

Abstract
The bifunctional trypanothione synthetase-amidase (TRYS) comprises two structurally distinct catalytic domains for synthesis and hydrolysis of trypanothione (N1,N8-bis(glutathionyl)spermidine). This unique dithiol plays a pivotal role in thiol-redox homeostasis and in defence against chemical and oxidative stress in trypanosomatids. A tetracycline-dependent conditional double knockout of TRYS (cDKO) was generated in bloodstream Trypanosoma brucei. Culture of cDKO parasites without tetracycline induction resulted in loss of trypanothione and accumulation of glutathione, followed by growth inhibition and cell lysis after 6 days. In the absence of inducer, cDKO cells were unable to infect mice, confirming that this enzyme is essential for virulence in vivo as well as in vitro. To establish whether both enzymatic functions were essential, an amidase-dead mutant cDKO line was generated. In the presence of inducer, this line showed decreased growth in vitro and decreased virulence in vivo, indicating that the amidase function is not absolutely required for viability. The druggability of TRYS was assessed using a potent small molecule inhibitor developed in our laboratory. Growth inhibition correlated in rank order cDKO, single KO, wild-type and overexpressing lines and produced the predicted biochemical phenotype. The synthetase function of TRYS is thus unequivocally validated as a drug target by both chemical and genetic methods.
Introduction
The protozoan parasite Trypanosoma brucei is the causative agent of human African trypanosomiasis (HAT), commonly known as sleeping sickness. This disease, thought to be largely controlled in the 1960s, has re-emerged as a major threat to human health due to a number of factors including lack of financial resources, conflict in affected countries and failure to adequately monitor infection. Currently, the World Health Organisation reports that over 400 000 people are infected with HAT, resulting in an annual death toll of more than 50 000 (Stuart et al., 2008). In the absence of an effective vaccine, treatment is solely dependent upon a pitifully small repertoire of drugs which suffer from a number of problems including severe toxic side-effects (Fairlamb, 2003) and acquired drug resistance (Barrett and Fairlamb, 1999). To compound these difficulties, many of the current chemotherapeutic treatments also require lengthy periods of hospitalization and are prohibitively expensive (Stuart et al., 2008). With no new drugs for the treatment of HAT in the pipeline, a concerted effort is being made to identify, characterize and validate novel molecular targets urgently required for drug discovery.
In the search for anti-parasitic drug targets, metabolic pathways that are both essential for parasite survival and absent from the host are logical starting points (El Sayed et al., 2005). One such pathway is the thiol metabolism of trypanosomes and Leishmania. These parasites are uniquely dependent upon trypanothione (N1,N8-bis(glutathionyl)spermidine, T[SH]2) as their principal thiol, in contrast to most other organisms (including their mammalian hosts) which utilize glutathione (γ-l-glutamyl-l-cysteinylglycine, GSH) (Fairlamb et al., 1985). This dithiol is primarily responsible for the maintenance of thiol-redox homeostasis within trypanosomatids, and is involved in a number of critical cellular processes including deoxyribonucleotide synthesis (Dormeyer et al., 2001), and defence against oxidative stress (Ariyanayagam and Fairlamb, 2001) and xenobiotics (Vickers et al., 2004). Trypanothione has also been implicated in the mode of action as well as the mechanism of resistance of antimonial drugs in Leishmania spp. (Mukhopadhyay et al., 1996; Wyllie et al., 2004; Croft et al., 2006) and in defence against chemical and oxidant stress induced by arsenicals and nifurtimox in T. brucei (Fairlamb et al., 1989; Ariyanayagam et al., 2005; Alibu et al., 2006). Therefore, in an attempt to exploit this essential and unique metabolite, T[SH]2-dependent and biosynthetic enzymes have become a major focus for drug discovery.
The pivotal role of T[SH]2 metabolism in the viability and virulence of trypanosomatids has been unequivocally demonstrated using genetic techniques such as classical gene knockout and RNA interference (RNAi). Trypanothione-dependent enzymes that have been validated as potential drug targets using these methodologies include: trypanothione reductase (Tovar et al., 1998; Krieger et al., 2000), tryparedoxin (Wilkinson et al., 2003) and tryparedoxin peroxidase (Wilkinson et al., 2003). Unsurprisingly, the enzyme responsible for the biosynthesis of T[SH]2, the bifunctional trypanothione synthetase-amidase (TRYS), has also been confirmed as essential for the viability and proliferation of T. brucei bloodstream (Comini et al., 2004) and procyclic forms (Ariyanayagam et al., 2005) by RNAi. In pathogenic trypanosomatids, TRYS catalyses the synthesis of T[SH]2 from GSH and spermidine in a two-step, ATP-dependent reaction via a glutathionylspermidine intermediate. In addition to this role in biosynthesis, the N-terminal domain of TRYS also functions as an amidase, hydrolysing T[SH]2 to GSH and spermidine, again via glutathionylspermidine. The biological significance of TRYS-amidase, classified as a cysteine–histidine-dependent aminohydrolase/peptidase amidase (Bateman and Rawlings, 2003; Rigden et al., 2003), is not fully understood. However, it has been suggested that the conflicting biosynthetic and hydrolytic functions of this enzyme may enable the parasite to respond to fluctuating cellular requirements for both T[SH]2 and polyamines, essential for proliferation and differentiation (Fairlamb and Cerami, 1992; Bacchi and Yarlett, 1995).
In the current study, we assessed the essentiality of TRYS by classical gene replacement to fulfil the stringent target validation criteria of the University of Dundee Drug Discovery Unit (Frearson et al., 2007). Gene replacement is the preferred method of genetic validation because ‘off-target’ effects cannot be ruled out using RNAi. Moreover, target knock-down by RNAi does not permit independent analysis of bifunctional enzymes such as DHFR-TS (Sienkiewicz et al., 2008) or TRYS. Using a combination of classical gene replacement, mutagenesis and chemical intervention methods, we dissect the opposing synthetic and hydrolytic functions of TRYS in order to independently study their respective roles in T. brucei cell virulence and viability.
Results and discussion
Generation of TRYS conditional null mutants
To date, the essentiality of T. brucei trypanothione synthetase (TRYS) has only been demonstrated by RNAi (Comini et al., 2004; Ariyanayagam et al., 2005), a methodology that lacks the robustness of classical gene replacement. With this in mind, we sought to definitively classify this enzyme as a drug target in bloodstream trypanosomes by replacing TRYS with drug-resistance genes (Fig. 1A). Previous studies have demonstrated that TRYS is present as a single-copy gene per haploid genome of T. brucei (Oza et al., 2003). The first TRYS gene copy was replaced with the puromycin N-acetyl transferase (PAC) gene by homologous recombination and subsequent selection for puromycin resistance, generating a single knockout (SKO) cell line. Attempts to create a null mutant by directly replacing the second allelic copy with the hygromycin-resistance gene (hygromycin phosphotransferase, HYG) proved unsuccessful, suggesting that TRYS is indeed essential for the viability of T. brucei bloodstream parasites. Consequently, a conditional double knockout (cDKO) cell line was generated by introducing an ectopic and tetracycline-inducible copy of TRYS prior to replacing the second copy with HYG. This ectopic copy of TRYS was inserted into the rDNA locus of the SKO cell line using a pLew 100 vector encoding a blasticidin-resistance gene (BSD) followed by deletion of the second genomic copy with HYG. As the modified T. brucei 427 cell line [wild-type (WT)] used in this study constitutively expresses the T7 RNA polymerase and the tetracycline repressor protein, the resulting cell line was a TRYS conditional null mutant where TRYS expression depends on the presence of tetracycline (cDKO). Southern blot analysis of genomic DNA from cell lines generated at each stage of this process confirmed the validity of the TRYS conditional null mutant (Fig. 1B).
Fig. 1.
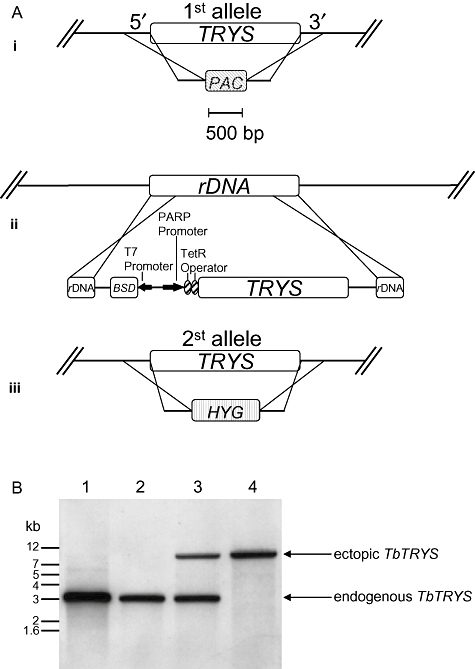
Genotypic analysis of WT, SKO and cDKO cell lines. A. Schematic representation of the stepwise generation of the TRYS cDKO cell line in T. brucei. (i) One allele of TRYS was replaced with the puromycin-resistance gene (PAC) by homologous recombination, generating ΔTRYS::PAC cell line (SKO); (ii) a tetracycline-inducible ectopic copy of TRYS was introduced into the rDNA, generating TRYSTiΔTRYS::PAC cell line; (iii) while tetracycline induces the expression of the ectopic copy, the remaining allele was replaced with a hygromycin resistance gene (HYG) by homologous recombination, resulting in conditional double knockout cell line TRYSTiΔTRYS::PAC/ΔTRYS::HYG (cDKO). B. Confirmation of genotype of T. brucei TRYS conditional double knockout cell line. Southern blot analysis of PstI-digested genomic DNA (∼5 μg) from wild-type T. brucei cells (lane 1), ΔTRYS::PAC (lane 2), TRYSTiΔTRYS::PAC (lane 3) and TRYSTiΔTRYS::PAC/ΔTRYS::HYG (lane 4) cells; the TRYS ORF probe shows allelic TbTRYS at 3 kb and the ectopic copy TbTRYSTi at ∼10 kb.
TRYS is essential in bloodstream T. brucei in vitro
In HMI9-T culture medium, no significant difference could be detected between the growth rates of the WT and SKO cells, with generation times of 9.6 ± 0.1 and 9.5 ± 0.2 h respectively (data not shown). Indeed, the cDKO cell line, grown in the presence of tetracycline, showed no marked growth phenotype with a doubling time of 9.9 ± 0.02 h (Fig. 2A). Following the removal of tetracycline from the medium, cDKO cells continued to grow exponentially for 3 days. Growth slowed by day 4 of culture with all cells finally dying by day 8. To determine the actual point where cDKO parasites grown in the absence of tetracycline completely lose viability, cells were subcultured each day into medium containing tetracycline. Up to the 6th day of culture cells remained viable in the absence of tetracycline; however, beyond this point no viable cDKO cells could be recovered on culture for a further 10 days. Failure of this cell line to grow in the absence of the tetracycline-induced expression of TRYS confirms that this enzyme is essential for bloodstream T. brucei viability. Interestingly, ectopic expression of the L. major TRYS was equally capable of complementing for the loss of endogenous TRYS in the cDKO cell line (Fig. S1). The fact that loss of TRYS activity is trypanocidal rather than cytostatic is highly advantageous from a drug discovery perspective because drug therapy is not dependent on a fully functional immune response (Frearson et al., 2007).
Fig. 2.
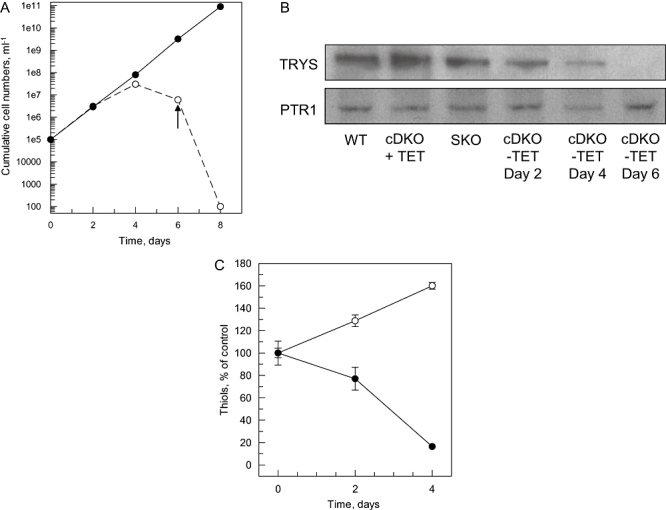
Growth characteristics and biochemical analysis of cDKO cells in vitro. A. The growth of the cDKO cell line in HMI9-T media was monitored in the presence (closed circles) and absence of tetracycline (open circles, dashed line). The arrow indicates the point where the viable cells are no longer recoverable when subcultured for up to 10 days in medium containing tetracycline. B. Immunoblots of cell extracts of WT, SKO and cDKO (plus and minus tetracycline) cells were probed with antiserum to T. brucei TRYS and to T. brucei PTR1 as a control (1 × 107 parasites in each lane). C. Intracellular T[SH]2 (closed circles) and GSH (open circles) levels in cDKO cells following the removal of tetracycline from cultures. Initial levels of T[SH]2 and GSH in untreated cells were 0.42 and 0.54 nmol(108 cells)−1 respectively. Each data point represents the means ± standard deviations from triplicate determinations.
Biochemical analyses of TRYS cDKO cells
The ‘slow death’ phenotype of cDKO cells following the removal of tetracycline may be partly explained by the low turnover of TRYS or its product, T[SH]2. Western blot analysis of whole cell extracts revealed that although the levels of this enzyme declined following the removal of tetracycline, it was not until day 6 that TRYS was no longer detectable (Fig. 2B). This observation suggests that the rate of turnover of TRYS (or T[SH]2) is very low in T. brucei and that TRYS (or T[SH]2) is only removed from the cell by dilution due to cell division in the absence of further protein synthesis. Nevertheless, the death of cDKO cells coinciding with the disappearance of TRYS once again confirms that this enzyme is essential in bloodstream trypanosomes.
The effect of TRYS depletion on intracellular thiols was studied by high-performance liquid chromatography (HPLC). Due to the number of cells required for this analysis, thiols could only be monitored in cultures for 4 days following the removal of tetracycline. The cessation of ectopic TRYS expression within these parasites had a pronounced effect on intracellular thiol levels (Fig. 2C). Glutathione, the substrate of TRYS, accumulated in cDKO cells in the absence of tetracycline, such that after 4 days, levels had reach 160% of those seen in control cells (cDKO cells plus tetracycline). In contrast, T[SH]2 and glutathionylspermidine, the products of this enzyme reaction, fell considerably. Indeed, T[SH]2 levels within these parasites fell to 16.5% of control levels. As 4 day cultures showed only minimally retarded growth in comparison with control cells, it would appear that bloodstream trypanosomes, at least in vitro, can survive with vanishingly small amounts of T[SH]2. However, it should be noted that the HMI9-T culture medium used in these studies is rich in reducing agents such as thioglycerol (56 μM) and cysteine (1.5 mM) which may protect these thiol-depleted cells from oxidant stress.
Virulence of cDKO parasites in mice
The oxidative environment in vivo is significantly different from in vitro culture conditions, underlining the importance of carrying out drug target validation studies in appropriate animal models (Frearson et al., 2007). With this in mind, groups of mice were inoculated with WT, cDKO or cDKO parasites which had been grown in the absence of tetracycline for 24 h. Mice infected with tetracycline-treated parasites were dosed with doxycycline in their drinking water 5 days prior to, and throughout infection to maintain expression of ectopic TRYS. Infections were monitored over a 30 day period and survival curves of each infection are shown in Fig. 3. Mice infected with WT parasites succumbed to infection on day 5 and this was also the case for mice infected with cells expressing ectopic TRYS (cDKO plus doxycycline). In contrast, four out of five mice infected with cDKO cells in the absence of doxycycline remained completely free of parasites beyond 30 days while the remaining mouse from this group only showed a lethal parasitaemia on day 23. Western blot analysis of cells recovered from this infected mouse confirmed that these parasites had regained expression of TRYS, suggesting that tetracycline control had been lost, a common phenomenon in conditional null mutants of essential genes in T. brucei (Chang et al., 2002; Roper et al., 2002). Collectively, these results confirm that TRYS is essential for parasite survival in a mammalian host.
Fig. 3.
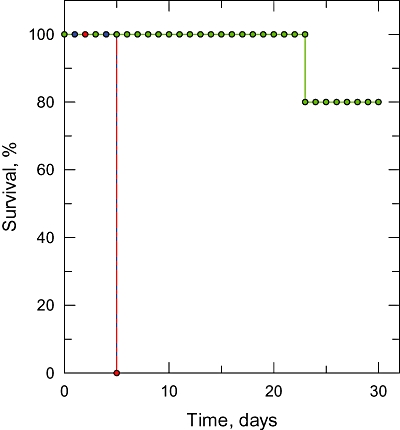
Virulence of cDKO parasites in mice. Groups of five mice were infected with either WT or cDKO cells (1 × 104 parasites). Mice infected with tetracycline-treated parasites were dosed with doxycycline in their drinking water 3 days prior to, and throughout, the infection. Data are presented in the form of a Kaplan–Meier survival plot. Symbols: WT, blue; cDKO in the presence of doxycycline, red; cDKO in absence of doxycycline, green.
Chemical validation of TRYS as a drug target in T. brucei
Genetic validation is a crucial first step in the identification of a drug target. However, the importance of demonstrating that a target is ‘druggable’ within a cell should not be underestimated. As a result of a high-throughput drug screening and medicinal chemistry campaign carried out by the Drug Discovery Unit in Dundee, DDU 86439 (N-(3-(dimethylamino)propyl)-2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamide, Fig. 4A) was identified as a potent inhibitor of recombinant T. brucei TRYS with an IC50 of 45 nM (full details are to be published elsewhere). This compound inhibited the growth of WT cells in HMI9-T media with an EC50 value of 7.0 ± 0.2 μm following a 72 h exposure. To confirm that TRYS was specifically targeted by this compound, TRYS SKO and overexpressing (OE) cell lines were compared with WT cells for their relative sensitivity to compound DDU 86439. Western analysis and densitometry demonstrated that TRYS protein levels were approximately twofold lower in SKO cells (Fig. 2B) and threefold higher in OE cells as compared with WT levels (Fig. S2). Changes in the level of TRYS in these cells correlated well with their relative sensitivity to compound DDU 86439 with EC50 values of 1.2 ± 0.04 and 23.0 ± 0.4 μM for SKO and OE cell lines respectively (Fig. 4B). cDKO cells, grown in the absence of tetracycline for 2 days prior to analysis, were found to be hypersensitive to this compound (EC50 0.46 ± 0.01 μM, Fig. 4B), providing further evidence that TRYS is the specific target of this inhibitor.
Fig. 4.
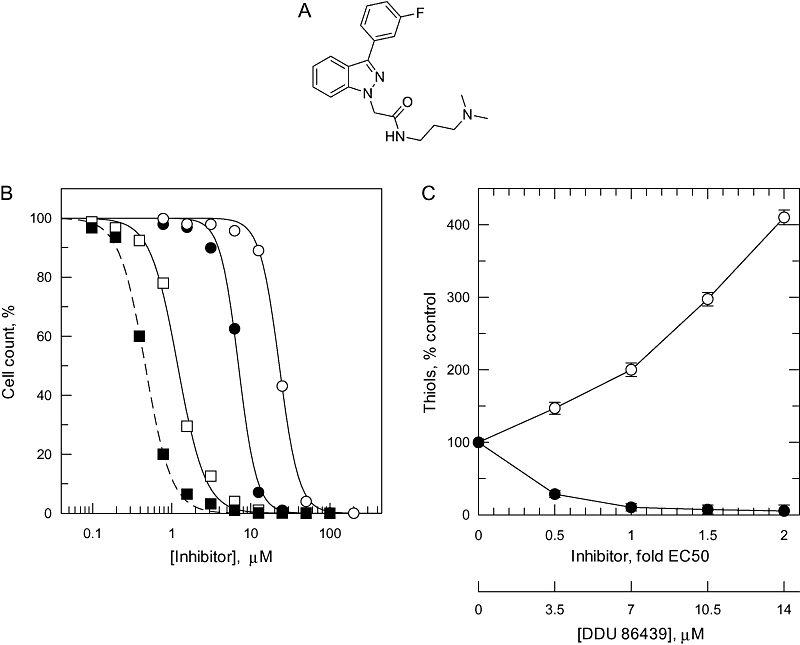
Chemical targeting of TRYS in T. brucei. A. Chemical structure of TRYS inhibitor DDU 86439 (N-(3-(dimethylamino)propyl)-2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamide). B. EC50 values were determined for DDU 86439 against WT (closed circles), SKO (open squares), TRYS-overexpressing (open circles) and cDKO cells grown in the absence of tetracycline for 2 days prior to analysis (closed squares, dotted line). The curves are the non-linear fits of data using a two-parameter EC50 equation provided by GraFit (see Experimental procedures). EC50 values of 7.1 ± 0.2, 1.2 ± 0.05, 23.3 ± 0.3 and 0.46 ± 0.01 μM were determined for WT, SKO, TRYS-overexpressing and cDKO cell lines respectively. Data are the mean of duplicate measurements. C. Analysis of intracellular T[SH]2 (closed circles) and GSH (open circles) levels in T. brucei bloodstream parasites (WT) following incubation with compound DDU 86439 (72 h). Data are represented as a percentage of the thiol levels of untreated cells (0.51 and 0.42 nmol(108 cells)−1 for GSH and T[SH]2 respectively; Table S1) and each measurement is the mean of three individual measurements.
Intracellular thiol levels were determined in WT parasites following incubation with concentrations of the TRYS inhibitor corresponding to 0.5, 1.0, 1.5 and 2.0 times the established EC50 value (7 μM) (Fig. 4C). Treatment with DDU 86439 resulted in a significant, dose-dependent decrease in the levels of intracellular T[SH]2. Trypanosomes incubated with the highest levels of this compound (14 μM) were found to maintain only 5% of their original T[SH]2 levels after 72 h. As in the case of cDKO cells (Fig. 2B), the absence of functional TRYS in drug-treated cells led to an accumulation of GSH, with levels reaching fourfold higher than those seen in untreated cells. The greater speed and magnitude of these effects are consistent with the acute effects of chemical inhibition compared with the slower effects of loss of target activity by genetic intervention (Fig. 2D). These observations also suggest that the minimum concentration of T[SH]2 compatible with survival is ∼3 μM [Table S3, assuming 108 cells = 5.8 μl (Opperdoes et al., 1984)].
Is the amidase activity of TRYS essential to bloodstream T. brucei?
Following the unequivocal chemical and genetic validation of the synthetase activity of TRYS as a drug target in bloodstream T. brucei, we posed the question: could this bifunctional enzyme actually embody two drug targets in one? Comparatively little is known about the amidase activity of TRYS and its biological function remains unclear. In previous kinetic and structural studies, a cysteine at position 59 of the equivalent Crithidia fasiculata (Oza et al., 2002a) and L. major (Fyfe et al., 2008) enzymes has been demonstrated as critical in the catalytic mechanism of the TRYS-amidase. Replacement of cysteine 59 with an alanine in the Crithidia enzyme resulted in a functional synthetase devoid of amidase activity. To determine the role of TRYS-amidase in parasite viability and virulence, this knowledge was exploited to generate a TRYS conditional null mutant cell line (cDKO(C57A)) expressing an amidase-‘dead’ enzyme under tetracycline control (Fig. S3).
The amidase dead cell line was viable in HMI9-T medium in the presence of tetracycline with a generation time of 11.1 ± 0.1 h (Fig. 5A), marginally slower than the original cDKO line (9.9 ± 0.02 h). Removal of tetracycline again resulted in the slow death of parasites over an 8 day period with viability being lost on day 6 of culture, confirming that TRYS synthetase, but not amidase activity is essential for growth in vitro. In an identical manner to the cDKO cell line, intracellular T[SH]2 levels fell following the removal of tetracycline from cDKO(C57A) cultures while GSH steadily accumulated (Fig. 5B). Parasites expressing the amidase-dead TRYS were markedly less virulent in mice than those expressing the functional enzyme (Fig. 5C). All five mice infected with cDKO(C57A) gradually succumbed to infection over a 9 day period, from day 6–15, while cDKO-infected animals died on day 5. The parasites recovered from cDKO(C57A)-infected mice continued to express the mutated form of the TRYS enzyme, as determined by PCR sequence analysis of genomic DNA (data not shown). Four out of five mice infected with cDKO(C57A) cells in the absence of doxycycline remained free of parasites for more than 20 days before becoming terminally infected, while one mouse remained parasite-free for the entire 30 days of the experiment. Western and PCR analysis of parasites recovered from the infected animals revealed that tetracycline control had once again been lost and the mutated TRYS was being freely expressed (data not shown). While it is evident that TRYS-amidase activity is not essential for the in vivo or in vitro viability of T. brucei, the absence of this activity retards growth in vitro and decreases virulence in the mouse model.
Fig. 5.
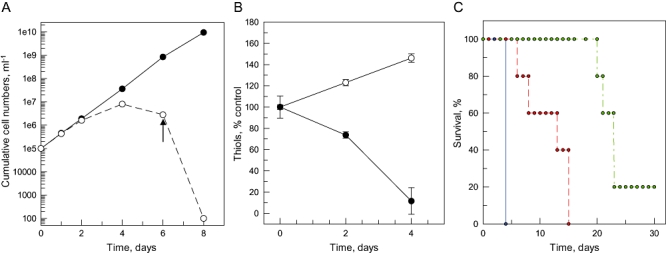
In vitro and in vivo characterization of a TRYS-amidase ‘dead’ cell line. A. The growth of the cDKO(C57A) cell line in HMI9-T medium was monitored in the presence (closed circles) and absence of tetracycline (open circles, dashed line).The arrow indicates the point where the viable cells are no longer recoverable when subcultured for up to 10 days in medium containing tetracycline. B. Intracellular T[SH]2 (closed circles) and GSH (open circles) levels in cDKO(C57A) cells following the removal of tetracycline from cultures. Each data point represents the means ± standard deviations from triplicate determinations. C. Groups of five mice were infected with either WT or cDKO(C57A) cells (1 × 104 parasites). Mice infected with tetracycline-treated parasites were dosed with doxycycline in their drinking water 3 days prior to, and throughout, the infection. Data are presented in the form of a Kaplan–Meier survival plot. Symbols: WT, blue; cDKO in the presence of doxycycline, red; cDKO in absence of doxycycline, green.
What is the biological function of TRYS-amidase in T. brucei?
As TRYS-amidase is not required for the viability or virulence of bloodstream T. brucei, the question of its biological function remains. Several researchers have suggested that the ability to hydrolyse T[SH]2 to its components, GSH and spermidine may enable cells to respond to conditions of polyamine deprivation (Oza et al., 2002b; Fyfe et al., 2008). To examine this hypothesis, cDKO and cDKO(C57A) cell lines were tested for their relative sensitivity to difluoromethylornithine (DFMO) (Fig. 6A), a suicide inhibitor of ornithine decarboxylase, the rate-determining enzyme in polyamine biosynthesis (Phillips and Wang, 1987; Phillips et al., 1988). Should the primary role of TRYS-amidase be to release polyamine stores from T[SH]2 in times of need, it would be reasonable to assume that cells without this ability would be more sensitive to DFMO. However, the cell line expressing the functional TRYS-amidase was just as sensitive to DFMO as the cell line expressing the ‘dead’ enzyme (EC50: 17.5 ± 0.5 and 16.6 ± 0.8 μM respectively).
Fig. 6.
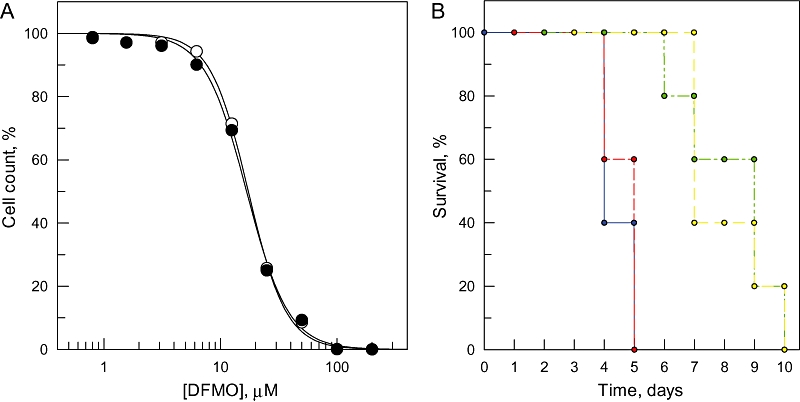
The effect of polyamines on the growth and virulence of cDKO(C57A) parasites. A. EC50 values were determined for DFMO against cDKO (closed circles) and cDKO(C57A) cells (closed circles). The curves are the non-linear fits of data using a two-parameter EC50 equation provided by GraFit (see Experimental procedures). EC50 values of 17.5 ± 0.5 and 16.6 ± 0.8 μM were determined for cDKO and cDKO(C57A) cell lines respectively. Data are the mean of duplicate measurements. B. Groups of five mice were infected with either cDKO or cDKO(C57A) cells (1 × 104 parasites). All mice were infected with tetracycline-treated parasites and dosed with doxycycline in their drinking water. One set of the cDKO and cDKO(C57A)-infected mice received daily intraperitoneal injections of spermidine (100 mg kg−1). Data are presented in the form of a Kaplan–Meier survival plot. Symbols: cDKO, blue; cDKO plus spermidine, red; cDKO(C57A), green; cDKO(C57A) plus spermidine, yellow.
To determine whether the reduced virulence of the cDKO(C57A) cell line could be attributed to an inability to access T[SH]2-conjugated polyamines, groups of cDKO(C57A)-infected mice were given a daily bolus of spermidine via intraperitoneal injection. Previous studies have shown that dosing of mice in this manner increased parasite polyamine levels to such an extent that DFMO could no longer cure infection (McCann et al., 1981; Nathan et al., 1981). Similarly, we hypothesized that increased polyamine levels in cDKO(C57A) cells may result in a return to the levels of virulence seen in cDKO infections. Mice inoculated with the cDKO cell line succumbed to infection on days 4 and 5, regardless of the presence or absence of additional spermidine (Fig. 6B). Once again, the cDKO(C57A) parasites were less virulent than those expressing a functional TRYS-amidase, with mice becoming heavily infected between days 6 and 10. Most significantly, daily dosing of infected mice with spermidine had absolutely no effect on the virulence of the cDKO(C57A) cells. The failure of additional spermidine to enhance the virulence of these cells could suggest that the primary function of TRYS-amidase is not to enable access to T[SH]2-conjugated polyamines.
These findings bring into question the hypothesis that TRYS-amidase plays a pivotal role in the regulation of cellular polyamines (Shim and Fairlamb, 1988; Tetaud et al., 1998; Oza et al., 2002b; Fyfe et al., 2008). If this is not the case, then what is the biological function of the amidase in T. brucei? Kinetic analysis of recombinant T. brucei and T. cruzi TRYS has shown that the amidase activity of these enzymes is equivalent to less than 1% of the synthetase activity under optimal assay conditions (Oza et al., 2002b; 2003). Should this also be true of the in vivo enzyme, it is difficult to see how the amidase could have any meaningful impact on intracellular polyamine levels. Alternatively, the low enzymatic activity of recombinant TRYS-amidase could be readily explained if T[SH]2 and GspdSH were not the natural substrates of this enzyme. In the Gram-positive bacteria Mycobacterium smegmatis, a thiol S-conjugate amidase has been characterized which specifically hydrolyses thiol-conjugated xenobiotics, resulting in detoxification of the xenobiotic and recycling of the thiol (Newton et al., 2000). It is tempting to suggest that TRYS-amidase may play a similar role in T. brucei and that this enzyme is highly selective for S-conjugates of T[SH]2 formed by trypanothione S-transferase (Vickers and Fairlamb, 2004). Studies are underway to test the validity of this hypothesis. What is irrefutable from this study is that TRYS-amidase is not a viable drug target in the African trypanosome.
Implications for parasite chemotherapy
In this study, TRYS has been chemically and genetically validated as a drug target in bloodstream T. brucei. We have shown that TRYS null trypanosomes are unable to establish an infection in mice and that chemical targeting of this enzyme leads to parasite death. The potency of our lead TRYS inhibitor against bloodstream trypanosomes (EC50 7 μM) compares favourably with that of eflornithine (EC50 17.5 μM, Fig. 6A), a current front-line drug used in the treatment of HAT. Like eflornithine, DDU 86439 is not rapidly cidal and the decrease of intracellular T[SH]2 content is compatible with dilution due to cell division rather than high metabolic turnover. Given that many existing drugs interact directly or indirectly with T[SH]2 metabolism (Fairlamb, 2003), inhibitors of TRYS could prove ideal candidates for combination therapy. RNAi studies have revealed that TRYS-depleted T. brucei procyclics are significantly more susceptible to trypanocides, such as melarsoprol and triostam, also known to act on T[SH]2 metabolism (Ariyanayagam et al., 2005). In addition, these procyclics were also more sensitive to the redox-cycling drug nifurtimox, currently in phase III clinical trials for use against African sleeping sickness. Collectively, these findings suggest that the use of drugs targeting TRYS, in combination with existing drugs, could prove an effective strategy in treating T. brucei infections.
Experimental procedures
Cell lines and culture conditions
Trypanosoma brucei bloodstream-form ‘single marker’ S427 (T7RPOL TETR NEO) and knockouts were cultured at 37°C in modified HMI9 medium (56 μM 1-thioglycerol was substituted for 200 μM 2-mercaptoethanol) supplemented with 2.5 μg ml−1 G418 to maintain expression of T7 RNA polymerase and the tetracycline repressor protein (Hirumi and Hirumi, 1989). Cultures were initiated with 1 × 105 cells ml−1 and subcultured when cell densities approached 1–2 (× 106) ml−1. Single marker (WT), SKO, cDKO and OE cell lines were also grown continuously in the presence of the appropriate drug selection (see below for further details).
In order to examine the effects of inhibitors on the growth of these parasites, triplicate cultures containing the inhibitor were seeded at 1 × 105 trypanosomes ml−1. Cell densities were determined using the CASY Model TT cell counter (Schärfe) after culture for 72 h. EC50 values were determined using the following two-parameter equation by non-linear regression using GraFit:
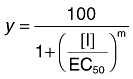 |
where the experimental data were corrected for background cell density and expressed as a percentage of the uninhibited control cell density. In this equation [I] represents inhibitor concentration and m is the slope factor.
Generation of knockout, overexpression and recovery constructs
The primers used are summarized in Table 1 and were designed using the T. brucei trypanothione synthetase (TRYS) sequence in GeneDB (Tb927.2.4370) as a template. The accuracy of all assembled constructs was verified by sequencing. TRYS gene replacement cassettes were generated by amplifying a region of DNA encompassing the 5′-UTR, open reading frame (ORF) and 3′-UTR of T. brucei TRYS from genomic DNA with primers 5′UTR-NotI_s and 3′UTR-NotI_as, using Pfu polymerase. This sequence was then used as a template for the amplification of the individual regions used in the assembly of replacement cassettes containing the selectable drug-resistance genes puromycin N-acetyl transferase (PAC) and hygromycin phosphotransferase (HYG), exactly as previously described (Martin and Smith, 2005).
Table 1.
Upper case letters refer to nucleotides corresponding to gene sequences in T. brucei; lower case refers to additional sequences used in generating constructs.
| Primer name | Primer sequence |
|---|---|
| 5′UTR-NotI_s | 5′-ataagaatgcggccgcTGTGGTTGTTGTTTC-3′ |
| 5′UTR-HindIII/PmeI_as | 5′-gtttaaacttacggaccgtcaagcttTTGTTTCAATTGCTTTTCC-3′ |
| 3′UTR-PmeI/BamHI_s | 5′-gacggtccgtaagtttaaacggatccCACAACCCTTCCGTTTTG-3′ |
| 3′UTR-NotI_as | 5′-ataagtaagcggccgcGGAATAAACATAAAGC-3′ |
| TbTRYS-HindIII_s | 5′-cggaaggagaaagcttatgggcagcagccat-3′ |
| TbTRYS-BamHI_as | 5′-ggatccCTACATTTGAATACGTACGGGACCAAACGGAGATTCGAGCCCAGTGATGAGCTT-3′ |
| TbTRYS(C57A) mut_s | 5′-cttttctgcggtttcaaataccaagctgtagaattt-3′ |
| TbTRYS(C57A) mut_as | 5′-aaattctacagcttggtatttgaaaccgcagaaaag-3′ |
Restriction endonuclease sites are underlined and mutations used to generate a amidase-dead TRYS are indicated in bold.
To generate the recovery construct pLew100_TbTRYS, the TRYS ORF from T. brucei was amplified from genomic DNA using TbTRYS-HindIII sense and TbTRYS-BamHI antisense primers, and cloned into pCR-Blunt II TOPO (Invitrogen). The pCR-Blunt II-TOPO-TbTRYS construct was then digested with HindIII and BamHI and the resulting fragment ligated into the HindIII/BamHI cloning site of a modified pLew100 tetracycline-inducible expression vector (Wirtz et al., 1999) which contains blasticidin S transferase (BSD) as the selectable gene marker (kindly supplied by Dr Kirstee Martin). The resulting rescue vector was also used as a TRYS overexpression vector in single marker T. brucei bloodstream parasites.
An additional recovery construct was generated containing a mutated version of the T. brucei TRYS gene. Site-directed mutagenesis was carried out following the QuikChange® protocol (Stratagene) and using the KOD HotStart DNA polymerase (Novagen). Using the pLew100_TbTRYS construct as a template, a C to A mutation was introduced at position 57 of T. brucei TRYS was generated by PCR resulting in the modified construct – pLew100_TbTRYS(C57A).
Generation of transgenic T. brucei cell lines
Knockout, recovery and overexpression constructs were prepared using QIAprep Miniprep Plasmid Kit (Qiagen). DNA was digested with NotI, ethanol precipitated and redissolved in sterile water at a final concentration of 1 μg μl−1. Trypanosomes were electroporated with 5 μg of DNA using reagents from the Human T cell Nucleofector kit as per manufacturers' instructions and using programme X-001 of the Nucleofector II electroporator (Amaxa, Cologne, Germany) (Burkard et al., 2007).
The cDKO of TRYS was generated by sequentially replacing the endogenous TRYS genes with drug-resistance genes. The first TRYS allele was replaced with the drug-resistance gene PAC resulting in a SKO cell line which was cultured in the continuous presence of 0.1 μg ml−1 puromycin. The SKO cells were then transformed with either the recovery vector pLew100_TbTRYS or the mutated version of this vector pLew100_TbTRYS(C57A) and cultured in the presence of 2.5 μg ml−1 blasticidin in addition to puromycin. Prior to removal of the remaining TRYS allele, expression of the recombinant TRYS proteins were induced by the addition of tetracycline (2 μg ml−1, 72 h) to the culture medium. A cDKO cell line was finally generated following replacement of the final TRYS allele with an HYG. The resulting cell line was cultured in the presence of hygromycin (4.0 μg ml−1) in addition to the previously described drugs. The TRYS overexpression cell line was generated by directly transforming single marker bloodstream parasites with the pLew100_TbTRYS vector and inducing recombinant protein expression by the addition of tetracycline. At each step in the generation of these transgenic cell lines, parasites were cloned by serial dilution.
Southern blot analyses of transgenic T. brucei cell lines
The ORF of T. brucei TRYS was amplified by PCR (using the primers previously described for the cloning of TbTRYS into pLew100-BSD) and the PCR DIG Probe Synthesis Kit (Roche). The resulting digoxigenin-labelled TbTRYS product was used as a probe. Samples of genomic DNA (5 μg) from single marker and transgenic cell lines were digested with the restriction endonuclease PstI, the digestion products were then separated on a 0.8% agarose gel and transferred on to a positively charged nylon membrane (Roche). The membrane was hybridized overnight in DIG Easy Hyb solution (Roche) at 42°C with the DIG-labelled TRYS ORF probe (2 μl of PCR product). Following hybridization, membranes were washed twice in low stringency conditions (25°C, 2 × 5 min, 2× SSC with 0.1% SDS) and twice in high stringency conditions (68°C, 2 × 15 min, 0.5× SSC with 0.1% SDS), where 1× SSC comprises 150 mM sodium chloride and 50 mM sodium acetate, pH 7.0. Bound probe was detected using the DIG immunological detection kit (Roche) as per manufacturers' instructions.
Western blot analysis of T. brucei cell lysates
Polyclonal antisera against T. brucei TRYS were raised in adult male Wistar rats. An initial injection of 100 μg of purified antigen, emulsified in complete Freund's adjuvant, was followed by two identical booster injections of antigen emulsified in Freund's incomplete adjuvant at 2 week intervals.
Trypanosoma brucei whole cell extracts (1 × 107 parasites per lane) were separated by SDS/PAGE and subsequently transferred onto nitrocellulose. After blocking with 7% skimmed milk in phosphate buffered saline (PBS) for 1 h, blots were incubated with either T. brucei TRYS (1:700 dilution) or T. brucei PTR1 (1:500) (Sienkiewicz et al., 2008) polyclonal antiserum for 1 h, washed in PBS containing 0.1% (v/v) Tween-20, and then incubated with a secondary antibody [rabbit anti(rat IgG)] (Dako, Ely, UK; 1:10 000 dilution). Immunoblots were developed using the ECL plus (enhanced chemiluminescence) system from GE Healthcare (Piscataway, NJ, USA).
Drug discovery
A high-throughput screening campaign of 63 000 compounds led to the identification of several novel chemical classes of T. brucei TRYS inhibitor. Chemical development of one of these lead compounds led to the identification of compound DDU 86439 (N-(3-(dimethylamino)propyl)-2-(3-(3-fluorophenyl)-1H-indazol-1-yl)acetamide) (patent pending) which inhibited recombinant TRYS with a potency of 0.045 μM and inhibited the growth of bloodstream trypanosomes with an EC50 of 6.9 ± 0.2 μM. DDU 86439 was prepared in five synthetic steps from indazole. Briefly, indazole was iodinated at the three position, as previously described (Edwards et al., 2003). The resultant aryl iodide served as the substrate for a Suzuki coupling reaction with 3-fluorophenylboronic acid in an analogous manner to that previously described (El Kazzouli et al., 2005). The indazole was then N1-alkylated by treatment with sodium hydride and ethyl bromoacetate. The resulting crude ester was hydrolysed by treatment with sodium hydroxide in water/tetrahydrofuran. Following acidic workup, the product carboxylic acid was purified by silica column chromatography. The amide synthesis was accomplished via standard carbodiimide chemistry utilizing N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide, 1-Hydroxybenzotriazole, N,N-Diisopropylethylamine and 3-(dimethylamino)-1-propylamine to furnish the title compound, which was purified by silica column chromatography. Full details of chemical synthesis of this and other inhibitors are to be published elsewhere.
Analysis of intracellular thiols
To determine the effect of inhibitor DDU 86439 on intracellular thiol levels, cultures containing 1 × 105 bloodstream trypanosomes ml−1 were incubated with varying concentrations of inhibitor DDU 86439 equivalent to 0.5, 1.0, 1.5 and 2× the previously determined EC50 (6.9 ± 0.2 μm). Following incubation for 72 h, cells (1 × 108) were collected by centrifugation (900 g, 10 min, 4°C), washed once in ice-cold PSG buffer (PBS, pH 8.0, 1.5% (w/v) glucose and 0.5 mg ml−1 BSA) and derivatised with monobromobimane, as described previously (Shim and Fairlamb, 1988). Acid-soluble thiols were separated by ion-paired, reverse phase HPLC on an ion-paired Ultrasphere C18 column using a Dionex Ultimate 3000 instrument fitted with a Dionex RF-2000 fluorometer.
In vivo studies
Wild-type and transgenic bloodstream T. brucei parasites were cultured in the absence of selectable drugs (hygromycin, blasticidin, puromycin and G418) for 24 h prior to infection of mice. During this time, cDKO cells were grown in the presence or absence of 1 μg ml−1 tetracycline. These parasites were then used to infect groups of five mice (dosed with and without doxycycline respectively) by a single intraperitoneal injection of 104 parasites in 0.2 ml of HMI9-T medium. The plus doxycycline group of animals were dosed with doxycycline in their drinking water (0.2 mg ml−1 in a 5% sucrose solution) for 5 days prior to infection and freshly prepared every second day for the duration of the experiment. Animals were inspected daily for clinical signs of infection and wet smears of tail blood were examined microscopically. Parasitaemia was determined using a Neubauer haemocytometer, as previously described (Sienkiewicz et al., 2008). Mice that exceeded a parasitaemia > 108 ml−1 were humanely killed, because prior experience indicated that animals would succumb to an overwhelming infection by the following day.
Acknowledgments
Our appreciation also goes to Adel Ibrahim of the University of Dundee Cloning Service for help with site-directed mutagenesis and cloning into pLew100 and Dr Natasha Sienkiewicz for helpful advice concerning the generation of transgenic organisms. We thank Dr Paul Wyatt, Director of Dundee Drug Discovery Unit for invaluable advice on the medicinal chemistry presented in this paper. This work was funded by grants from the Wellcome Trust (WT 079838; WT 077705; WT 083481).
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article
References
- Alibu VP, Richter C, Voncken F, Marti G, Shahi S, Renggli CK, et al. The role of Trypanosoma brucei MRPA in melarsoprol susceptibility. Mol Biochem Parasitol. 2006;146:38–44. doi: 10.1016/j.molbiopara.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Ariyanayagam MR, Fairlamb AH. Ovothiol and trypanothione as antioxidants in trypanosomatids. Mol Biochem Parasitol. 2001;115:189–198. doi: 10.1016/s0166-6851(01)00285-7. [DOI] [PubMed] [Google Scholar]
- Ariyanayagam MR, Oza SL, Guther ML, Fairlamb AH. Phenotypic analysis of trypanothione synthetase knockdown in the African trypanosome. Biochem J. 2005;391:425–432. doi: 10.1042/BJ20050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchi CJ, Yarlett N. Polyamine metabolism. In: Marr JJ, Müller M, editors. Biochemistry and Molecular Biology of Parasites. London: Academic Press; 1995. pp. 119–131. [Google Scholar]
- Barrett MP, Fairlamb AH. The biochemical basis of arsenical-diamidine cross-resistance in African trypanosomes. Parasitol Today. 1999;15:136–140. doi: 10.1016/s0169-4758(99)01414-3. [DOI] [PubMed] [Google Scholar]
- Bateman A, Rawlings ND. The CHAP domain: a large family of amidases including GSP amidase and peptidoglycan hydrolases. Trends Biochem Sci. 2003;28:234–237. doi: 10.1016/S0968-0004(03)00061-6. [DOI] [PubMed] [Google Scholar]
- Burkard G, Fragoso CM, Roditi I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol Biochem Parasitol. 2007;153:220–223. doi: 10.1016/j.molbiopara.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Chang T, Milne KG, Guther MLS, Smith TK, Ferguson MAJ. Cloning of Trypanosoma brucei and Leishmania major genes encoding the GlcNAc-phosphatidylinositol de-n-acetylase of glycosylphosphatidylinositol biosynthesis that is essential to the african sleeping sickness parasite. J Biol Chem. 2002;277:50176–50182. doi: 10.1074/jbc.M208374200. [DOI] [PubMed] [Google Scholar]
- Comini MA, Guerrero SA, Haile S, Menge U, Lunsdorf H, Flohé L. Validation of Trypanosoma brucei trypanothione synthetase as drug target. Free Radic Biol Med. 2004;36:1289–1302. doi: 10.1016/j.freeradbiomed.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Croft SL, Sundar S, Fairlamb AH. Drug Resistance in Leishmaniasis. Clin Microbiol Rev. 2006;19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormeyer M, Reckenfelderbaumer N, Ludemann H, Krauth-Siegel RL. Trypanothione-dependent synthesis of deoxyribonucleotides by Trypanosoma brucei ribonucleotide reductase. J Biol Chem. 2001;276:10602–10606. doi: 10.1074/jbc.M010352200. [DOI] [PubMed] [Google Scholar]
- Edwards ML, Cox PJ, Amendola S, Deprets SD, Gillespy TA, Edlin CD, et al. Benzimidazoles and analogues and their use as protein kinases inhibitors. Aventis Pharmaceuticals Inc. Patent WO/2003/035065.
- El Kazzouli S, Bouissane L, Khoulli M, Guillaumet G. Synthesis of 4-substituted and 3,4-disubstituted indazole derivatives by palladium-mediated cross-coupling reactions. Tetrahedron Lett. 2005;46:6163–6167. [Google Scholar]
- El Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, et al. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- Fairlamb AH. Chemotherapy of Human African Trypanosomiasis: current and future prospects. Trends Parasitol. 2003;19:488–494. doi: 10.1016/j.pt.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Fairlamb AH, Cerami A. Metabolism and functions of trypanothione in the Kinetoplastida. Annu Rev Microbiol. 1992;46:695–729. doi: 10.1146/annurev.mi.46.100192.003403. [DOI] [PubMed] [Google Scholar]
- Fairlamb AH, Blackburn P, Ulrich P, Chait BT, Cerami A. Trypanothione: a novel bis (glutathionyl) spermidine cofactor for glutathione reductase in trypanosomatids. Science. 1985;227:1485–1487. doi: 10.1126/science.3883489. [DOI] [PubMed] [Google Scholar]
- Fairlamb AH, Henderson GB, Cerami A. Trypanothione is the primary target for arsenical drugs against African trypanosomes. Proc Natl Acad Sci USA. 1989;86:2607–2611. doi: 10.1073/pnas.86.8.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frearson JA, Wyatt PA, Gilbert IH, Fairlamb AH. Target assessment for antiparasitic drug discovery. Trends Parasitol. 2007;23:589–595. doi: 10.1016/j.pt.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe PK, Oza SL, Fairlamb AH, Hunter WN. Leishmania trypanothione synthetase-amidase structure reveals a basis for regulation of conflicting synthetic and hydrolytic activities. J Biol Chem. 2008;283:17672–17680. doi: 10.1074/jbc.M801850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol. 1989;75:985–989. [PubMed] [Google Scholar]
- Krieger S, Schwarz W, Ariyanayagam MR, Fairlamb AH, Krauth-Siegel RL, Clayton C. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol Microbiol. 2000;35:542–552. doi: 10.1046/j.1365-2958.2000.01721.x. [DOI] [PubMed] [Google Scholar]
- McCann PP, Bacchi CJ, Clarkson AB, Seed JR, Nathan HC, Amole BO, et al. Further studies on difluoromethylornithine in African trypanosomes. Med Biol. 1981;59:434–440. [PubMed] [Google Scholar]
- Martin K, Smith TK. The myo-inositol-1-phosphate synthase gene is essential in Trypanosoma brucei. Biochem Soc Trans. 2005;33:983–985. doi: 10.1042/BST0330983. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R, Dey S, Xu N, Gage D, Lightbody J, Ouellette M, Rosen BP. Trypanothione overproduction and resistance to antimonials and arsenicals in Leishmania. Proc Natl Acad Sci USA. 1996;93:10383–10387. doi: 10.1073/pnas.93.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan HC, Bacchi CJ, Hutner SH, Rescigno D, Sjoerdsma A. Antagonism by polyamines of the curative effects of α-difluoromethylornithine in Trypanosoma brucei brucei infections. Biochem Pharmacol. 1981;30:3010–3013. doi: 10.1016/0006-2952(81)90270-7. [DOI] [PubMed] [Google Scholar]
- Newton GL, Av-Gay Y, Fahey RC. A novel mycothiol-dependent detoxification pathway in mycobacteria involving mycothiol S-conjugate amidase. Biochemistry. 2000;39:10739–10746. doi: 10.1021/bi000356n. [DOI] [PubMed] [Google Scholar]
- Opperdoes FR, Baudhuin P, Coppens I, de Roe C, Edwards SW, Weijers PJ, Misset O. Purification, morphometric analysis, and characterization of the glycosomes (microbodies) of the protozoan hemoflagellate Trypanosoma brucei. J Cell Biol. 1984;98:1178–1184. doi: 10.1083/jcb.98.4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza SL, Ariyanayagam MR, Fairlamb AH. Characterization of recombinant glutathionylspermidine synthetase/amidase from Crithidia fasciculata. Biochem J. 2002a;364:679–686. doi: 10.1042/BJ20011370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza SL, Tetaud E, Ariyanayagam MR, Warnon SS, Fairlamb AH. A single enzyme catalyses formation of trypanothione from glutathione and spermidine in Trypanosoma cruzi. J Biol Chem. 2002b;277:35853–35861. doi: 10.1074/jbc.M204403200. [DOI] [PubMed] [Google Scholar]
- Oza SL, Ariyanayagam MR, Aitcheson N, Fairlamb AH. Properties of trypanothione synthetase from Trypanosoma brucei. Mol Biochem Parasitol. 2003;131:25–33. doi: 10.1016/s0166-6851(03)00176-2. [DOI] [PubMed] [Google Scholar]
- Phillips MA, Wang CC. A Trypanosoma brucei mutant resistant to α-difluoromethylornithine. Mol Biochem Parasitol. 1987;22:9–17. doi: 10.1016/0166-6851(87)90064-8. [DOI] [PubMed] [Google Scholar]
- Phillips MA, Coffino P, Wang CC. Trypanosoma brucei ornithine decarboxylase: enzyme purification, characterization, and expression in Escherichia coli. J Biol Chem. 1988;263:17933–17941. [PubMed] [Google Scholar]
- Rigden DJ, Jedrzejas MJ, Galperin MY. Amidase domains from bacterial and phage autolysins define a family of gamma-D,1-glutamate-specific amidohydrolases. Trends Biochem Sci. 2003;28:230–234. doi: 10.1016/s0968-0004(03)00062-8. [DOI] [PubMed] [Google Scholar]
- Roper JR, Guther ML, Milne KG, Ferguson MA. Galactose metabolism is essential for the African sleeping sickness parasite Trypanosoma brucei. Proc Natl Acad Sci USA. 2002;99:5884–5889. doi: 10.1073/pnas.092669999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Fairlamb AH. Levels of polyamines, glutathione and glutathione-spermidine conjugates during growth of the insect trypanosomatid Crithidia fasciculata. J Gen Microbiol. 1988;134:807–817. doi: 10.1099/00221287-134-3-807. [DOI] [PubMed] [Google Scholar]
- Sienkiewicz N, Jaroslawski S, Wyllie S, Fairlamb AH. Chemical and genetic validation of dihydrofolate reductase-thymidylate synthase as a drug target in African trypanosomes. Mol Microbiol. 2008;69:520–533. doi: 10.1111/j.1365-2958.2008.06305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart KD, Brun R, Croft SL, Fairlamb AH, Gurtler RE, McKerrow JH, et al. Kinetoplastids: related protozoan pathogens, different diseases. J Clin Invest. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetaud E, Manai F, Barrett MP, Nadeau K, Walsh CT, Fairlamb AH. Cloning and characterization of the two enzymes responsible for trypanothione biosynthesis in Crithidia fasciculata. J Biol Chem. 1998;273:19383–19390. doi: 10.1074/jbc.273.31.19383. [DOI] [PubMed] [Google Scholar]
- Tovar J, Cunningham ML, Smith AC, Croft SL, Fairlamb AH. Down-regulation of Leishmania donovani trypanothione reductase by heterologous expression of a trans-dominant mutant homologue: effect on parasite intracellular survival. Proc Natl Acad Sci USA. 1998;95:5311–5316. doi: 10.1073/pnas.95.9.5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers TJ, Fairlamb AH. Trypanothione S-transferase activity in a trypanosomatid ribosomal elongation factor 1B. J Biol Chem. 2004;279:27246–27256. doi: 10.1074/jbc.M311039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers TJ, Wyllie SH, Fairlamb AH. Leishmania major elongation factor 1B complex has trypanothione S-transferase and peroxidase activity. J Biol Chem. 2004;279:49003–49009. doi: 10.1074/jbc.M407958200. [DOI] [PubMed] [Google Scholar]
- Wilkinson SR, Horn D, Prathalingam SR, Kelly JM. RNA interference identifies two hydroperoxide metabolizing enzymes that are essential to the bloodstream form of the African trypanosome. J Biol Chem. 2003;278:31640–31646. doi: 10.1074/jbc.M303035200. [DOI] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross GAM. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- Wyllie S, Cunningham ML, Fairlamb AH. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J Biol Chem. 2004;279:39925–39932. doi: 10.1074/jbc.M405635200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.