

Abstract
The pathogenicity of mycobacterial infections depends on virulence factors that mediate survival inside host macrophages. These virulence factors are generally believed to be specific for pathogenic species and absent or mutated in non-pathogenic strains. The serine/threonine protein kinase G (PknG) mediates survival of mycobacteria within macrophages by blocking lysosomal delivery. Here we describe a gene of the non-pathogenic species Mycobacterium smegmatis that is 78% identical with pknG of Mycobacterium tuberculosis and M. bovis bacillus Calmette–Guérin (BCG). When cloned into expression vectors, the M. smegmatis pknG orthologue produced an active kinase and performed the same function as its M. bovis BCG counterpart in intracellular survival. In addition, similar levels of pknG transcripts were found in M. bovis BCG and M. smegmatis. However, virtually no translation product of chromosomal pknG could be detected in M. smegmatis both after in vitro growth and after macrophage infection. This lack of efficient translation was shown to be caused by regulatory elements in the upstream region of the M. smegmatis gene. The data reveal dramatically increased translational efficiency of a virulence gene in a pathogenic mycobacterium compared with a non-pathogenic mycobacterium suggesting that changes in expression levels may underlie evolution of pknG and other pathogenicity genes in mycobacterium.
Introduction
Pathogenic mycobacteria, such as Mycobacterium tuberculosis, utilize an array of different strategies to survive inside mammalian host cells. Survival strategies are employed during the initial phases of the infection when the bacteria acquire access to their host cells, as well as during later stages of infection as the bacteria convert into a so-called dormant or latent state (Cosma et al., 2003). In the initial phases of infection, it is important that mycobacteria gain entry to macrophages, as they have the unique capacity to avoid antibacterial effectors of the immune system in this intracellular niche (Russell, 2001; Vergne et al., 2004; Houben et al., 2006). To ensure survival inside macrophages, pathogenic mycobacteria avoid their degradation by actively inhibiting fusion of the mycobacterial phagosome with lysosomes (Armstrong and Hart, 1971; Russell, 2001; Houben et al., 2006).
The molecular mechanisms that are responsible for diverting the traffic of pathogenic mycobacteria from the lysosomal degradative pathway are beginning to be understood and both host factors and mycobacterial molecules are involved in this process (Vergne et al., 2004; Houben et al., 2006). The eukaryotic-like serine/threonine kinase, protein kinase G (PknG), is one of the mycobacterial factors involved in blocking lysosomal delivery (Walburger et al., 2004; Scherr et al., 2007). Although it has been reported that PknG in M. tuberculosis and a related microorganism (Corynebacterium glutamicum) is required for optimal in vitro growth and is linked to intracellular glutamine/glutamate levels (Cowley et al., 2004; Niebisch et al., 2006), PknG in Mycobacterium bovis bacillus Calmette–Guérin (BCG) has been shown not to be required for mycobacterial growth outside macrophages in a complete liquid medium and not to be involved in glutamine metabolism (Walburger et al., 2004; Nguyen et al., 2005). After their uptake within macrophage phagosomes however, pathogenic mycobacteria lacking PknG are readily transported to lysosomes and destroyed (Walburger et al., 2004 and data not shown).
Interestingly, also the non-pathogenic Mycobacterium smegmatis genome contains a gene that is very similar to M. tuberculosis and M. bovis pknG. We here characterize the gene product from M. smegmatis pknG, showing that it encodes an active serine/threonine kinase and is able to block the lysosomal delivery of mycobacteria inside macrophages when expressed from expression vectors. However, while PknG transcripts were found in M. smegmatis, chromosomal pknG was not effectively translated in this non-pathogenic mycobacterium. The data show that PknG expression is blocked on a translational level in M. smegmatis and suggest that translational efficiency is an important factor in assessing candidate virulence genes.
Results
Mycobacterial PknG orthologues
The mycobacterial serine/threonine kinase PknG is required for survival of pathogenic mycobacteria inside macrophages (Walburger et al., 2004). To analyse whether the presence of PknG is specific for pathogenic mycobacteria, all available mycobacterial genome sequences, i.e. of M. tuberculosis, M. bovis (BCG), M. microti, M. marinum, M. avium (paratuberculosis), M. leprae and M. smegmatis, were analysed for the presence of a locus encoding pknG orthologues (Fig. 1). All of these genomes encoded an open reading frame that was highly homologous to PknG of M. tuberculosis. Strikingly, the genome of the non-pathogenic M. smegmatis also encodes a pknG orthologue (named MSMEG_0786) showing 78% identity and 87% similarity to M. tuberculosis PknG. In addition, the postulated operon structure around the pknG gene is conserved in M. smegmatis as well (see also Fig. 8A). For all PknGs, the homology extends over the kinase domain (residues 141–398 of M. tuberculosis PknG) as well as the C-terminal region, with the only deviation from homology within the N-terminus. On a phylogenetic perspective, we conclude that the pknG gene was present in mycobacteria before the divergence of the fast-growing mycobacterial species (M. smegmatis), from the slow-growing species (pathogenic mycobacteria) (Shinnick and Good, 1994).
Fig. 1.

Alignment of mycobacterial protein kinase G orthologues. PknG sequences from M. tuberculosis H37Rv (Rv0410c, from the Pasteur Institute), M. marinum (MMAR_0713, from the Sanger Institute), M. avium paratuberculosis (MAP3893c, from TIGR), M. leprae (ML0304, from the Sanger Institute) and M. smegmatis (MSMEG_0786, from TIGR) were aligned using clustalw. The PknG orthologues of M. bovis and M. microti were omitted since they were identical to M. tuberculosis PknG. The M. smegmatis PknG coding sequence starts at the ATG that aligns to the start codon of M. tuberculosis PknG (see Fig. S1 online). Identical residues are shown in grey and the kinase domain is underlined.
Fig. 8.
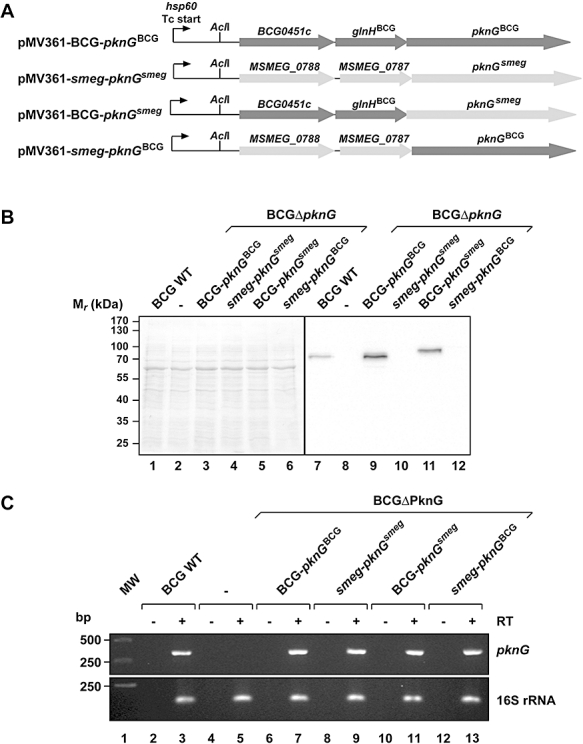
Analysis of the influence of the upstream region of M. smegmatis and M. bovis BCG pknG on pknG expression. A. Schematic representation of the fusion constructs used in this experiment. All constructs were introduced in the pMV361 vector at the AclI restriction site in the hsp60 promoter leaving the transcription (Tc) start intact (see also Experimental Procedures). pMV361-BCG-PknGBCG, pMV361 with M. bovis BCG pknG and its own upstream region; pMV361-smeg-PknGsmeg, pMV361 with M. smegmatis pknG and its own upstream region; pMV361-BCG-PknGsmeg, pMV361 with pknGsmeg fused to the M. bovis BCG pknG upstream region; pMV361-smeg-PknGBCG, pMV361 with pknGBCG fused to the M. smegmatis pknG upstream region. B. Lysates from M. bovis BCG (lane 7) and M. bovis BCGDΔpknG transformed with empty vector (lane 8) or the plasmids shown under (A) (lanes 9–12) were separated on a 10% SDS-PAGE gel and immunoblotted using anti-PknGBCG antiserum (right). The total protein pattern was analysed by Ponceau Red staining (left). C. cDNA was prepared from total RNA isolated from M. bovis BCG wt and the M. bovis BCGΔpknG transformants analysed under (B) by using reverse transcriptase and random primers. cDNA of pknG was subsequently amplified using gene-specific primers. As controls the same reactions were carried out without reverse transcriptase (−RT) or in stead of pknG primers with 16S rRNA-specific primers. Representative data from three experiments are shown.
Kinase activity of PknG from M. smegmatis
In several non- or low-virulent bacteria, virulence genes have been lost through deletion or acquisition of mutations that either prevent the expression of a full-length protein or give rise to an inactive product (Denecker et al., 2002; Higashi et al., 2002; Roche et al., 2005). To determine whether the M. smegmatis open reading frame encodes a functional kinase, as is predicted by its sequence, His-tagged versions of M. smegmatis PknG as well as M. bovis BCG PknG, which is identical to M. tuberculosis PknG (Walburger et al., 2004), were expressed in Escherichia coli, purified and their kinase activity was analysed. As shown in Fig. 2, M. smegmatis PknG was phosphorylated to a similar degree as M. bovis BCG PknG. Furthermore, inclusion of the PknG specific inhibitor AX20017 (Walburger et al., 2004; Scherr et al., 2007) blocked autophosphorylation of both PknGs. We conclude that the M. smegmatis pknG gene encodes a orthologue of M. bovis BCG PknG that is an enzymatically active serine/threonine kinase.
Fig. 2.
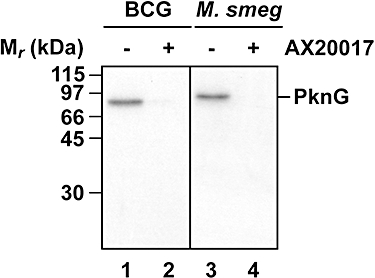
Kinase activity of PknG. The kinase activity of PknG was determined by incubation of 0.1 μg of purified M. bovis BCG and M. smegmatis His-PknG in the absence or presence of the specific PknG inhibitor AX20017 (1 mM) for 30 min at 37°C in a kinase buffer (25 mM Tris/HCl pH 7.4, 2 mM MnCl2, 1 mM DTT and 3.7 × 105 Bq [γ-32P]-ATP). The reactions were terminated by the addition of SDS sample buffer and proteins were separated by 10% SDS-PAGE, fixed, dried and autoradiographed to determine the phosphorylation of the PknGs. Results are representative data from one experiment.
In vivo function of M. smegmatis PknG
We then investigated whether M. smegmatis PknG was able to carry out the same in vivo function as PknG of pathogenic mycobacteria, i.e. in blocking the fusion of mycobacterial phagosomes with lysosomes. For this, a PknG knockout of M. bovis BCG was transformed with the plasmids pMV361-pknGsmeg or pMV361-pknGBCG, in which PknG expression was directed by the mycobacterial hsp60 promoter and translational initiation sequences. The intracellular trafficking of the transformants was subsequently analysed after infection of bone marrow-derived macrophages (Fig. 3). As observed previously, M. bovis BCGΔpknG colocalized predominantly with the lysosomal marker LAMP1, while wild-type mycobacteria largely remained in LAMP1-negative vacuoles (Walburger et al., 2004). Importantly, this defect of the mutant was complemented with either pMV361-pknGBCG (Houben et al., 2006) or pMV361-pknGsmeg. We conclude that PknG from M. smegmatis is not only an enzymatically active serine/threonine kinase, but is also able to carry out the same biological function as PknG of M. bovis BCG in blocking lysosomal delivery of mycobacteria.
Fig. 3.
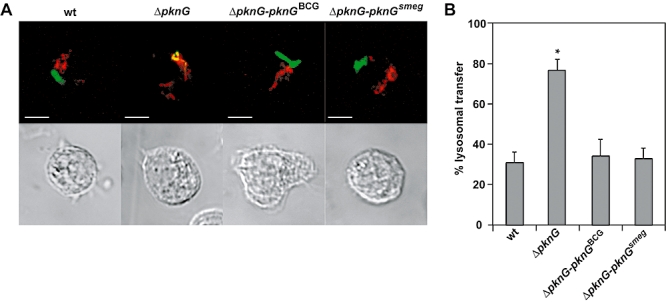
Subcellular localization of M. bovis BCG in the absence or presence of PknG. A. Bone marrow-derived macrophages were infected with M. bovis BCG (wt), M. bovis BCGΔpknG (ΔpknG), M. bovis BCGΔpknG-pMV361-pknGBCG (ΔpknG-pknGBCG) or M. bovis BCGΔpknG-pMV361-pknGsmeg (ΔpknG-pknGsmeg) for 1 h, followed by a 2 h chase, fixed, permeabilized, and immunodecorated with antibodies raised against LAMP1 (rat) and M. bovis (rabbit). Anti-rat Alexa Fluor 568 (red) and anti-rabbit Alexa Fluor 488 (green) were used as secondary antibodies. Scale bar, 5 μm. B. For quantifications, cells containing mycobacteria in LAMP-positive vacuoles were scored. Results are mean values (± SD) from three independent experiments (50 mycobacteria containing vacuoles were scored per experiment). Compared with M. bovis BCG (wt) only non-complemented M. bovis BCGΔpknG (ΔpknG) colocalized significantly more with LAMP1 (*P < 0.05).
Expression of M. smegmatis PknG during in vitro growth
Initial experiments to investigate the expression of chromosomal M. smegmatis pknG by immunoblotting using antibodies against M. bovis BCG did not detect any endogenous PknG in M. smegmatis (Walburger et al., 2004). To determine whether this represented specificity of the antibody for M. bovis BCG PknG, M. smegmatis was transformed with the plasmids pMV361-pknGsmeg or pMV361-pknGBCG. Exponentially growing cultures of these M. smegmatis transformants and M. bovis BCG cultures were analysed by Western blotting using an antiserum raised against PknGBCG (Fig. 4A). PknGsmeg expressed in M. smegmatis from pMV361 was readily recognized by the PknG antibodies (lane 10), to a similar degree as PknG from M. bovis BCG expressed endogenously (lane 6) or in M. smegmatis from pMV361-pknGBCG (lane 9). Furthermore, recombinant PknG from M. bovis BCG and M. smegmatis that was produced in E. coli and purified generated comparable signals after immunoblotting (not shown). Therefore, the failure to detect PknG in M. smegmatis was not due to a defect in the cross-reactivity of the PknGBCG-specific antisera.
Fig. 4.
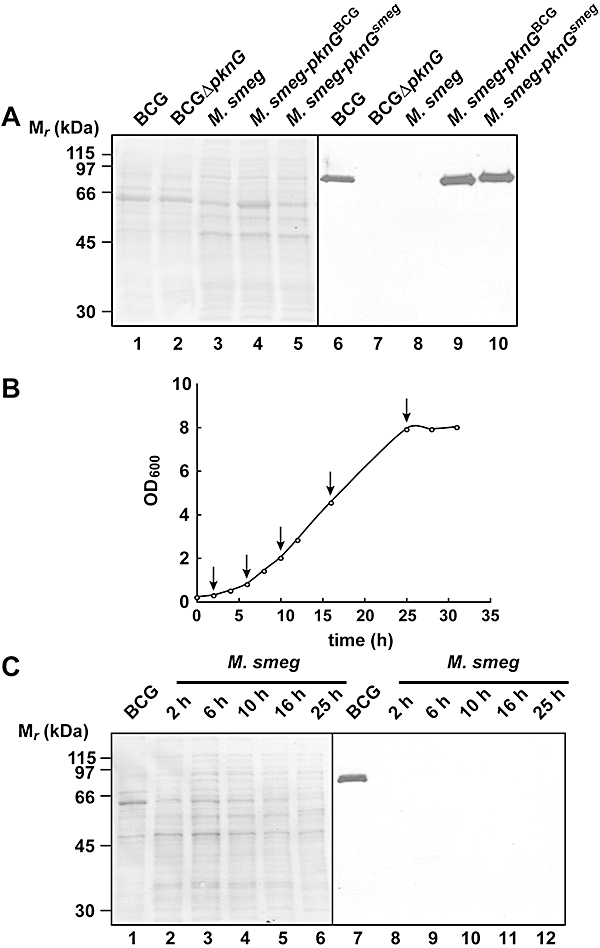
Expression of M. smegmatis PknG during in vitro growth. A. Lysates from M. bovis BCG (lane 1), M. bovis BCGΔpknG (lane 2), M. smegmatis (lane 3), M. smegmatis-pknGBCG (lane 4), M. smegmatis-pknGsmeg (lane 5) were separated on a 10% SDS-PAGE gel and immunoblotted using anti-PknGBCG antiserum (right). The total protein pattern was analysed by Ponceau Red staining (left). B and C. M. smegmatis was grown in 7H9-OADC medium until stationary phase. Bacterial samples were collected at different time points (arrows in B) and analysed for the presence of PknG by immunoblotting as under A (C). BCG lysate was loaded in parallel as a positive control (lane 1). Again, the total protein pattern was analysed by Ponceau Red staining (left). Results are representative data from one experiment.
To explore the possibility that expression of PknG in M. smegmatis was growth phase dependent, an overnight culture of M. smegmatis was diluted to OD600 0.1 and grown until the culture reached stationary phase (Fig. 4B). Samples were taken for immunoblotting of bacterial lysates at different time points during growth. As seen in Fig. 4C, no PknG could be detected using anti-PknGBCG at any stage during growth of M. smegmatis, showing that PknG expression in M. smegmatis is not induced at a particular growth phase.
Expression of M. smegmatis PknG after macrophage infection
Although PknG is not detected in M. smegmatis growing in vitro, it is possible that expression is upregulated when M. smegmatis is phagocytosed by macrophages. To analyse this, bone marrow-derived macrophages were infected with either M. smegmatis wild type or M. smegmatis-expressing PknGBCG from pMV361. After 16 h of infection, cells were harvested, homogenized and a post-nuclear supernatant was prepared and separated by SDS-PAGE followed by immunoblotting. As can be seen in Fig. 5, while PknG was readily observed in macrophages that had been infected with M. smegmatis expressing PknGBCG controlled by the hsp60 promoter, no PknG was detected in cells that had phagocytosed M. smegmatis. We therefore conclude that PknG is not upregulated in M. smegmatis upon infection of macrophages.
Fig. 5.
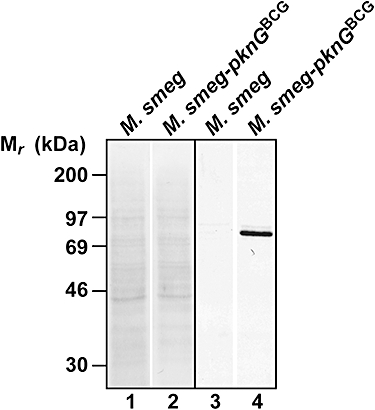
Expression of M. smegmatis PknG during macrophage infection. PknG immunoblot of macrophages infected for 16 h with M. smegmatis or M. smegmatis expressing PknGBCG. After infection, cells were harvested, homogenized and a post-nuclear supernatant was submitted to immunoblotting (lanes 3 and 4). The total protein pattern was analysed by Ponceau Red staining (lanes 1 and 2). Results are representative data from one experiment.
Selected ion monitoring analysis of total M. smegmatis lysates
Finally, a more sensitive mass spectrometry-based approach was adopted to detect PknG in M. smegmatis. Selected ion monitoring analysis (SIM) of defined peptide masses was employed to analyse the presence of PknG-derived peptides in M. smegmatis lysates. Total proteins from M. smegmatis and M. smegmatis transformed with pMV361-pknGsmeg were separated by SDS-PAGE. Proteins between 65 and 115 kDa were excised from the gel (Fig. 6A), subjected to protease digestion and analysed by mass spectrometry. Selected ion monitoring allowed the detection of two selected peptides 51 times in the wild-type M. smegmatis sample, while six peptides (see Experimental procedures) were identified 3289 times in a lysate from M. smegmatis expressing PknGsmegin trans (Fig. 6B). These data, together with the absence of a PknG signal following immunoblotting, show that PknG is translated at extremely low levels in M. smegmatis.
Fig. 6.
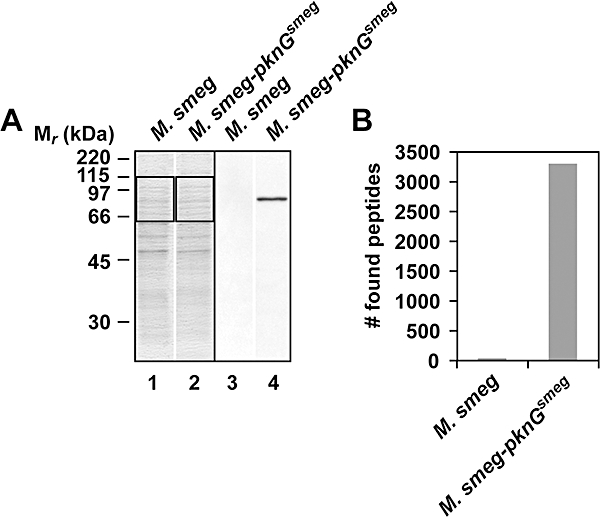
Selected ion monitoring analysis of M. smegmatis lysates. A. Lysates (10 μg) from M. smegmatis and M. smegmatis-pknGsmeg were separated on a 10% SDS-PAGE gel, followed by Coomassie Blue staining (lanes 1 and 2) or immunoblotting using anti-PknGBCG antiserum (lanes 3 and 4). B. Thirty micrograms of the same lysates as analysed under (A) were separated on a 10% SDS-PAGE gel and stained with Colloidal Blue. The region between 65 and 115 kDa was excised (indicated by the rectangles in A), digested with trypsin and the obtained peptides were identified using selected ion monitoring (see Experimental procedures). Representative data from two independent experiments are shown.
Transcription of pknG from M. smegmatis
The extremely low levels of PknG from M. smegmatis could be explained by inefficient transcription of the pknG gene. To analyse pknG transcription, total RNA was isolated from M. smegmatis or M. bovis BCG and analysed for the presence of pknG transcripts by RT-PCR (Fig. 7A). Strikingly, a specific band could be amplified by RT-PCR using M. smegmatis as well as M. bovis BCG RNA with pknG-specific primers (only after reverse transcription), indicating the presence of pknG RNA in M. smegmatis. Sequencing confirmed that this product was the anticipated amplified fragment of the PknG transcripts (not shown).
Fig. 7.
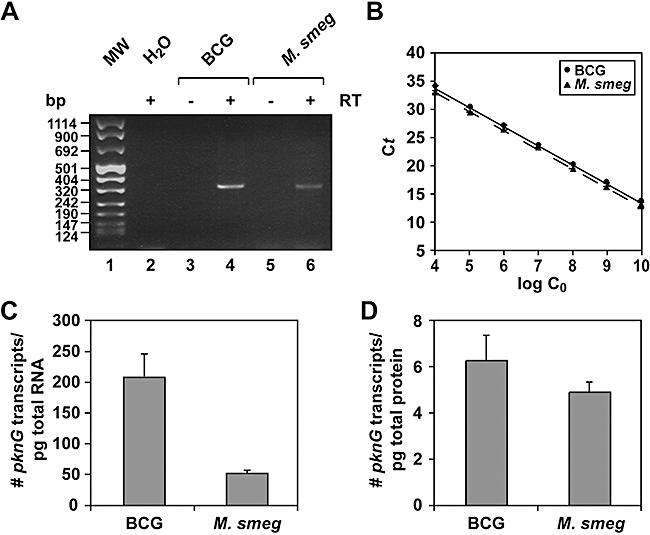
Analysis of pknG transcription in M. smegmatis. A. cDNA was prepared from total M. bovis BCG and M. smegmatis RNA by using reverse transcriptase and random primers. cDNA of pknG was amplified using gene-specific primers. As a control the same reactions were carried out without reverse transcriptase (−RT) or without RNA (H2O). B. Standard curves for M. bovis BCG and M. smegmatis pknG TaqMan primer/probe sets were constructed from serial dilutions of known quantities of in vitro synthesized transcripts. Threshold cycle (Ct) values were plotted against copies of transcripts. C and D. PknG mRNA expression in M. bovis BCG and M. smegmatis per pg of total RNA (C) and per pg of total protein (D). Copies pg−1 total RNA were calculated from the standard curves shown in (B). These values were subsequently corrected by the RNA : protein ratio (see Experimental procedures) to calculate the number of PknG mRNA copies pg−1 total protein. Results are mean values (±SD) from three RNA isolations. M. smegmatis PknG mRNA levels were not significantly different from M. bovis BCG PknG mRNA levels (P > 0.05).
To accurately measure the levels of PknG transcripts in M. bovis BCG and M. smegmatis, quantitative real-time RT-PCR was carried out using in vitro synthesized PknGBCG and PknGsmeg transcripts as standards (Fig. 7B). Results are expressed relative to total cellular RNA or protein. This allowed comparison of the differences in pknG transcription relative to stable RNA (rRNA, tRNA) or mRNA translation products in these cells (see Experimental procedures). When normalized to stable RNA, M. bovis BCG had a four times higher level of pknG transcript (Fig. 7C). When normalized to cellular protein however, pknG mRNA levels in M. smegmatis were not significantly different (P = 0.23) compared with M. bovis BCG (Fig. 7D). We conclude that the sharply decreased levels of PknG in M. smegmatis as analysed by immunoblotting or MS analyses are mainly due to a reduced translational efficiency.
Regulation of pknG translation
To address whether the severe difference in the translational efficiency of PknG between BCG and M. smegmatis could be due to regulatory elements upstream of the two pknG genes, fusion constructs were produced. The ribosome binding site (RBS) in the hsp60 promoter of pMV361-pknGBCG and pMV361-pknGsmeg was replaced by the upstream region of the corresponding pknG genes. Since pknG has been proposed to be the last gene of an operon of three genes, the two upstream genes of pknG were introduced (Fig. 8A; see also Experimental procedures). In addition, the upstream regions were exchanged between the constructs; the upstream region of M. smegmatis pknG was cloned upstream of BCG pknG and visa versa. The constructs were subsequently introduced in the ‘PknG minus’ background of M. bovis BCGΔpknG and PknG expression was analysed via Western blotting and RT-PCR (Fig. 8B and C).
Exponentially growing cultures of the M. bovis BCG transformants were analysed by Western blotting using an antiserum raised against PknGBCG (Fig. 8B). PknGBCG translationally controlled by its own upstream region in pMV361 was clearly expressed in M. bovis BCGΔpknG (Fig. 8B, lane 9), and the product migrated similarly to PknG from M. bovis BCG expressed endogenously (Fig. 8B, lane 7). In contrast, no PknG signal was detectable after introduction of the construct containing M. smegmatis pknG with its own upstream region in M. bovis BCGΔpknG (lane 10). This shows that the low translation of PknGsmeg is not related to any M. smegmatis-specific factors. However, introduction of the construct, in which the M. smegmatis gene was fused to the M. bovis BCG upstream region, resulted in clear expression of M. smegmatis PknG (lane 11), while after introduction of the construct containing BCG pknG with the M. smegmatis upstream region no PknG signal could be detected (lane 12). From this we conclude that the upstream region of M. smegmatis pknG is solely responsible for the virtual lack of expression of the M. smegmatis gene.
To verify that the lack of expression of the pknG genes fused to the M. smegmatis upstream region occurred on a translational level, total RNA was isolated of M. bovis BCG wt and the various BCG transformants and subsequently analysed for the presence of pknG transcripts by RT-PCR (Fig. 8C). A specific band could be amplified by RT-PCR only in the presence of the reverse trancriptase (RT) using RNA from M. bovis BCG wt and all M. bovis BCGΔpknG tranformants, except M. bovis BCGΔpknG that was transformed with the empty vector (lane 5). We can therefore conclude that the lack of PknG expression in M. bovis BCGΔpknG transformed with pknG genes fused to the upstream region of M. smegmatis is caused by a expression failure on a translational level.
Discussion
Pathogenic mycobacteria survive within macrophages by preventing fusion of mycobacterial phagosomes with lysosomes (Armstrong and Hart, 1971; Russell, 2001; Houben et al., 2006). A crucial molecule involved in the modulation of phagosome lysosome fusion is PknG, a eukaryotic-like mycobacterial serine/threonine kinase (Walburger et al., 2004). Notably, while PknG functions as an important virulence factor in pathogenic mycobacteria, a pknG gene is also found in a saprophytic species, M. smegmatis. Here we show that when expressed from expression vectors, M. smegmatis pknG encodes an enzymatically active serine/threonine kinase that provides normal biological activity in the survival of mycobacteria inside macrophages. However, virtually no translation product of endogenous pknG was detected either when M. smegmatis was grown in vitro or within macrophages under the conditions examined here. Quantitative transcriptional assays showed that the sharply reduced expression in M. smegmatis could not be attributed to mRNA levels, showing that PknG expression is blocked on a post-transcriptional level in this non-pathogenic mycobacterium.
Replacement of the upstream regions of M. smegmatis and M. bovis BCG pknG by vector-encoded transcriptional and translational expression sequences resulted in similar levels of PknGsmeg and PknGBCG expression in M. smegmatis. This led to the conclusion that the differences in accumulation of pknG gene products could be caused neither by post-translational mechanisms such as host-specific proteases, nor by the codon usage within the pknG genes. The latter is in agreement with the observation that the codon biases of M. smegmatis and M. bovis BCG pknG, represented by their codon adaptation indices (de Miranda et al., 2000), do not significantly differ (not shown). However, replacing the vector-encoded translational expression sequences by their own or each others upstream regions of the pknG genes, while keeping the transcriptional sequences of the vector intact, clearly showed that elements in the upstream regions of the genes cause the differences in translation efficiencies. A typical upstream translational regulatory element is the ribosome binding site and it is a possibility that the ribosome binding site of M. smegmatis pknG is very inefficient. Alternatively, recognition of the upstream region by regulatory RNA, proteins or metabolites might cause the differences in translational efficiencies between mycobacterial species.
Western blot analysis for the expression of PknG in a wide range of mycobacterial species grown under similar growth conditions revealed that only some of the slow-growing pathogenic species, mainly the members of the M. tuberculosis complex and species closely related to the M. tuberculosis complex, expresses PknG, while none of the analysed non-pathogenic, fast-growing species showed PknG expression (Fig. 9). Notably, all mycobacterial genomes, of which sequence data are available at this moment (from both pathogenic and non-pathogenic species), contain a pknG gene. Therefore, pknG might be conserved in all mycobacterial species, which implies that not only in M. smegmatis, but in more mycobacterial species PknG expression is suppressed.
Fig. 9.
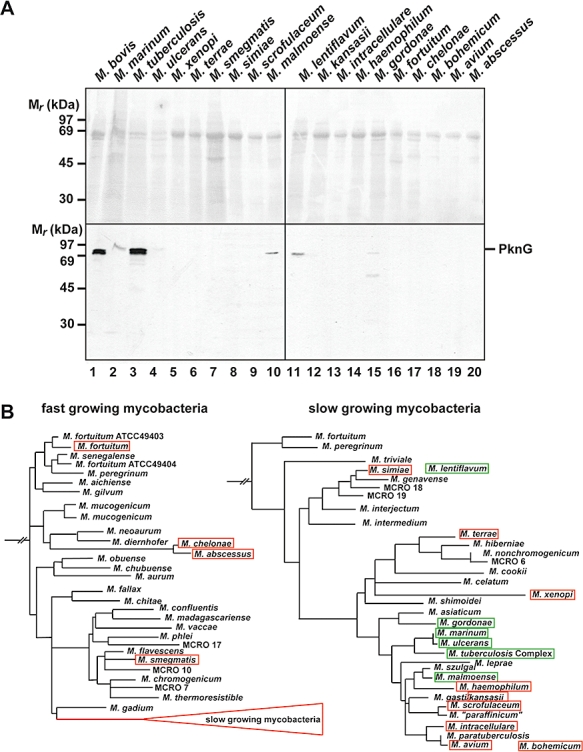
Analysis of pknG expression in a wide range of mycobacterial species. A. Lysates of various mycobacterial species were separated on a 10% SDS-PAGE gel and immunoblotted using anti-PknG antiserum (bottom). The total protein pattern was analysed by Ponceau Red staining (top). For sources and growth conditions of the mycobacterial species see Table S2. B. Phylogenic tree of mycobacteria based on 16S rRNA gene sequences. Species that show a PknG signal in (A) are boxed in green, the ones that do not show expression are boxed in red. Tree adapted from Springer et al. (1996).
The availability of a growing number of mycobacterial genome sequences allows comparisons that may help to identify genes corresponding to strain-specific pathogenicity functions (Cole et al., 2001). Our studies suggest the existence of another subset of genes whose relative levels of expression, and not their presence or absence, defines virulence in mycobacterium. It is important to recognize that the changes on the level of expression that determine pknG activity in different species might be achieved by the evolution of regulatory systems that target subsets of mycobacterial virulence genes. Identification and analysis of these regulatory systems may provide new strategies to combat important mycobacterial diseases such as tuberculosis and leprosy.
Experimental procedures
Cell lines, bacterial strains and antibodies
Mycobacterium bovis BCG Pasteur was provided by the Institut Pasteur (Paris, France) and M. smegmatis mc2155 was from ATCC (ATCC 700084). Disruption of pknG in the M. bovis BCG Pasteur was carried out as described (Walburger et al., 2004). Mycobacteria were grown in 7H9 mycobacterial medium supplemented with 10% OADC Middlebrook supplement (Difco Laboratories). Bone marrow-derived macrophage isolation from C57/BL6 mice, anti-LAMP1 monoclonal antibodies, secondary reagents and the PknG inhibitor AX20017 have been described (Ferrari et al., 1999; Gatfield and Pieters, 2000; Walburger et al., 2004). Antibodies against PknG were raised by Eurogentec in New Zealand white rabbits against His-tagged recombinant PknG that was expressed in E. coli and purified as described (Walburger et al., 2004). PknG-specific IgGs were further purified using a HiTrap Protein A column, followed by a HiTrap NHS-activated column coupled with His-tagged recombinant PknG (both supplied by GE Healthcare Bio-Sciences). Polyclonal rabbit antibodies against M. bovis were purchased from Dako Corporation.
Cloning of PknG expression vectors
Anchored primers (EcoRI–NdeI for the 5′ primer and NdeI–HindIII for the 3′ primer) were used to amplify pknG from M. smegmatis genomic DNA by PCR, cloned in pGEM-T-Easy (Promega) and sequenced (Applied Biosystems). After sequencing, the genes were cloned as EcoRI–HindIII fragments in pMV361 (Stover et al., 1991) and in pBAD/His B (Invitrogen Life Technologies). The resulting plasmids were designated pMV361-pknGsmeg and pBad-PknGsmeg. An anchored 5′ primer (AclI-BCG0451c.fw containing an AclI site; see Table S1) and a 3′ primer recognizing a sequence downstream of the AclI site in M. bovis BCG pknG (BCG-Pk-EcoRV.rev) were used to amplify pknG together with 2345 bp upstream region (containing BCG0431c and glnH including 40 bp upstream of BCG0431c) from M. bovis BCG genomic DNA by PCR and cloned AclI–AclI in pMV361-pknGBCG (Walburger et al., 2004), designated pMV361-BCG-pknGBCG (see also Fig. 8A). Since the AclI site in the hsp60 promoter region of pMV361 is located upstream of the ribosome binding site, but downstream of the transcription start (Stover et al., 1991), only the transcription start of hsp60 is left in this construct. Similarly, an anchored 5′ primer with an AclI site (AclI-MSMEG_0788.fw) was used together with a 3′ primer recognizing the XmnI site in M. smegmatis pknG (Smeg-XmnI.rev) to amplify M. smegmatis pknG together with the 2423 bp upstream region (containing MSMEG_0788, MSMEG_0787 and 40 bp upstream of MSMEG_0788) from M. smegmatis genomic DNA and cloned AclI–XmnI into pMV361-pknGsmeg (this study), resulting in pMV361-smeg-pknGsmeg. In addition, the upstream regions of the two genes were exchanged by a nested PCR approach (by using the 5′ anchor primers, the 3′ AclI or XmnI primer and internal overlapping primers; for primer sequences see Table S1), resulting in pMV361-BCG-pknGsmeg for pknGsmeg with the upstream region of pknGBCG and pMV361-smeg-pknGBCG for pknGBCG together with the upstream region of pknGsmeg. All constructs were sequenced (Applied Biosystems) and electroporated into M. smegmatis mc2155 and/or M. bovis BCGΔpknG Pasteur (Parish and Stoker, 1998).
Expression and purification of M. smegmatis PknG
For expression and purification of M. smegmatis PknG, E. coli strain BL21 was transformed with pBAD-PknGsmeg. Transformants were grown in LB medium containing 100 μg ml−1 ampicillin and 0.2% glucose at 37°C until the OD600 reached 0.45. Bacteria were pelleted (4200 g for 10 min) and re-suspended in LB medium containing 100 μg ml−1 ampicillin and 0.2% arabinose. Bacteria were grown for another 4 h at 37°C and harvested (4200 g for 10 min at 4°C). (His)6-PknGsmeg was purified via a Ni2+-loaded HisTrap chelating column from GE Healthcare Bio-Sciences using an imidazole gradient as described (Walburger et al., 2004).
Biochemical methods
Homogenization of mycobacterial lysates was performed and kinase activity of purified kinase was measured as described (Walburger et al., 2004).
RT-PCR and qRT-PCR
Isolation of RNA samples from M. bovis BCG and M. smegmatis, DNase treatments, cDNA synthesis and the amplification of pknG cDNA has been described (Nguyen et al., 2005). The protein concentration of the mycobacterial lysates and the amount of RNA isolated from these lysates were determined using a Bradford protein assay and the NanoDrop spectrophotometer respectively. Protein and RNA concentrations were additionally verified on SDS-PAGE and agarose gels respectively. Amplification of pknG cDNA from M. bovis BCG and M. smegmatis was carried out using the same primers (RT-PknG.fw and RT-PknG.rev in Table S1). As a control, transcription of the 16S rRNA gene (16S-RNA.fw and 16S-RNA.rev in Table S1) was analysed as well.
The same RNA samples were used for quantitative RT-PCR. In addition, M. bovis BCG and M. smegmatis PknG transcripts were synthesized from HindIII linearized pET15b-PknGBCG (Walburger et al., 2004) and pGEM-T-Easy-PknGsmeg using the T7 RiboMAX express large-scale RNA production system (Promega) and used as standards. Quantitative, real-time, one-step RT-PCR was carried out using the Applied Biosystems 7500 Sequence Detection Systems (SDS). Sets of primer pairs and a TaqMAN probe (Table S1) were designed and qRT-PCR was performed using the QuantiTect probe RT-PCR kit (QIAGEN). Fluorescence data were analysed using the Applied Biosystems Sequence Detection software version 1.2.3. Each reaction was run in duplicates.
Sample preparation for mass spectrometric analysis
The region between 65 and 100 kDa of each entire lane was excised followed by in-gel trypsin digestion as described (Granvogl et al., 2006).
Nano-LC-IT-SIM-MS analysis
The resulting peptides from each gel piece were analysed by liquid chromatography (LC) coupled to a LTQ ion trap mass spectrometer (Thermo Electronics) equipped with a nano-LC electrospray ionization source. Peptides were dissolved in buffer A (1% formic acid, 0.5% acetic acid, 0.012% heptafluorobutyric acid) and concentrated and desalted online on a C18 PepMap 100 micro precolumn (5 μm particle size, 300 μm × 1 mm; Dionex Corporation) that was coupled to a self-packed (7 cm, 3 μm; ProntoSil C18-ACE-EPS, Bischoff Chromatography) and pulled (P-2000 laser puller, Sutter Instrument) fused silica capillary (100 μm inner diameter × 365 μm outer diameter). The chromatographic separation was then performed by a 100 min non-linear gradient from 5–55% buffer B (80% acetonitrile/0.5% acetic Acid/0.012% HFBA) with a constant flow rate of 0.20 μl min−1. The mass spectrometric data acquisition was performed with a survey scan (450–1500 m/z) followed by five selected ion monitoring scans (SIM). The selected masses correspond to the following peptides: LGGGLVEIPR [MH2+ 505.8], LIDLGAVSR [MH2+ 472.28], TGPTVATDIYTVGR [MH2+ 725.88], LTSAVTLLSGR [MH2+ 559.33], TLAALTLDLPTR [MH2+ 642.88] and ALLDLGDVAK [MH2+ 507.28]. The collision energy was set to 35%.
Database searching for protein identification
SEQUEST (Bioworks, Thermo Electron Corporation) was used to search the SWISSPROT database (UniprotKB/Swiss-Prot) supplemented with the corresponding pknG sequence of M. smegmatis for peptide sequence and protein identification. Search parameters included differential mass modification to methionine due to possible oxidation and static mass modification to cysteine due to alkylation by iodoacetamide. Furthermore, one missed cleavage of trypsin was accepted.
Statistical analysis
Data were statistically analysed using the Student's t-test and differences were considered significant if the P-value was < 0.05.
Acknowledgments
We thank Roland Brosch (Institute Pasteur) for kindly providing M. bovis BCG Pasteur and Gerd Pluschke and Thomas Bodmer (Swiss Tropical instituite, Basel, Switzerland) for providing mycobacterial lysates. This work was supported by FEBS fellowships (to E.N.G.H. and A.W.) and grants from the World Health Organization, the Swiss National Science Foundation, the Swiss Life Jubileum Foundation and the Olga Mayenfisch Foundation (to J.P.).
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Armstrong JA, Hart PD. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med. 1971;134:713–740. doi: 10.1084/jem.134.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, et al. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–1011. doi: 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- Cosma CL, Sherman DR, Ramakrishnan L. The secret lives of the pathogenic mycobacteria. Annu Rev Microbiol. 2003;57:641–676. doi: 10.1146/annurev.micro.57.030502.091033. [DOI] [PubMed] [Google Scholar]
- Cowley S, Ko M, Pick N, Chow R, Downing KJ, Gordhan BG, et al. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol Microbiol. 2004;52:1691–1702. doi: 10.1111/j.1365-2958.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- Denecker G, Totemeyer S, Mota LJ, Troisfontaines P, Lambermont I, Youta C, et al. Effect of low- and high-virulence Yersinia enterocolitica strains on the inflammatory response of human umbilical vein endothelial cells. Infect Immun. 2002;70:3510–3520. doi: 10.1128/IAI.70.7.3510-3520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari G, Langen H, Naito M, Pieters J. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell. 1999;97:435–447. doi: 10.1016/s0092-8674(00)80754-0. [DOI] [PubMed] [Google Scholar]
- Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 2000;288:1647–1650. doi: 10.1126/science.288.5471.1647. [DOI] [PubMed] [Google Scholar]
- Granvogl B, Reisinger V, Eichacker LA. Mapping the proteome of thylakoid membranes by de novo sequencing of intermembrane peptide domains. Proteomics. 2006;6:3681–3695. doi: 10.1002/pmic.200500924. [DOI] [PubMed] [Google Scholar]
- Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci USA. 2002;99:14428–14433. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben ENG, Nguyen L, Pieters J. Interaction of pathogenic mycobacteria with the host immune system. Curr Opin Microbiol. 2006;9:76–85. doi: 10.1016/j.mib.2005.12.014. [DOI] [PubMed] [Google Scholar]
- de Miranda AB, Alvarez-Valin F, Jabbari K, Degrave WM, Bernardi G. Gene expression, amino acid conservation, and hydrophobicity are the main factors shaping codon preferences in Mycobacterium tuberculosis and Mycobacterium leprae. J Mol Evol. 2000;50:45–55. doi: 10.1007/s002399910006. [DOI] [PubMed] [Google Scholar]
- Nguyen L, Walburger A, Houben ENG, Koul A, Muller S, Morbitzer M, et al. Role of protein kinase G in growth and glutamine metabolism of Mycobacterium bovis BCG. J Bacteriol. 2005;187:5852–5856. doi: 10.1128/JB.187.16.5852-5856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niebisch A, Kabus A, Schultz C, Weil B, Bott M. Corynebacterial protein kinase G controls 2-oxoglutarate dehydrogenase activity via the phosphorylation status of the OdhI protein. J Biol Chem. 2006;281:12300–12307. doi: 10.1074/jbc.M512515200. [DOI] [PubMed] [Google Scholar]
- Parish T, Stoker NG. Electroporation of mycobacteria. Methods Mol Biol. 1998;101:129–144. doi: 10.1385/0-89603-471-2:129. [DOI] [PubMed] [Google Scholar]
- Roche SM, Gracieux P, Milohanic E, Albert I, Virlogeux-Payant I, Temoin S, et al. Investigation of specific substitutions in virulence genes characterizing phenotypic groups of low-virulence field strains of Listeria monocytogenes. Appl Environ Microbiol. 2005;71:6039–6048. doi: 10.1128/AEM.71.10.6039-6048.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- Scherr N, Honnappa S, Kunz G, Mueller P, Jayachandran R, Winkler F, et al. Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2007;104:12151–12156. doi: 10.1073/pnas.0702842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinnick TM, Good RC. Mycobacterial taxonomy. Eur J Clin Microbiol Infect Dis. 1994;13:884–901. doi: 10.1007/BF02111489. [DOI] [PubMed] [Google Scholar]
- Springer B, Stockman L, Teschner K, Roberts GD, Böttger EC. Two-laboratory collaborative study on identification of mycobacteria: molecular versus phenotypic methods. J Clin Microbiol. 1996;34:296–303. doi: 10.1128/jcm.34.2.296-303.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, et al. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- Vergne I, Chua J, Singh SB, Deretic V. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol. 2004;20:367–394. doi: 10.1146/annurev.cellbio.20.010403.114015. [DOI] [PubMed] [Google Scholar]
- Walburger A, Koul A, Ferrari G, Nguyen L, Prescianotto-Baschong C, Huygen K, et al. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science. 2004;304:1800–1804. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.