DOI] [PubMed] [Google Scholar]
- Herder C, Schneitler S, Rathmann W, Haastert B, Schneitler H, Winkler H, Bredahl R, Hahnloser E, Martin S. (2007) Low-grade inflammation, obesity and insulin resistance in adolescents. J Clin Endocrinol Metab 92:4569–4574 [DOI] [PubMed] [Google Scholar]
- Imig JD. (2005) Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol 289:F496–F503 [DOI] [PubMed] [Google Scholar]
- Imig JD, Dimitropoulou C, Reddy DS, White RE, Falck JR. (2008) Afferent arteriolar dilation to 11,12-EET analogs involves PP2A activity and Ca2+-activated K+ channels. Microcirculation 15:137–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaide JI, Zhang F, Wei Y, Jiang H, Yu C, Wang WH, Balazy M, Abraham NG, Nasjletti A. (2001) Carbon monoxide of vascular origin attenuates the sensitivity of renal arterial vessels to vasoconstrictors. J Clin Invest 107:1163–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karara A, Dishman E, Falck JR, Capdevila JH. (1991) Endogenous epoxyeicosatrienoyl-phospholipids. A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. J Biol Chem 266:7561–7569 [PubMed] [Google Scholar]
- Kruger AL, Peterson SJ, Schwartzman ML, Fusco H, McClung JA, Weiss M, Shenouda S, Goodman AI, Goligorsky MS, Kappas A, et al. ( 2006) Up-regulation of heme oxygenase provides vascular protection in an animal model of diabetes through its antioxidant and antiapoptotic effects. J Pharmacol Exp Ther 319:1144–1152 [DOI] [PubMed] [Google Scholar]
- L'Abbate A, Neglia D, Vecoli C, Novelli M, Ottaviano V, Baldi S, Barsacchi R, Paolicchi A, Masiello P, Drummond GS, et al. ( 2007) Beneficial effect of heme oxygenase-1 expression on myocardial ischemia-reperfusion involves an increase in adiponectin in mildly diabetic rats. Am J Physiol Heart Circ Physiol 293:H3532–H3541 [DOI] [PubMed] [Google Scholar]
- Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. (2006) 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol 26:5336–5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen BT, Gutterman DD, Sato A, Toyama K, Campbell WB, Zeldin DC, Manthati VL, Falck JR, Miura H. (2008) Hydrogen peroxide inhibits cytochrome P450 epoxygenases: interaction between two endothelium-derived hyperpolarizing factors. Circ Res 102:59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kim DH, Tsenovoy PL, Peterson SJ, Rezzani R, Rodella LF, Aronow WS, Ikehara S, Abraham NG. (2008) Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes 57:1526–1535 [DOI] [PubMed] [Google Scholar]
- Luria A, Weldon SM, Kabcenell AK, Ingraham RH, Matera D, Jiang H, Gill R, Morisseau C, Newman JW, Hammock BD. (2007) Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem 282:2891–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller DN, Theuer J, Shagdarsuren E, Kaergel E, Honeck H, Park JK, Markovic M, Barbosa-Sicard E, Dechend R, Wellner M, et al. ( 2004) A peroxisome proliferator-activated receptor-alpha activator induces renal CYP2C23 activity and protects from angiotensin II-induced renal injury. Am J Pathol 164:521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa K, Holla VR, Wei Y, Wang WH, Gatica A, Wei S, Mei S, Miller CM, Cha DR, Price E, Jr, et al. ( 2006) Salt-sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J Clin Invest 116:1696–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng VY, Huang Y, Reddy LM, Falck JR, Lin ET, Kroetz DL. (2007) Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor α. Drug Metab Dispos 35:1126–1134 [DOI] [PubMed] [Google Scholar]
- Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T, Walsh K. (2004) Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem 279:1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SJ, Kim DH, Li M, Positano V, Vanella L, Rodella LF, Piccolomini F, Puri N, Gastaldelli A, Kusmic C, et al. ( 2009) The L-4F mimetic peptide prevents insulin resistance through increased levels of HO-1, pAMPK, and pAKT in obese mice. J Lipid Res 50:1293–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman RJ. (2002) P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82:131–185 [DOI] [PubMed] [Google Scholar]
- Sacerdoti D, Bolognesi M, Di Pascoli M, Gatta A, McGiff JC, Schwartzman ML, Abraham NG. (2006) Rat mesenteric arterial dilator response to 11,12-epoxyeicosatrienoic acid is mediated by activating heme oxygenase. Am J Physiol Heart Circ Physiol 291:H1999–H2002 [DOI] [PubMed] [Google Scholar]
- Sacerdoti D, Colombrita C, Di Pascoli M, Schwartzman ML, Bolognesi M, Falck JR, Gatta A, Abraham NG. (2007) 11,12-Epoxyeicosatrienoic acid stimulates heme-oxygenase-1 in endothelial cells. Prostaglandins Other Lipid Mediat 82:155–161 [DOI] [PubMed] [Google Scholar]
- Sambuceti G, Morbelli S, Vanella L, Kusmic C, Marini C, Massollo M, Augeri C, Corselli M, Ghersi C, Chiavarina B, et al. ( 2009) Diabetes impairs the vascular recruitment of normal stem cells by oxidant damage, reversed by increases in pAMPK, heme oxygenase-1, and adiponectin. Stem Cells 27:399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seta F, Bellner L, Rezzani R, Regan RF, Dunn MW, Abraham NG, Gronert K, Laniado-Schwartzman M. (2006) Heme oxygenase-2 is a critical determinant for execution of an acute inflammatory and reparative response. Am J Pathol 169:1612–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, et al. ( 2008) Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 118:1645–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. (2000) Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem 275:40504–40510 [DOI] [PubMed] [Google Scholar]
- Spector AA, Norris AW. (2007) Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol 292:C996–C1012 [DOI] [PubMed] [Google Scholar]
- Spiecker M, Liao JK. (2005) Vascular protective effects of cytochrome P450 epoxygenase-derived eicosanoids. Arch Biochem Biophys 433:413–420 [DOI] [PubMed] [Google Scholar]
- Tomas E, Tsao TS, Saha AK, Murrey HE, Zhang Cc C, Itani SI, Lodish HF, Ruderman NB. (2002) Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci U S A 99:16309–16313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao CC, Coulter SJ, Chien A, Luo G, Clayton NP, Maronpot R, Goldstein JA, Zeldin DC. (2001) Identification and localization of five CYP2Cs in murine extrahepatic tissues and their metabolism of arachidonic acid to regio- and stereoselective products. J Pharmacol Exp Ther 299:39–47 [PubMed] [Google Scholar]
- Wang ZV, Scherer PE. (2008) Adiponectin, cardiovascular function, and hypertension. Hypertension 51:8–14 [DOI] [PubMed] [Google Scholar]
- Zeldin DC. (2001) Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem 276:36059–36062 [DOI] [PubMed] [Google Scholar]