

Abstract
We have designed and synthesized a cyclic, bivalent Smac mimetic (compound 3) and characterized its interaction with the X-linked inhibitor of apoptosis protein (XIAP). Compound 3 binds to XIAP containing both BIR2 and BIR3 domains with a biphasic dose–response curve representing two binding sites with IC50 values of 0.5 and 406 nM, respectively. Compound 3 binds to XIAPs containing the BIR3-only and BIR2-only domain with Ki values of 4 nM and 4.4 μM, respectively. Gel filtration experiments using wild-type and mutated XIAPs showed that 3 forms a 1:2 stoichiometric complex with XIAP containing the BIR3-only domain. However, it forms a 1:1 stoichiometric complex with XIAP containing both BIR2 and BIR3 domains, and both BIR domains are involved in the binding. Compound 3 efficiently antagonizes inhibition of XIAP in a cell-free functional assay and is >200 times more potent than its corresponding monovalent compound 2. Determination of the crystal structure of 3 in complex with the XIAP BIR3 domain confirms that 3 induces homodimerization of the XIAP BIR3 domain and provides a structural basis for the cooperative binding of one molecule of compound 3 to two XIAP BIR3 molecules. On the basis of this crystal structure, a binding model of XIAP containing both BIR2 and BIR3 domains and 3 was constructed, which sheds light on the ability of 3 to relieve the inhibition of XIAP with not only caspase-9 but also caspase-3/-7. Compound 3 is cell-permeable, effectively activates caspases in whole cells, and potently inhibits cancer cell growth. Compound 3 is a useful biochemical and pharmacological tool for further elucidating the role of XIAP in regulation of apoptosis and represents a promising lead compound for the design of potent, cell-permeable Smac mimetics for cancer treatment.
Inhibitors of apoptosis proteins (IAPs) make up an important class of regulators of apoptosis (1, 2). Due to its potent biological functions, the X-linked inhibitor of apoptosis protein (XIAP) is the best-characterized IAP. XIAP suppresses apoptosis, at least in part, through binding to and inhibition of an initiator, caspase-9, and two effector caspases, caspase-3 and -7 (1, 2). XIAP contains three baculoviral IAP repeat (BIR) domains (BIR1, BIR2, and BIR3). The XIAP BIR3 domain directly binds to the small subunit of caspase-9 (3) and blocks the formation of active caspase-9 (4, 5). The XIAP BIR2 domain, together with the immediately preceding linker, binds to caspase-3 and caspase-7 and inhibits the activity of these two caspases (6–8).
Smac/DIABLO (second mitochondrion-derived activator of caspase or direct IAP-binding protein with low pI) is a potent pro-apoptotic protein (9, 10) which functions as an endogenous antagonist of XIAP. Smac forms a homodimer, antagonizes XIAP by binding to both the BIR2 and BIR3 domains, and promotes the activity of caspase-9 and caspase-3/-7. Previous studies have clearly demonstrated that Smac interacts with the XIAP BIR3 domain in the same site where caspase-9 binds via its AVPI motif, thus removing the inhibition of XIAP to caspase-9 by direct competition (11). The precise mechanism by which Smac antagonizes the inhibition of XIAP to caspase-3/-7 is not completely clear (4, 10). Modeling studies suggest that Smac binds to the XIAP BIR2 domain also through its AVPI motif and prevents the binding of XIAP to caspase-3/-7 (6, 7, 12). In this manner, a dimeric Smac protein effectively removes the inhibition of XIAP to caspase-9 and to caspase-3/-7 by concurrently binding to both the BIR2 and BIR3 domains in XIAP (13).
XIAP has been considered as a highly attractive molecular target for the development of new classes of anticancer drugs. It is found to be highly expressed in many human tumor cell lines and tumor samples from patients (14) and plays an important role in conferring cancer cells resistance to a variety of anticancer drugs (15). Because XIAP blocks apoptosis at a downstream point, where multiple signaling pathways converge, strategies targeting XIAP may prove to be especially effective in overcoming the resistance of cancer cells to apoptosis. In the last several years, intense effort has been spent on the design of small molecule inhibitors to target the XIAP BIR3 (16–20) or BIR2 domain (21, 22).
Although most of the research efforts have been focused on the design of small molecule inhibitors targeting either the BIR2 or BIR3 domain in XIAP, one study has shown that a bivalent, small molecule Smac mimetic antagonizes XIAP with a potency equal or greater than that of the Smac protein in a cell-free functional assay (23). The precise mode of action for this bivalent Smac mimetic was not completely delineated, but Li et al. (23) hypothesized that its extremely high potency in antagonizing XIAP could be attributed to its bivalency and possible concurrent targeting of both BIR2 and BIR3 domains in XIAP. Recently, our group has reported the design and characterization of a novel bivalent Smac mimetic (SM-164) (24) and showed that SM-164 achieves a high affinity for XIAP and potently induces apoptosis in human cancer cells.
In this study, we have designed a cyclized, conformationally constrained, bivalent Smac mimetic and characterized in detail its interaction with XIAPs containing either the BIR2 or BIR3 domain or both the BIR2 and BIR3 domains. Our results show that this bivalent Smac mimetic binds to the XIAP BIR3-only protein with a high affinity and induces protein dimerization. In comparison, when presented with XIAPs containing both BIR2 and BIR3 domains, the bivalent Smac mimetic interacts concurrently with both BIR2 and BIR3 domains in XIAP and achieves an even higher affinity than the XIAP BIR3-only protein. Our determination of a high-resolution crystal structure of this bivalent Smac mimetic with the XIAP BIR3-only protein provides a structural basis for the high-affinity interaction and a template with which to model its binding to XIAP containing both BIR2 and BIR3 domains. This cyclized, bivalent Smac mimetic was found to potently inhibit cell growth in cancer cells with high levels of XIAP and represents a promising lead compound for further optimization.
Experimental Procedures
Chemistry
All peptides were synthesized manually using standard solid phase peptide chemistry with Fmoc1 protected amino acids on 2-Cl-Trt resin at a 0.1 mmol scale. The acid sensitive 2-Cl-Trt resin was purchased from Novabiochem (Torrance, CA). Fmoc derivatives of standard amino acids were obtained from Anaspec, Inc. (San Jose, CA). HBTU/HOBt activation of Nα protected amino acids was employed for coupling, and 20% piperidine/DMF was used for Fmoc deprotection. A HATU/HOAt/DIEA mixture in DMF was used for backbone cyclization. The crude peptide was purified by semipreparative reverse phase HPLC: Vydac C18 column (22 mm × 250 mm); solvent system, 0.05% TFA in water (A) and 0.05% TFA in 90% acetonitrile in water (B); gradient, 10 to 80% B over 30 min; flow rate, 10.0 mL/min; UV detector at 225 nm. The purity of products was characterized by analytical HPLC and MALDI-TOF-MS. HPLC using two different conditions: (a) (I) a Merck Chromolith Performance RP-18e column (100 mm × 4.6 mm) with a gradient from 10 to 60% B over 9 min, a flow rate of 5.0 mL/min, and an UV detector at 225 nm and (II) a Merck Chromolith Performance RP-18e column (100 mm × 4.6 mm) with a gradient from 0 to 20% B over 9 min, a flow rate of 5.0 mL/min, and an UV detector at 225 nm; (b) (I) an Agilent Zorbax 3.5 μm SB-CN column (75 mm × 4.6 mm) with a gradient from 0 to 60% B over 12 min, a flow rate of 2.0 mL/min, and an UV detector at 225 nm and (II) an Agilent Zorbax 3.5 μm SB-CN column (75 mm × 4.6 mm) with a gradient from 0 to 20% B over 12 min, a flow rate of 2.0 mL/min, and an UV detector at 225 nm. 1H (400 MHz) NMR spectra were recorded in D2O. MALDI-TOF-MS spectra (Kratos Axima-CFR instrument; matrix, α-cyano-4-hydroxycinnamic acid) verified molecular masses of all peptides.
Cyclized Smac mimetics (compounds 3 and 4) were synthesized using the scheme presented in Figure 1B. In a solid phase reaction vessel, a solution of Fmoc-Phe-OH (619 mg, 1.6 mmol) and DIEA (1.11 mL, 6.4 mmol) in DCM (5.0 mL) was added to the 2-Cl-Trt resin (0.8 mmol) preswollen in DCM (30 min). The reaction mixture was stirred under nitrogen for 2 h. The mixture was drained, and the resin was washed with 3× DCM/MeOH/DIPEA mixture (17:2:1), CH2Cl2 (3 × 5 mL), DMF (2 × 5 mL), and DCM (2 × 5 mL). The Fmoc-Phe-2-Cl-Trt resin was dried in vacuo and treated as a preloaded resin for automated peptide synthesis with an ABI 433A peptide synthesizer, using the chemical protocols based on Fmoc chemistry (coupling with a HBTU/HOBt/DIPEA mixture in NMP). On completion of the (Boc)KPF(Boc)KPF-2-Cl-Trt resin sequence, the side chain protected peptide was cleaved from the resin with 10 mL of a 1% TFA/DCM mixture (2 min). The solution was filtered by applying nitrogen pressure to the contents of a flask containing 10% NMM in methanol (2 mL) in an ice bath. This procedure was repeated 10 times; then the residual protected peptide was washed from the resin with 3 × 30 mL of DCM, 3 × 30 mL of MeOH, 2 × 30 mL of DCM, and 3 × 30 mL of MeOH, and the filtrates were checked by TLC. The combined solution was evaporated in vacuo. The residue was a white oil and used as such in the subsequent synthesis step. For backbone cyclization, the linear crude peptide was dissolved in DCM (250 mL, 0.4 mM) under nitrogen, and the solution was cooled at 0 °C. DIPEA (0.6 mmol) and a HATU/HOAt mixture (0.3 mmol each) were added, and the reaction mixture was stirred at ambient temperature for 24 h. The mixture was neutralized with 30% AcOH and evaporated under reduced pressure. The crude protected cyclic peptide was treated with TFA (2 mL) and DCM (2 mL) to deprotect the Boc group. After 2 h, the solvent was evaporated under reduced pressure to yield compound 4, which was used in the subsequent synthesis step. To a solution of Boc-N-methylalanine (0.203 g, 1 mmol), EDCI·HCl (0.192 g, 1 mmol), and HOBt (0.135 g, 1 mmol) in DCM (5 mL) was added DIPEA (0.349 mL, 2 mmol) at 0 °C. Subsequently, cyclic peptide 4 in a DCM (1 mL) solution was added dropwise to this solution, and the resulting solution was stirred at room temperature for 12 h. The solvent was evaporated, and the residue was dissolved in EtOAc, washed with H2O and brine, and dried over Na2SO4. The solvent was removed, and the residue was dissolved in DCM (2 mL). TFA (2 mL) was added to the solution described above at 0 °C, and the resulting solution was stirred at room temperature for 2 h. After the solvent had been removed, the residue was dissolved in water and lyophilized. The crude compound 3 was purified by RP-HPLC: tR = 18.8 min (gradient from 10 to 80% B over 35 min); MALDI-MS (M + H)+ 915.5 (calcd 915.6); tR = 4.10 min [condition a (I), purity of 98%]; tR = 9.42 min [condition b (II), purity of 97%]; 1H NMR (400 MHz, D2O) δ 7.70 (NH, d, J = 7.6 Hz), 7.23–7.06 (10H, m), 4.54 (2H, dd, J = 5.2, 8.0 Hz), 4.41 (2H, t, J = 7.6 Hz), 4.15 (2H, dd, J = 5.6, 8.6 Hz), 3.83 (2H, q, J = 6.8 Hz), 3.65–3.52 (4H, m), 3.06 (2H, m), 2.96–2.83 (6H, m), 2.54 (6H, s), 2.04 (2H, m), 1.87–1.81 (4H, m), 1.62–1.44 (6H, m), 1.30 (6H, d, J = 6.8 Hz), 1.12–1.33 (8H, m). The crude compound 4 was purified by RP-HPLC: tR = 16.3 min (gradient from 10 to 80% B over 35 min); MALDI-MS (M + H)+ 745.3 (calcd 745.4).
Figure 1.
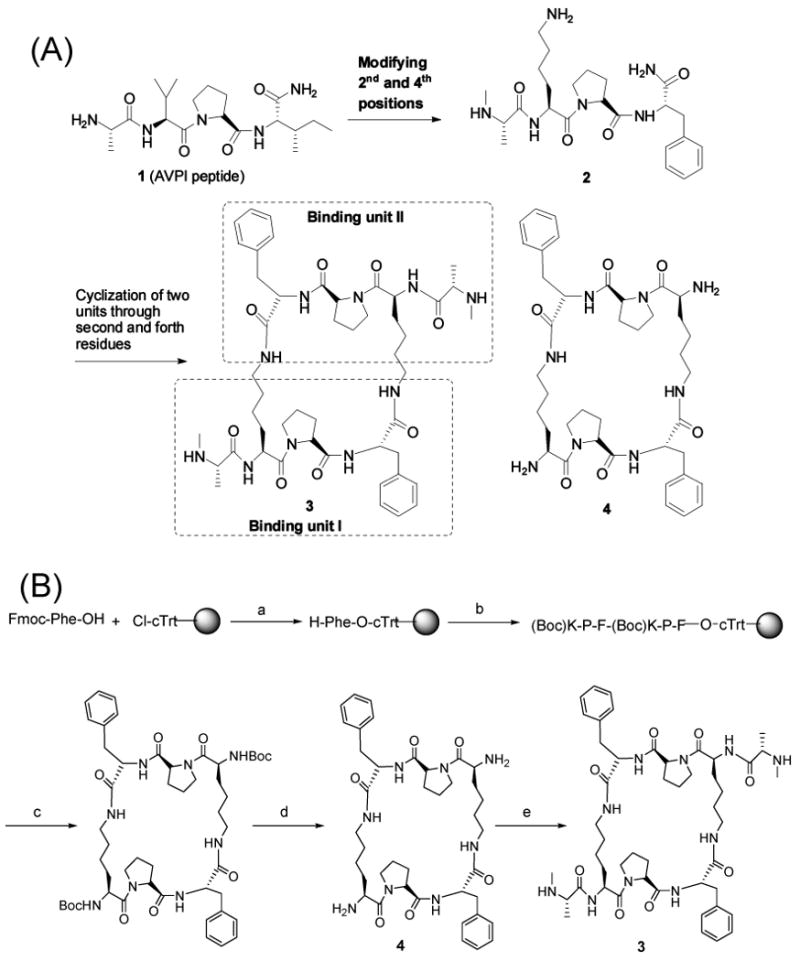
(A) Design of a potent cyclized, bivalent Smac peptidomimetic, compound 3. (B) Synthesis of compounds 3 and 4. Reagents and conditions: (a) (1) DIPEA/CH2Cl2/MeOH, (2) piperidine/20% DMF; (b) five cycles of Fmoc SPPS with HBTU/HOBt/DIPEA coupling; (c) (1) 1% TFA in DCM for 2 min 20 times, (2) HATU/HOAt/DIPEA in DCM; (d) 50% TFA in DCM for 2 h; (e) (1) Boc-N-methylalanine with EDC/HOAt/DCM for 12 h, (2) 50% TFA in DCM for 2 h.
For compound 2, the Fmoc-Phe-2-Cl-Trt resin was treated as a preloaded resin for automated peptide synthesis with an ABI 433A peptide synthesizer, using chemical protocols based on Fmoc chemistry (coupling with HBTU/HOBt/DIPEA in NMP). On completion of the N-methyl-AK(Boc)PF-2-Cl-Trt resin sequence, the side chain protected peptide was cleaved from the resin and deprotected using the method described above. The crude compound 2 was purified via RP-HPLC: tR = 11.3 min (gradient from 10 to 80% B over 35 min); MALDI-MS (M + H)+ 475.3 (calcd 475.3); tR = 3.88 min [condition a (II), purity of 99%]; tR = 5.42 min [condition b (II), purity of 98%]; 1H NMR (400 MHz, D2O) δ 8.07 (NH, d, J = 7.2 Hz), 7.25–7.13 (5H, m), 4.47 (1H, dd, J = 5.6, 8.4 Hz), 4.36 (1H, m), 4.23 (1H, dd, J = 6.0, 8.4 Hz), 3.81 (1H, q, J = 7.2 Hz), 3.66 (1H, m), 3.48 (1H, m), 2.95 (2H, d, J = 7.6 Hz), 2.84 (2H, t, J = 7.6 Hz), 2.54 (3H, s), 2.08 (1H, m), 1.85 (2H, m), 1.71–1.53 (5H, m), 1.36 (3H, d, J = 7.2 Hz), 1.38–1.30 (2H, m).
Protein Expression and Purification
Different constructs of human XIAPs, including linker–BIR2–BIR3 protein (residues 120–356), BIR2–BIR3 protein without the linker proceeding BIR2 (residues 156–356), BIR3-only (residues 241–356), BIR2-only (residues 120–240), mutated BIR2 (E219R)–BIR3 (residues 156–356), were cloned into the pET28 vector (Novagen) with an N-terminal six-His tag. Proteins were produced in Escherichia coli BL21(DE3) cells grown as previously described (24).
Fluorescence Polarization-Based Binding for XIAP BIR3, BIR2, and Linker–BIR2–BIR3 Proteins
A sensitive in vitro binding assay using the fluorescence polarization (FP)-based method (25) was used to determine the binding affinity of Smac mimetics for the XIAP BIR3 protein. In this assay, 5-carboxyfluorescein was coupled to the lysine side chain of a mutated Smac peptide with the sequence AbuRPFK-Fam. This fluorescently tagged peptide (named SM5F) was used as the fluorescent tracer in both FP-based binding assays with XIAP BIR2 (residues 120–240) and BIR3 (residues 241–356) proteins.
In the competitive binding experiments, the tested compound was incubated with 2 μM XIAP BIR2 protein and 50 nM SM5F in assay buffer [100 mM potassium phosphate (pH 7.5), 100 μg/mL bovine gamma globulin, and 0.02% sodium azide, purchased from Invitrogen]. For testing their binding affinities against XIAP BIR3 protein, 30 nM XIAP BIR3 protein and 5 nM SM5F peptide were used.
A FP-based competitive binding assay was established for quantitative determination of the binding affinities of our designed Smac mimetics for XIAP containing both BIR2 and BIR3 domains (26). In the competitive binding experiments, the tested compound was incubated with 3 nM XIAP linker–BIR2–BIR3 protein (residues 120–356) and 1 nM Smac-1F in the same assay buffer. Polarization values were measured after incubation for 2–3 h, using the Ultra plate reader. The IC50 value, the inhibitor concentration at which 50% of the bound tracer was displaced, is determined from the plot using nonlinear least-squares analysis. For each assay, the Smac AVPI peptide was used as the control. Curve fitting was performed using GRAPHPAD PRISM (GraphPad Software, Inc., San Diego, CA).
Analytical Gel Filtration Experiments
To probe the mode of binding of our designed cyclized Smac peptides to XIAPs, we performed analytical gel filtration experiments containing BIR3-only (residues 241–356) protein, BIR2–BIR3 (residues 156–356) protein, or BIR2(E219R)–BIR3 (residues 156–356) protein. Analytical gel filtration experiments were performed on a Superdex 75 column (GE Healthcare) attached to an AKTA Purifier-10 system in 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 50 μM Zn(OAc)2, and 1 mM DTT. The recombinant protein was run on the column alone or after incubation with a tested Smac mimetic. Molecular weight standards from Amersham-Pharmacia were used to calibrate the column.
Cell-Free Caspase Functional Assays
MDA-MB-231 cell lysates were prepared by solubilizing cells in ice-cold buffer, 50 mM KCl, 5 mM EGTA, 2 mM MgCl2, 1 mM DTT, 0.2% CHAPS, and 50 mM HEPES (pH 7.5), containing cocktail protease inhibitors, incubating the cells on ice for 10 min, and then freezing them in liquid nitrogen. Cytochrome c and dATP were added to the cell lysates, which were then incubated at 30 °C in a water bath for 60 min to activate caspase-3/-7. Addition of recombinant XIAP BIR3 protein (500 nM) or XIAP linker–BIR2–BIR3 protein (50 nM) to the cell lysates completely suppressed the activity of caspase-3/-7. Different concentrations of a tested Smac mimetic (from 1 nM to 100 μM) were added to determine the restoration of the activity of these caspases.
For determination of caspase activity, 25 μM caspase-3/-7 substrate (Z-DEVD-AFC) (BioVision Inc.) was added. Fluorescence detection of substrate cleavage by caspase-3/-7 was carried out on a TECAN ULTRA plate reader using an excitation wavelength of 400 nm and an emission wavelength of 505 nm. The reaction was monitored for 1–2 h.
Cell Growth Assay
The effect of cyclized Smac peptides on the growth of MDA-MB-231 human breast cancer cells, purchased from the American type Culture Collection (ATCC), was evaluated with a WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] assay (Dojindo Molecular Technologies, Inc.). Cells (3–4 × 103 cells in each well) were cultured in 96-well tissue culture plates in 200 μL of medium containing various concentrations of Smac mimetics for the indicated time. At the end of the incubation, 20 μL of WST-8 dye was added to each well, the contents were incubated for an additional 1–3 h, and the absorbance was measured in a microplate reader (Molecular Devices, Sunnyvale, CA) at 450 nm. Cell growth inhibition was evaluated as the ratio of the absorbance of the sample to that of the control.
Cell-Based Caspase Activity
The effect of Smac peptides or mimetics on induction of caspase-3 activity after a 24 h treatment of MDA-MB-231 human breast cancer cells was evaluated quantitatively using a caspase-3 fluorometric assay kit from BioVision.
Crystallization of XIAP BIR3 Protein and Data Collection
Diffraction quality crystals were grown using vapor diffusion by mixing 2 μL of 10 mg/mL XIAP BIR3 protein in a 1:1.5 molar ratio of compound 3 with 2 μL of mother liquor containing 5% Peg 6000 and 1.4 M NaCl. Crystals grew in the tetragonal I422 space group with one molecule in the asymmetric unit and compound 3 positioned on a crystallographic 2-fold axis. Crystals were cyroprotected, prior to being flash-frozen in liquid nitrogen, with 5% Peg 6000, 1.4 M NaCl, and 25% ethylene glycol. Diffraction data for the crystals were collected at COMCAT 32-ID-B at the Advanced Photon Source equipped with a MAR165 CCD plate and processed with HKL2000 (27).
Structure Determination and Refinement
The BIR3–compound 3 complex structure was determined by molecular replacement methods using MolRep from the CCP4 program suite (CCP4). Another BIR3 domain crystal structure determined in our laboratory was used as the search model. After molecular replacement, the resulting coordinates were entered into CNS (28) for refinement. Difference Fourier maps (Fo – Fc) calculated after CNS rigid-body refinement of BIR3 against the BIR3–compound 3 diffraction data revealed the clear presence of compound 3 bound to protein. A model of compound 3 was built using O (29) and refined in CNS. Multiple rounds of CNS refinement and model building resulted in a structure refined to 2.1 Å resolution (Table 1). Simulated annealing omit maps calculated in CNS were used to check for the correct placement of atoms in the structure. The final structure was analyzed with PROCHECK (30). All residues are in the allowed regions of the Ramachandran plot.
Table 1.
Data Collection, Phasing, and Refinement Statistics
| Data Collection | |
| space group | I422 |
| unit cell | a = b = 102.478 Å, c = 65.281 Å |
| wavelength (Å) | 0.9685 |
| resolution (Å) | 2.1 (2.18–2.10)a |
| Rsym (%)b | 5.6 (19.0)a |
| 〈I/σI〉c | >20 (20)a |
| completeness (%)d | 99.9 (100)a |
| redundancy | 14.3 (14.7)a |
| Refinement | |
| resolution (Å) | 2.1 |
| R-factor (%)e | 21.7 |
| Rfree (%)f | 22.4 |
| no. of protein atoms | 784 |
| no. of water molecules | 60 |
| no. of unique reflections | 10431 |
| rmsdg | |
| bonds (Å) | 0.005 |
| angles (deg) | 1.1 |
Statistics for the highest-resolution bin of reflections in parentheses.
Rsym = ΣhΣj|Ihj − 〈Ih〉|/ΣhΣjIhj, where Ihj is the intensity of observation j of reflection h and 〈Ih〉 is the mean intensity for multiply recorded reflections.
Intensity of the signal-to-noise ratio.
Completeness of the unique diffraction data.
R-factor = Σh‖Fo| − |Fc‖/Σh|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively, for reflection h.
Rfree is calculated against a 10% random sampling of the reflections that were removed before structure refinement.
Root-mean-square deviation.
Molecular Modeling of Compound 3 in Complex with XIAP Containing BIR2 and BIR3 Domains
The model of compound 3 in complex with XIAP containing both BIR2 and BIR3 domains (residues 127–345) was built using the crystal structure of 3 in complex with XIAP BIR3 protein and a crystal structure of the XIAP BIR2 domain in complex with caspase-3 (PDB entry 1I3O). In the modeling, one BIR3 monomer was replaced with a BIR2 molecule (F127–I237) after structural alignment using Swiss PDB viewer (31). The initial model lacked a 12-residue segment (R238–N249) that connects the BIR2 and BIR3 domains in the native protein. To incorporate the missing segment into the model, we used a loop from the XIAP BIR3 domain determined by NMR structure (PDB entry 1TFQ), in which the loop segment of S241–N249 was determined. After F250 had been aligned between the XIAP BIR3 protein in 1TFQ and our template structure, the loop segment (S241–N249) oriented toward the XIAP BIR2 protein did not have steric clashes with either domain of the protein. However, the shortest distance between I237 and S241 was 18 Å. Since Gly has a flexible backbone motion, the backbone amide bond of G232 in XIAP BIR2 protein was rotated to bring I237 closer to S241 while preventing collisions of the two protein domains. Finally, a distance of 7.7 Å between I237 and S241 was obtained; three additional missing residues (R238–E240) were added using Sybyl (32), and a local minimization between I237 and S241 was performed. The resulting complex was further refined through extensive molecular dynamics (MD) simulations using the Amber program suite (33).
Results
Design of a Cyclized, Bivalent Smac Mimetic
Structural and biological studies have demonstrated that Smac binds to both the BIR2 and BIR3 domains through its AVPI tetrapeptide binding motif (3, 4). It was shown that replacement of the second residue (valine) in the Smac AVPI peptide with a lysine residue did not have a detrimental effect on binding to the XIAP BIR3 protein (4) and replacement of the fourth residue (isoleucine) with phenylalanine improved the binding affinity for XIAP BIR3 protein (34). Accordingly, in our design, we used the AKPF tetrapeptide as the basic monovalent template. In our previous study, we showed that introduction of an N-methyl group into the Ala residue can dramatically improve the cellular activity of our resulting Smac mimetics without a detrimental effect on their binding affinities for XIAP BIR3 protein (18). Accordingly, we replaced the alanine in AKPF with an N-methylated alanine. Using CH3-AKPF as the basic monovalent template, we designed and synthesized a cyclized, conformationally constrained bivalent Smac mimetic (compound 3), which contains two AKPF binding units (Figure 1A). Compound 3 was designed and synthesized by chemically tethering the amino group of the lysine side chain from one binding unit through an amide bond to the backbone carbonyl of a phenylalanine from the other binding motif.
The alanine residue in the Smac AVPI peptide plays a key role in its binding to XIAP through both hydrogen bonding and hydrophobic contacts (4, 5). To test the specificity, compound 4 was designed as an inactive control by removing the both N-methylated alanines from compound 3 (Figure 1A).
Determination of Binding Affinities for XIAP BIR2-Only and BIR3-Only Proteins
The binding affinities of monovalent compound 2, bivalent compound 3, and the inactive control 4 for XIAP BIR3-only protein were evaluated using a previously established sensitive and robust fluorescence polarization (FP) binding assay (25), and the results are shown in Figure 2A.
Figure 2.
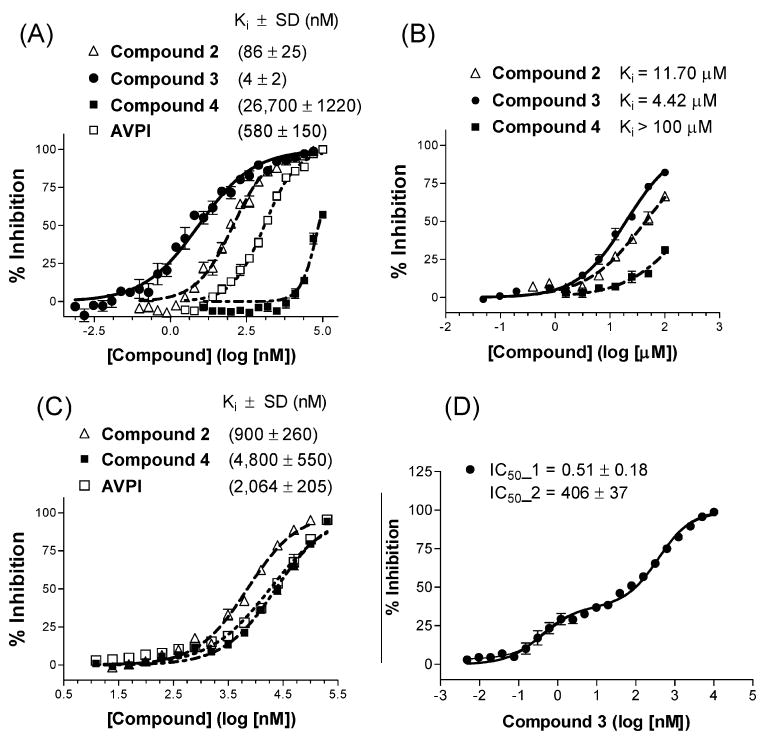
Competitive binding curves for binding of compounds 2, 3, and 4 to (A) the BIR3 domain, (B) the BIR2 domain, and (C and D) the linker–BIR2–BIR3 protein determined using a fluorescence polarization-based binding assay. A synthetic mutated Smac peptide (AbuRPFK), C-terminally labeled with 5-carboxyfluorescein (5-Fam), and its complex with recombinant XIAP BIR3 protein (residues 241–356) and XIAP BIR2 protein (residues 120–240) were used to evaluate competitive BIR3 domain binding of tested peptides. A synthetic bivalent Smac mimetic peptide, C-terminally labeled with 5-carboxyfluorescein (5-Fam), and its complex with recombinant XIAP linker–BIR2–BIR3 protein (residues 120–356) were used to evaluate competitive binding of tested peptides against full-length XIAP.
Compound 2 binds to XIAP BIR3 with a Ki value of 86 nM. In comparison, bivalent compound 3 has a Ki value of 4 nM under the same assay conditions (Figure 2A) and is thus 22 times more potent than the monovalent compound 2. As predicted, compound 4 binds to the XIAP BIR3 protein with a very weak affinity (Ki value of 27 μM) and is >6000 times less potent than compound 3, demonstrating the binding specificity of 3 for XIAP BIR3 protein.
We next determined the binding affinities of compounds 2, 3, and 4 for XIAP BIR2-only protein using the same fluorescently labeled tracer (25). Compounds 2, 3, and 4 have Ki values of 11.7, 4.4, and >100 μM, respectively, for binding to XIAP BIR2-only protein (Figure 2B). Thus, although compound 3 binds to both XIAP BIR2-only and BIR3-only proteins, it binds to BIR3-only protein with an ∼1000 times higher affinity than to BIR2-only protein.
Determination of Binding Affinities for XIAP Containing both BIR2 and BIR3 Domains
To measure the binding affinities of these compounds for XIAP containing both BIR2 and BIR3 domains (L–BIR2–BIR3), we have established a new FP-based assay using a fluorescently labeled Smac-based peptide tracer (26). Using this new FP assay, we have determined the binding affinities of compounds 2, 3, and 4 and the AVPI peptide for the XIAP L–BIR2–BIR3 protein (Figure 2C,D). Compounds 2 and 4 and the AVPI peptide bind to XIAP with Ki values of 900, 4800, and 2064 nM, respectively. Analysis of the binding data for compound 3 using the Klotz plot revealed several characteristics of multiple binding sites, such as a Hill slope of ≠1 (∼0.4) and multiple inflection points on the obtained curve. Since we employed XIAP containing BIR2 and BIR3 domains, a two-site binding equation was used to fit the data. A biphasic dose–response curve shows the best fit (Figure 2D), the first, tighter binding site with an IC50 of 0.5 nM and the second, weaker binding site with an IC50 of 406 nM.
To test if the linker preceding the BIR2 domain is involved in the binding of XIAP containing both BIR domains, we also evaluated the binding of compound 3 to XIAP BIR2–BIR3 protein without the linker (residues 156–356). Our data showed that compound 3 binds to the XIAP BIR2–BIR3 protein with essentially the same affinity as it does to the L–BIR2–BIR3 protein, indicating that the linker does not interact with compound 3 (data not shown).
Probing the Modes of Binding of Monovalent Compound 2 and Bivalent Compound 3 to XIAPs by Analytical Gel Filtration
To further understand the mode of binding of bivalent compound 3, we have carried out analytical gel filtration analysis using XIAP BIR3-only (residues 241–356) and BIR2–BIR3 (residues 156–356) proteins. XIAP BIR2–BIR3 protein was used in these experiments because it has a solubility, superior to that of XIAP L–BIR2–BIR3 protein, and compound 3 binds to these two proteins with identical affinities.
In the gel filtration experiments, XIAP BIR3-only protein elutes as a monomer with an apparent molecular mass of 17.4 kDa and stays as a monomer when incubated with compound 2 or 4. In contrast, compound 3 clearly induces dimerization of the XIAP BIR3-only protein with a molecular mass of 31.4 kDa (Figure 3A). These data suggest that each binding unit in compound 3 binds to one BIR3 protein molecule and forms a 1:2 stoichiometric complex between compound 3 and the protein while the monovalent compound 2 forms a 1:1 stiochiometric complex with the protein.
Figure 3.
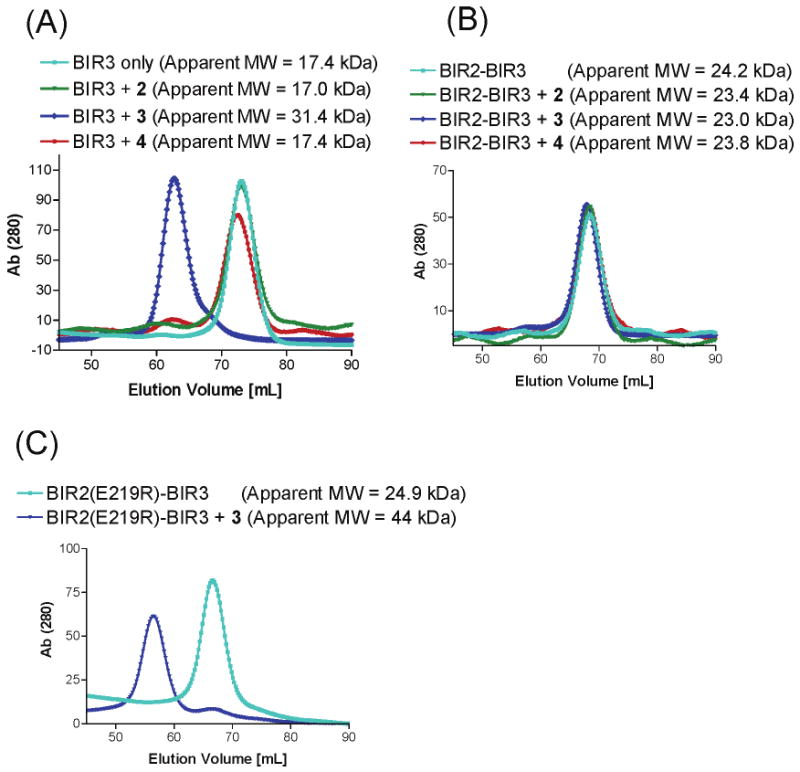
Analytical gel filtration elution profiles of compounds 2, 3, and 4 in complex with XIAP (A) BIR3, (B) BIR2–BIR3, and (C) BIR2(E219R)–BIR3 proteins. XIAPs were run on the Superdex 75 column equilibrated with 20 mM Tris-HCl (pH 7.5) containing 200 mM NaCl, 50 mM zinc acetate, and 1 mM dithiothreitol, alone and after incubation with a 1.5-fold molar excess of tested compounds. Molecular mass standards from Amersham Pharmacia were used to calibrate the column. The corresponding absorbance scale is indicated (absorbance units at 280 nm).
We next determined if bivalent compound 3 can also induce dimerization of XIAP containing both BIR2 and BIR3 domains. Incubation of compound 3 with XIAP BIR2–BIR3 protein does not induce dimerization (Figure 3B). Instead, compound 3 appears to make the protein become slightly more compact, as judged by the apparent molecular mass in the presence or absence of compound 3. Both compounds 2 and 4 fail to induce dimerization of the XIAP BIR2–BIR3 protein. These gel filtration data, together with the binding affinities, show that when interacting with XIAP BIR3-only protein, each molecule of bivalent compound 3 interacts with two protein molecules and induces dimerization. In contrast, when interacting with XIAP containing both BIR2 and BIR3 domains, each molecule of compound 3 binds to one protein molecule and does not cause protein dimerization.
Although our binding and gel filtration data strongly suggest that bivalent compound 3 concurrently interacts with both BIR2 and BIR3 domains, these data alone did not provide conclusive evidence that BIR2 is involved in the interaction. To confirm that the BIR2 domain is directly involved in the binding of XIAP BIR2–BIR3 protein to compound 3, we mutated Glu219 into Arg (E219R) in the BIR2 domain to generate the BIR2(E219R)–BIR3 mutant. The rationale for this mutation is that in our modeled structure, E219 in the BIR2 domain has an extensive charge–charge interaction with the positively charged N-methylated amino group in compound 3. The gel filtration experiments using this mutated protein showed that in the absence of 3, the protein behaves as a monomer (Figure 3C) but in the presence of compound 3, dimerization of the XIAP BIR2(E219R)–BIR3 protein is observed. These data provide evidence that BIR2 is directly involved in the interaction of XIAP BIR2–BIR3 protein with bivalent compound 3. The gel filtration experiments using both native and mutated proteins, together with the binding data, provide clear evidence that bivalent compound 3 interacts simultaneously with both the BIR2 and BIR3 domains in XIAP.
Functional Antagonism against XIAPs
We established cell-free functional assays using both XIAP BIR3-only and XIAP L–BIR2–BIR3 proteins and evaluated compounds 2 and 3 for their ability to antagonize XIAP BIR3-only or L–BIR2–BIR3 protein on a functional level. Compound 4 was used as an inactive control in these assays.
Addition of cytochrome c and dATP to cellular extracts leads to robust activation of caspase-3/-7 in a time-dependent manner (Figure 4A). XIAP BIR3-only protein dose-dependently inhibits the activity of these caspases and achieves complete inhibition of these caspases at 500 nM (Figure 4A). Since XIAP BIR3 protein is known not to interact with caspase-3/-7 directly, these data indicate that XIAP BIR3-only protein inhibits the activity of caspase-3/-7 via binding to and inhibition of caspase-9.
Figure 4.
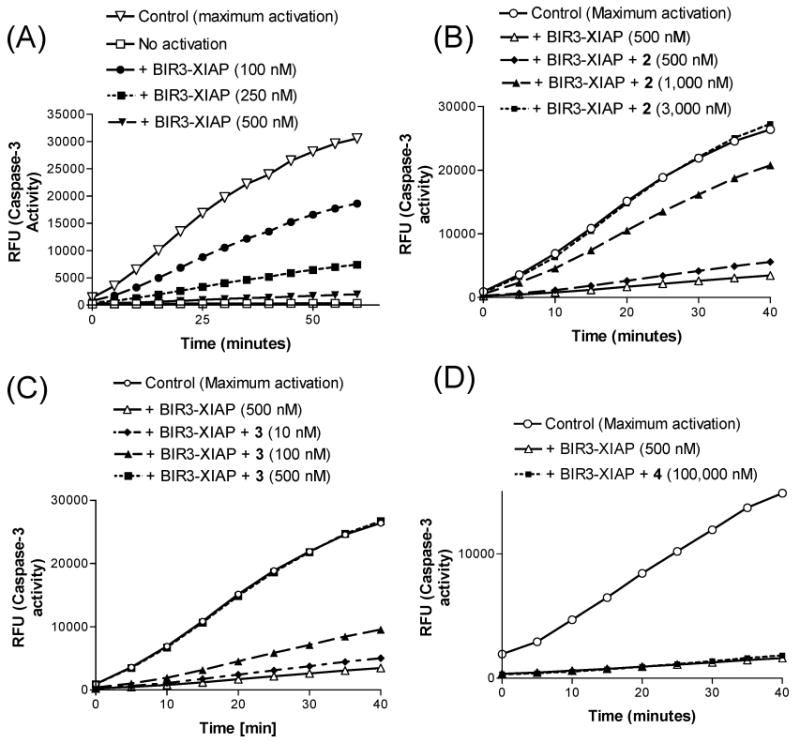
(A) XIAP BIR3 protein abolishes caspase-3 activity in a dose-dependent manner. (B–D) Kinetic analysis for the relief of XIAP BIR3 protein-mediated caspase-3 by compounds 2, 3, and 4. MDA-MB-231 cell extract was activated with bovine cytochrome c and dATP. The caspase activity was inhibited by adding 500 nM recombinant XIAP BIR3, and varying concentrations of tested compounds were added. The onset of caspase-3 activity was monitored as a fluorogenic substrate (DEVD-AFC, BioVision) was cleaved in situ (rfu, relative fluorescence units).
Compound 3 at an equal molar concentration (500 nM) of the XIAP BIR3-only protein fully restores the activity of caspase-3/-7 (Figure 4B). In comparison, compound 2 at 500 nM has a minimal effect but completely restores the activity of these caspases at 3000 nM (Figure 4C). The inactive control 4 has no effect at 100 μM (Figure 4D). These functional data thus show that both compounds 2 and 3 antagonize XIAP BIR3 protein, but 3 is 6 times more potent than 2, consistent with their binding affinities for XIAP BIR3 protein.
In the functional assays, XIAP L–BIR2–BIR3 protein functions as a much more potent antagonist of caspase-3/-7 than XIAP BIR3 protein. The L–BIR2–BIR3 protein at 50 nM is sufficient to completely inhibit the activity of these caspases (Figure 5A). Both compounds 2 and 3 can restore the activities of caspase-3/-7 by antagonizing XIAP L–BIR2–BIR3 protein, but compound 3 is much more potent than compound 2 (Figure 5B,C). While compound 3 at a concentration of 500 nM fully restores the activity of these caspases, a 200-fold higher concentration (100000 nM) of compound 2 is needed to do so. Compound 4 has no effect at 100000 nM (Figure 5D).
Figure 5.
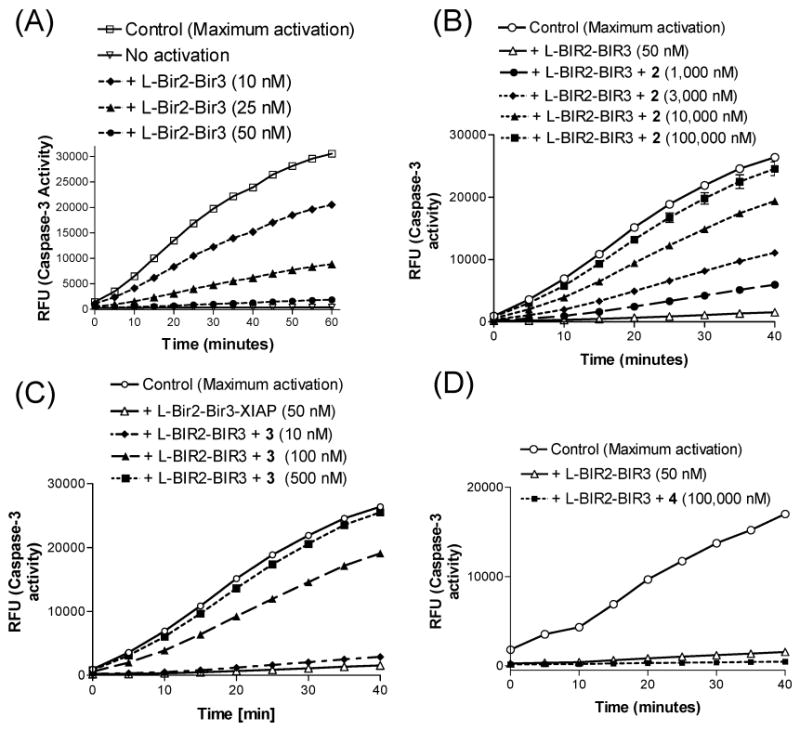
(A) XIAP L–BIR2–BIR3 protein abolishes caspase-3 activity in a dose-dependent manner. (B–D) Kinetic analysis for the relief of XIAP L–BIR2–BIR3 protein-mediated caspase-3 inhibition by compounds 2, 3, and 4. MDA-MB-231 cell extract was activated with bovine cytochrome c and dATP. The caspase activity was inhibited by adding 50 nM recombinant XIAP L–BIR2–BIR3 protein, and varying concentrations of tested compounds were added. The onset of caspase-3 activity was monitored as a fluorogenic substrate (DEVD-AFC, BioVision) was cleaved in situ (rfu, relative fluorescence units).
Taken together, these functional data provide evidence that bivalent compound 3 functions as a potent antagonist of both XIAP BIR3-only and L–BIR2–BIR3 proteins. However, while compound 3 is only 6 times more potent than compound 2 in antagonizing XIAP BIR3-only protein, compound 3 is 200 times more potent than compound 2 in antagonizing XIAP L–BIR2–BIR3 protein. Hence, these functional data show that bivalent compound 3 is a far more effective antagonist of XIAP L–BIR2–BIR3 protein than its corresponding monovalent compound 2, consistent with their binding affinities for XIAP L–BIR2–BIR3 protein.
Evaluation of Bivalent Compound 3 in Inhibition of Cell Growth and Induction of Caspase Activity in Cancer Cells
Another major motivation for our design of compound 3 is that a cyclized peptide would be more resistant to protease degradation and more cell-permeable than its linear counterparts. We thus evaluated 2, 3, and 4 for their activity in inhibition of cell growth in the MDA-MB-231 human breast cancer cell line, which has been shown to be sensitive to other Smac mimetics in previous studies (18, 19) (Figure 6A). Our results show that 3 is effective in inhibition of cell growth and has an IC50 value of 2.2 μM, indicating that compound 3 is indeed cell-permeable. In comparison, 2 and 4 fail to show any significant activity at concentrations as high as 100 μM.
Figure 6.
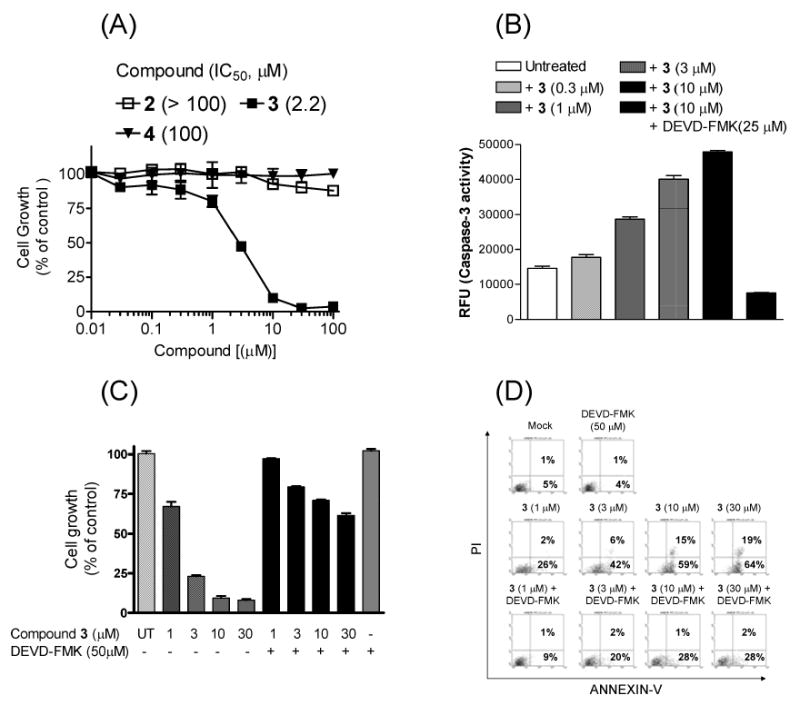
(A) Inhibition of cell growth by compounds 2, 3, and 4 in the MDA-MB-231 human breast cancer cell line. Cells were treated with compounds for 4 days, and cell growth was assessed using a WST assay. (B) Caspase-3 activity in MDA-MB-231 cell lysates upon treatment with different concentrations of compound 3 for 16 h, with or without Z-DEVD-FMK, an inhibitor of caspase-3. The relative fluorescence intensity units (RFU) were determined after a 1 h incubation with a caspase-3 specific fluorogenic substrate (Z-DEVD-AFC). (C) Cell growth inhibition of compound 3 in the MDA-MB-231 cancer cell line with or without Z-DEVD-FMK. Cells were treated for 4 days with compound 3 alone, Z-DEVD-FMK alone, or both in combination. Cell growth was assessed using a WST assay. (D) Induction of apoptosis by compound 3 in the MDA-MB-231 cell line with or without Z-DEVD-FMK. Cells were treated for 24 h, and apoptosis was assessed using annexin V/propodium iodide (PI) double staining and analyzed by flow cytometry.
Since compound 3 effectively antagonizes XIAP, promotes activation of caspase-3/-7 in cell-free functional assays, and inhibits cell growth, we determined whether 3 can activate caspase-3/-7 in the MDA-MB-231 cell line (Figure 6B). Compound 3 indeed induces activation of caspase-3/-7. The induced activation of caspase-3/-7 by compound 3 can be abolished by Z-DEVD-FMK, a cell-permeable, specific inhibitor of caspase-3/-7.
To determine if the cell growth inhibition of compound 3 in the MDA-MB-231 cancer cell line depends upon caspase-3/-7, we treated the cells with compound 3 alone or in the presence of Z-DEVD-FMK (Figure 6C). Our data showed that while compound 3 inhibits cell growth in a dose-dependent manner, Z-DEVD-FMK can effectively attenuate the activity of compound 3, clearly indicating that the cell inhibitory activity of compound 3 is mediated by caspase-3/-7.
To determine if compound 3 induces apoptosis in the MDA-MB-231 cancer cell line, we performed annexin V/propodium iodide (PI) analysis by flow cytometry (Figure 6D). As one can see, compound 3 effectively induces the MDA-MB-231 cells to undergo apoptosis in a dose-dependent manner. Furthermore, the induction of apoptosis by compound 3 is caspase-3/-7-dependent since Z-DEVD-FMK can effectively inhibit the activity. Of note is the fact that Z-DEVD-FMK alone has no effect on cells.
Determination of a High-Resolution Crystal Structure of XIAP BIR3 Protein in a Complex with Compound 3
To gain more insight into the interactions of 3 with XIAPs, we attempted to determine the crystal structures of compound 3 in a complex with XIAP BIR3-only and BIR2–BIR3 proteins. Although we were unable to obtain crystals of XIAP BIR2–BIR3 protein alone or in the presence of compound 3, we were successful in obtaining diffraction quality crystals of XIAP BIR3-only protein in a complex with compound 3. The crystal structure was determined to 2.1 Å resolution (Figure 7).
Figure 7.
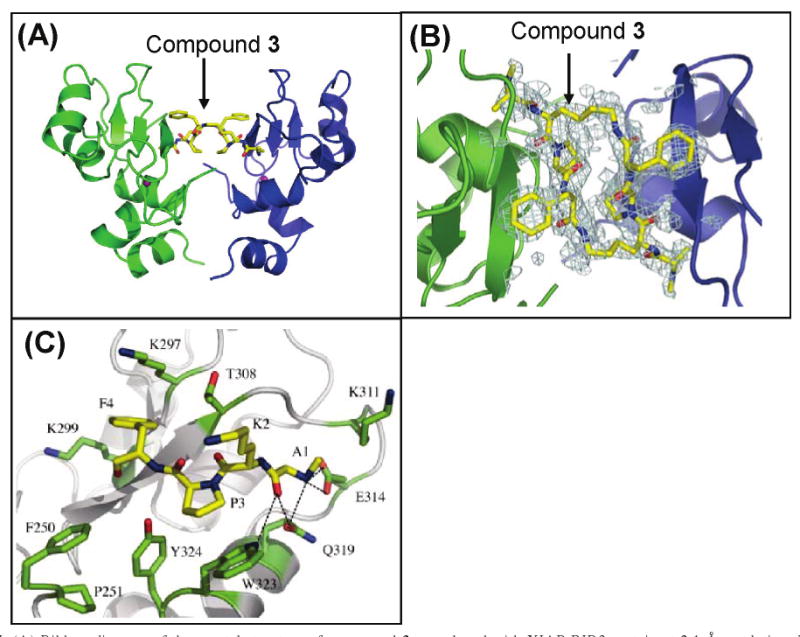
(A) Ribbon diagram of the crystal structure of compound 3 complexed with XIAP BIR3 protein at 2.1 Å resolution. Zinc ions are colored purple. (B) Initial difference electron density of compound 3 in the XIAP BIR3 binding site. Two XIAP BIR3 molecules are displayed in blue and green, and compound 3 is shown as sticks with carbons colored yellow, oxygens red, and nitrogens blue. (C) Interactions between one BIR3 domain and one binding unit of compound 3. Compound 3 (residues A1, K2, P3, and F4) is shown as sticks with carbons colored yellow, oxygens red, and nitrogens blue; side chains of the BIR3 domain are shown with green carbons.
In this high-resolution crystal structure, two XIAP BIR3 molecules bind one molecule of 3, which is consistent with our gel filtration experiments in solution. In the structure, the binding pockets of the two BIR3 molecules come together to form one large pocket that surrounds a single molecule of 3. The binding pocket is lined with hydrophobic residues, including four residues (Phe250, Pro251, Trp323, and Tyr324) from both protein molecules. The total buried surface area of the two BIR3 molecules and 3 is approximately 1500 Å2. The interaction between XIAP BIR3 and one binding unit of 3 is very similar to that observed previously in two other structures of XIAP BIR3 complexed with Smac protein or peptide (4, 5) (Figure 7C).
Several key interactions occur between 3 and one XIAP BIR3 molecule. The positively charged methylated amino group in the first alanine residue of 3 has strong charge–charge interactions with the negatively charged Glu314 residue of BIR3, whereas its methyl side chain inserts into the small pocket formed by Trp310, Gln319, and Leu307. The amino and carbonyl groups of the second lysine residue form two strong hydrogen bonds with Thr308 backbone amino and carbonyl groups. The proline ring of the third residue is in close contact with Trp323. The phenyl ring in the fourth residue inserts into a hydrophobic pocket formed by the side chains of Leu292, Lys297, and Lys299 with its amino group forming a strong hydrogen bond with the carbonyl group of Gly306.
Hence, this high-resolution crystal structure clearly shows that bivalent Smac mimetic 3 induces dimerization of XIAP BIR3 and provides a structural basis for high-affinity interaction.
Modeling Studies of Compound 3 Complexed with XIAP L–BIR2–BIR3 Protein
Because of the high degree of sequence and structural homology between XIAP BIR2 and BIR3, the high-resolution crystal structure of XIAP BIR3 complexed with compound 3 also provides an ideal template for modeling of XIAP L–BIR2–BIR3 protein complexed with compound 3. In our modeling, one BIR3 molecule in the crystal structure was replaced with a BIR2 structure and the connecting loop between BIR2 and BIR3 domains was modeled. The resulting complex was further refined through extensive molecular dynamics (MD) simulations (Figure 8A).
Figure 8.
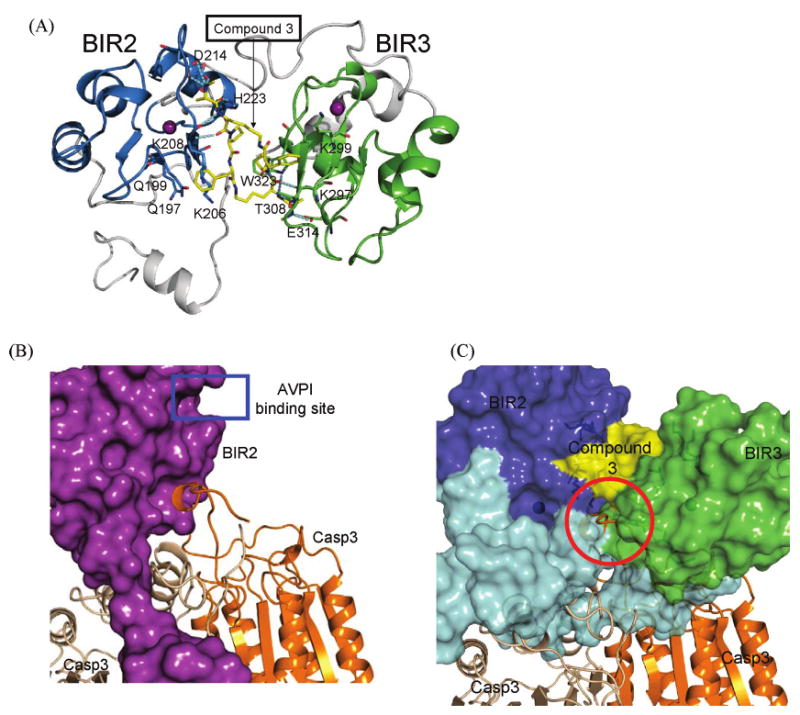
(A) Proposed model between compound 3 and XIAP L–BIR2–BIR3 protein. Segments colored blue and green are the BIR2 and BIR3 domains, respectively. Segments colored gray include both the linker preceding BIR2 and the loop region between BIR2 and BIR3. Compound 3 is colored yellow. Zinc ions are colored purple. Key residues interacting with compound 3 are displayed, and the hydrogen bonds are depicted as cyan dashed lines. (B) Inhibition of XIAP L–BIR2 (purple) with caspase-3 from the crystal structure (PDB entry 1I3O). (C) Proposed model of inhibition of compound 3 with XIAP L–BIR2–BIR3 against caspase-3. The refined model of XIAP L–BIR2–BIR3 is superimposed with one BIR2 domain in the crystal structure between XIAP BIR2 and caspase-3. Segments colored blue and green are the BIR2 and BIR3 domains, respectively. Segments colored cyan include both the linker preceding BIR2 and the loop region between BIR2 and BIR3. The yellow region is compound 3. Segments colored wheat and orange are the dimeric caspase-3.
The modeled structure (Figure 8A) shows that the BIR2 and BIR3 domains come together to form a large binding pocket, similar to that observed in the crystal structure of XIAP BIR3 complexed with compound 3. The BIR2 and BIR3 domains are brought together by the strong interactions between each binding unit in compound 3 with each BIR domain.
On the BIR2 side, for the first alanine residue, the positively charged methylated amino group has strong charge–charge interactions with the negatively charged Asp214 and Glu219 residues, whereas the methyl group in the side chain lodges into a small hydrophobic pocket formed by Leu207 and Trp210. For the second lysine residue, its amino and carbonyl groups form two optimal hydrogen bonds with Lys208 backbone amino and carbonyl groups. For the proline residue, its five-membered ring is in close contact with the side chain of His223 in BIR2 and also with that of Trp323 in BIR3. For the fourth residue, the phenyl group inserts into a pocket formed by the hydrophobic portion of the side chains of Lys208, Gln197, and Gln199, while its amino group forms a weak hydrogen bond with the carbonyl group of Lys206. On the BIR3 side, all the interactions observed in the crystal structure of XIAP BIR3-only protein complexed with compound 3 are preserved after the MD refinement.
Our crystal and modeled structures also reveal critical differences in the interactions of one binding unit in compound 3 with XIAP BIR2 and BIR3 domains, respectively. First, Trp323 in XIAP BIR3 is replaced with a histidine residue (His223) in XIAP BIR2, which weakens the hydrophobic interaction with the proline residue in compound 3. Second, Glu314 in XIAP BIR3 forms a strong charge–charge interaction with the positively protonated methylated amino group in compound 3. This glutamate residue is replaced with an aspartate residue (Asp214) in XIAP BIR2. Since Asp has a shorter side chain than glutamate, the charge–charge interaction of the positively protonated methylated amino group in 3 with Asp214 in XIAP BIR2 appears to be weaker than that in XIAP BIR3.
Our modeled structure of compound 3 in complex with XIAP containing both BIR2 and BIR3 domains also sheds light on the functional antagonism of compound 3 against XIAP to activate caspase-9 and caspase-3/-7. XIAP inhibits caspase-9 by binding to the ATPF motif in the small subunit of caspase-9 via its BIR3 domain where Smac and compound 3 bind. Thus, compound 3, like the Smac protein, nullifies the inhibition of XIAP to caspase-9 through direct competition. In the crystal structure of XIAP BIR2 complexed with caspase-3, the major interactions are observed at two different sites (Figure 8B). At the first site, the linker immediately preceding the BIR2 domain in XIAP binds to a well-defined binding pocket in caspase-3, which is known to be the major energetic determinant for binding and is critical for the inhibition of caspase-3 activity. The second binding site is mediated by (a) the SGVDD segment of caspase-3 and the AVPI binding site in XIAP BIR2 and (b) a small helix in caspase-3 and a pocket near the AVPI binding site in XIAP BIR2. Superposition of the crystal structure of XIAP BIR2 in the complex with caspase-3 and our modeled structure of compound 3 in the complex with XIAP BIR2–BIR3 protein showed that the simultaneous binding of compound 3 with BIR2 and BIR3 domains precludes the small helix in caspase-3 from binding to XIAP BIR2 due to steric clashes (Figure 8C). Although the interaction at the second binding site is less extensive than that at the first site, it serves a cooperative and crucial role for the inhibition of XIAP to caspase-3 (12, 13). Thus, our modeled structure suggests that compound 3 can efficiently antagonize XIAP by preventing the interaction between the XIAP BIR2 domain and caspase-3 but not by blocking the interaction of the linker preceding BIR2 with caspase-3.
Discussion
XIAP functions as an efficient apoptosis suppressor in cells by concurrently binding to an initiator caspase-9 and two effectors, caspase-3/-7. The binding and inhibition of XIAP with these caspases are mediated by two different BIR domains, namely, the BIR2 and BIR3 domains. While the BIR3 domain of XIAP binds to caspase-9 and inhibits its activity, the BIR2 domain, together with the immediately preceding short linker, binds to caspase-3/-7, inhibiting their activity. Smac, the endogenous antagonist of XIAP, forms a dimer and binds to both the BIR2 and BIR3 domains in XIAP and efficiently removes the inhibition of XIAP with caspase-9 and caspase-3/-7.
In this study, we have designed and synthesized a cyclized, conformationally constrained Smac mimetic (compound 3) containing two AKPF binding units to mimic the mode of the binding of the dimeric Smac protein to XIAP. We have characterized the interaction of compound 3 with XIAP BIR2-only, BIR3-only, and L–BIR2-BIR3 proteins.
Our studies showed that compound 3 binds to XIAP BIR2 and BIR3-only domains with very different binding affinities, 4.4 μM and 4 nM, respectively. Furthermore, compound 3 binds to XIAP L–BIR2–BIR3 protein with a biphasic dose–response curve characteristic of two binding sites, with an IC50 value of 0.5 nM for the first site and 406 nM for the second site. Gel filtration experiments demonstrated that compound 3 induces clear dimerization of XIAP BIR3-only protein, suggesting that each of the two binding units in compound 3 binds to one molecule of the BIR3-only protein. However, XIAP BIR2–BIR3 protein remains a monomer in the presence of compound 3, indicating a lack of dimerization. When a key binding residue (E219) in the BIR2 domain was mutated to arginine (E219R), the mutated BIR2(E219R)–BIR3 protein remains a monomer without compound 3 but dimerizes in the presence of compound 3. These data show that one molecule of the bivalent compound 3 interacts with two molecules of XIAP BIR3-only protein, forming a 1:2 stoichiometric complex. In contrast, bivalent compound 3 concurrently interacts with both BIR2 and BIR3 domains in the presence of XIAP containing both BIR domains and does not induce dimerization of the protein. Our data are consistent with two recent studies (35, 36), which also showed that bivalent Smac-based peptide ligands interact with both BIR2 and BIR3 domains in XIAP and do not induce dimerization of XIAP containing both BIR domains. Interestingly, another study (37) using a different set of bivalent Smac-based peptide ligands showed that such compounds interact with only the BIR3 domain and not the BIR2 domain in XIAP. Since these studies did not employ the same bivalent Smac-based ligands, it remains possible that different types of bivalent ligands may indeed have different modes of interaction with XIAP containing both BIR2 and BIR domains. Further studies need to be performed to clarify the discrepancy.
In the cell-free functional assays, compound 3 acts as a potent antagonist against both XIAP BIR3-only and L–BIR2–BIR3 proteins. While compound 3 is 6 times more potent than its monovalent counterpart compound 2 in antagonizing XIAP BIR3 protein, it is >200 times more potent than 2 in antagonizing XIAP L–BIR2–BIR3. These results show that a bivalent Smac mimetic, which concurrently binds to both BIR domains and removes the inhibition of XIAP with caspase-3/-7, is a much more efficient antagonist against XIAP L–BIR2–BIR3 protein than the corresponding monovalent Smac mimetic.
The determination of a high-resolution crystal structure of compound 3 in a complex with the XIAP BIR3 domain confirmed that one molecule of compound 3 interacts with two molecules of XIAP BIR3 protein, as shown by the gel filtration results obtained in solution. This crystal structure also provides a structural basis for the high-affinity binding of compound 3 to XIAP BIR3.
Using the crystal structure, we have modeled the structure of compound 3 in a complex with XIAP L–BIR2–BIR3. Our modeled structure shows that compound 3, through its bivalent binding units, has strong interactions with both BIR2 and BIR3 domains and brings BIR2 and BIR3 domains together in the same XIAP molecule to form a large, single binding site. Importantly, this modeled structure sheds light on the functional antagonism of compound 3 against XIAP L–BIR2–BIR3. A previous study demonstrated that the BIR2 domain in XIAP works cooperatively with the immediate preceding linker region for binding to and inhibition of caspase-3 (12). However, this study, together with a recent report (36), clearly shows that binding of Smac mimetics to the AVPI binding site in the BIR2 domain of XIAP can effectively remove its inhibition with caspase-3/-7, without the need for interacting with the linker preceding the BIR2 domain. Our study strongly suggests that small molecules that bind to the AVPI binding pocket in XIAP BIR2 can function as effective antagonists to remove the inhibition of XIAP with caspase-3/-7 without interacting with the linker preceding BIR2.
Because of its cyclized structure, compound 3 is cell-permeable and effectively activates caspases in cancer cells. Compound 3 is potent and effective in inhibition of cell growth and induction of apoptosis in the MDA-MB-231 breast cancer cell line in a casaspe-3/-7-dependent manner. Hence, compound 3 serves as a useful biochemical and pharmacological tool with which to further elucidate the function of XIAP in vitro and its role in apoptosis and other cellular processes. Since compound 3 is designed to mimic Smac protein, we predict that it may also bind to other IAP proteins, such as cIAP-1/2 and ML-IAP, with high affinities. A study aiming to characterize its detailed interaction with other IAP proteins is underway and will be reported in due course. Compound 3 represents a promising lead compound for the design of ultrapotent, cell-permeable antagonists of XIAP and other IAP proteins for the treatment of human cancers.
Acknowledgments
We thank Dr. G. W. A. Milne for critical reading of the manuscript.
Footnotes
Financial support from the Breast Cancer Research Foundation and the National Cancer Institute (NIH Grant R01CA109025).
Diffraction data and structural coordinates for the XIAP–compound 3 complex have been deposited in the Protein Data Bank as entry 2VSL.
Abbreviations: Boc, tert-butoxycarbonyl; DCC, N,N′-dicyclohexylcarbodiimide; DCM, dichloromethane; DCU, N,N′-dicyclohexylurea; DIEA, N,N-diisopropylethylamine; DMF, N,N-dimethylformamide; EDCI, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide; Fmoc, fluorenylmethoxycarbonyl; HATU, O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate; HOAt, 1-hydroxy-7-azabenzotriazole; HOBt, 1-hydroxybenzotriazole; HBTU, N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate; MBHA, 4-methylbenzhydrylamine; Mtt, 4-methyltrityl; NMP, N-methylpyrrolidine; TFA, trifluoroacetic acid; Trt, trityl; DTT, dithiothreitol.
References
- 1.Deveraux QL, Reed JC. IAP family proteins: Suppressors of apoptosis. Genes Dev. 1999;1:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 2.Salvesen GS, Duckett CS. IAP proteins: Blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 3.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 4.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 5.Liu Z, Sun C, Olejniczak ET, Meadows R, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–1008. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 6.Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H. Structural basis of caspase inhibition by XIAP: Differential roles of the linker versus the BIR domain. Cell. 2001;104:781–790. [PubMed] [Google Scholar]
- 7.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Dataa P, Alnemri ES, Shi Y. Structural Basis of Caspase-7 Inhibition by XIAP. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 8.Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP inhibits caspase-3 and -7 using two binding sites: Evolutionarily conserved mechanism of IAPs. EMBO J. 2005;24:645–655. doi: 10.1038/sj.emboj.7600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 10.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 11.Shiozaki EN, Shi Y. Caspases, IAPs and Smac/DIABLO: Mechanisms from structural biology. Trends Biochem Sci. 2004;29:486–494. doi: 10.1016/j.tibs.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y, Rich RL, Myszka DG, Wu H. Requirement of both the second and third BIR domains for the relief of X-linked inhibitor of apoptosis protein (XIAP)-mediated caspase inhibition by Smac. J Biol Chem. 2003;278:49517–49522. doi: 10.1074/jbc.M310061200. [DOI] [PubMed] [Google Scholar]
- 14.Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, Scudiero DA, Tudor G, Qui YH, Monks A, Andreeff M, Reed JC. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6:1796–1803. [PubMed] [Google Scholar]
- 15.Holcik M, Gibson H, Korneluk RG. XIAP: Apoptotic brake and promising therapeutic target. Apoptosis. 2001;6:253–261. doi: 10.1023/a:1011379307472. [DOI] [PubMed] [Google Scholar]
- 16.Sun H, Nikolovska-Coleska Z, Yang CY, Xu L, Liu M, Tomita Y, Pan H, Yoshioka Y, Krajewski K, Roller PP, Wang S. Structure-based design of potent, conformationally constrained Smac mimetics. J Am Chem Soc. 2004;126:16686–16687. doi: 10.1021/ja047438+. [DOI] [PubMed] [Google Scholar]
- 17.Sun H, Nikolovska-Coleska Z, Yang CY, Xu L, Tomita Y, Krajewski K, Roller PP, Wang S. Structure-based design, synthesis, and evaluation of conformationally constrained mimetics of the second mitochondria-derived activator of caspase that target the X-linked inhibitor of apoptosis protein/caspase-9 interaction site. J Med Chem. 2004;47:4147–4150. doi: 10.1021/jm0499108. [DOI] [PubMed] [Google Scholar]
- 18.Sun H, Nikolovska-Coleska Z, Lu J, Qiu S, Yang CY, Gao W, Meagher J, Stuckey J, Wang S. Design, synthesis, and evaluation of a potent, cell-permeable, conformationally constrained second mitochondria derived activator of caspase (Smac) mimetic. J Med Chem. 2006;49:7916–7920. doi: 10.1021/jm061108d. [DOI] [PubMed] [Google Scholar]
- 19.Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak ET, Oleksijew A, Oltersdorf T, Rosenberg SH, Shoemaker AR, Tomaselli KJ, Zou H, Fesik SW. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem. 2004;47:4417–4426. doi: 10.1021/jm040037k. [DOI] [PubMed] [Google Scholar]
- 20.Zobel K, Wang L, Varfolomeev E, Franklin MC, Elliott LO, Wallweber HJ, Okawa DC, Flygare JA, Vucic D, Fairbrother WJ, Deshayes K. Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem Biol. 2006;1:525–533. doi: 10.1021/cb600276q. [DOI] [PubMed] [Google Scholar]
- 21.Wu TY, Wagner KW, Bursulaya B, Schultz PG, Deveraux QL. Development and characterization of nonpeptidic small molecule inhibitors of the XIAP/caspase-3 interaction. Chem Biol. 2003;10:759–767. doi: 10.1016/s1074-5521(03)00157-1. [DOI] [PubMed] [Google Scholar]
- 22.Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, Pedersen IM, Kitada S, Scott FL, Bailly-Maitre B, Glinsky G, Scudiero D, Sausville E, Salvesen G, Nefzi A, Ostresh JM, Houghten RA, Reed JC. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell. 2004;5:25–35. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]
- 23.Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFα-mediated cell death. Science. 2004;305:1471–1474. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 24.Sun H, Nikolovska-Coleska Z, Lu J, Meagher JL, Yang CY, Qiu S, Tomita Y, Ueda Y, Jiang S, Krajewski K, Roller PP, Stuckey JA, Wang S. Design, Synthesis, and Characterization of a Potent, Nonpeptide, Cell-Permeable, Bivalent Smac Mimetic That Concurrently Targets Both the BIR2 and BIR3 Domains in XIAP. J Am Chem Soc. 2007;129:15279–15294. doi: 10.1021/ja074725f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PR, Krajewski K, Saito NA, Stuckey J, Wang S. Development and Optimization of a Binding Assay for the XIAP BIR3 Domain Using Fluorescence Polarization. Anal Biochem. 2004;332:261–273. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 26.Nikolovska-Coleska Z, Meagher JL, Jiang S, Kawamoto SA, Gao W, Yi H, Qin D, Roller PR, Stuckey J, Wang S. Design and characterization of bivalent Smac-based peptides as antagonists of XIAP and development and validation of a fluorescence polarization assay for XIAP containing both BIR2 and BIR3 domains. Anal Biochem. 2008;374:87–98. doi: 10.1016/j.ab.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW, Sweet MR Jr, editors. Methods in Enzymology, Volume 276: Macromolecular Crystallography, Part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]; Collaborative Computational Project, Number 4 (1994) The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Brunger AT, Admas PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewsji J, Nigles N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 30.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 31.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 32.Sybyl, a molecular modeling system, is supplied by Tripos, Inc., St. Louis, MO
- 33.Case DA, Pearlman DA, Caldwell JW, Cheatham TE, III, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJY, Duan PJ, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER7. University of California; San Francisco: 2002. [Google Scholar]
- 34.Kipp RA, Case MA, Wist AD, Cresson CM, Carrell M, Griner E, Wiita A, Albiniak PA, Chai J, Shi Y, Semmelhack MF, McLendon GL. Molecular targeting of inhibitors of apoptosis proteins based on small molecule mimics of natural binding partners. Biochemistry. 2002;41:7344–7349. doi: 10.1021/bi0121454. [DOI] [PubMed] [Google Scholar]
- 35.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJA, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. IAP Antagonists Induce Autoubiquitination of c-IAPs, NF-κB Activation, and TNFα-Dependent Apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 36.Gao Z, Tian Y, Wang J, Yin Q, Wu H, Li YM, Jiang X. A Dimeric Smac/Diablo Peptide Directly Relieves Caspase-3 Inhibition by XIAP. J Biol Chem. 2007;282:30718–30727. doi: 10.1074/jbc.M705258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Splan KE, Allen JE, McLendon GL. Biochemical Basis for Enhanced Binding of Peptide Dimers to X-Linked Inhibitor of Apoptosis Protein. Biochemistry. 2007;46:11938–11944. doi: 10.1021/bi061938t. [DOI] [PubMed] [Google Scholar]