

Summary
As in many other hemoglobins, no direct route for migration of ligands between solvent and active site is evident from crystal structures of Scapharca inaequivalvis dimeric HbI. Xenon (Xe) and organic halide binding experiments along with computational analysis presented here reveal protein cavities as potential ligand migration routes. Time-resolved crystallographic experiments show that photodissociated carbon monoxide (CO) docks within 5ns at the distal pocket B-site and at more remote Xe4 and Xe2 cavities. CO rebinding is not affected by the presence of dichloroethane within the major Xe4 protein cavity, demonstrating that this cavity is not on the major exit pathway. The crystal lattice has a substantial influence on ligand migration, suggesting that significant conformational rearrangements may be required for ligand exit. Taken together, these results are consistent with a distal histidine gate as one important ligand entry and exit route, despite its participation in the dimeric interface.
Introduction
Enzyme catalysis and molecular transport require the translocation of small molecules from the bulk solvent to a specific site that is often buried within a relatively large protein molecule. The migration of ligands within proteins often requires conformational changes that facilitate moving between internal cavities to their binding sites (Brunori, 2000; Brunori and Gibson, 2001). Ligand migration has been extensively studied in myoglobin (Mb), which reversibly binds small molecules such as molecular oxygen, nitric oxide, and carbon monoxide at a heme group within the distal pocket with no obvious channel for ligand diffusion. Alternate routes have been proposed for the ligand entry to and exit from the distal pocket including the distal His gate (Perutz and Mathews, 1966) and a cavity network route (Huang and Boxer, 1994). Experimental data from numerous investigations suggest that the distal His gate model, in which ligand entry and escape are facilitated by the rotation of the distal His to transiently open a direct pathway between the solvent and active site, provides the primary migration route: 1) The distal His occupies multiple conformations within atomic resolution structures at low temperatures (Vojtechovsky et al., 1999); 2) The distal His swings out in low pH structures (Tian et al., 1993; Yang and Phillips, 1996); 3) Kinetic analyses of tryptophan mutants at the distal Histidine and internal cavities suggest that at least 75 % of the ligands leave by the swinging His pathway (Olson et al., 2007; Scott et al., 2001); 4) Ligand rebinding studies as a function of temperature in the presence of xenon (Xe) indicate the distal His route to be a prevailing pathway (Scott et al., 2001; Tetreau et al., 2004); 5) Time-resolved crystallographic studies showed no major direct ligand exit from the Xe1 site to the solvent (Schmidt et al., 2005). However, the distal histidine gate is not universally used among globins. Smaller truncated (2/2) and mini-globins exhibit apolar channels (Milani et al., 2001; Pesce et al., 2002); the large core channel in Cerebratulus hemoglobin has recently been shown to be the major pathway for O2 migration (Salter et al., 2008). Thus, variability exists in ligand migration pathways among globins.
Protein assemblies larger than monomers present additional complexities for ligand transport, particularly when the predominant route for ligand transport into a monomer now participates in intersubunit contacts. This is the case in all known invertebrate, cooperative hemoglobin structures, which have sub-assemblies formed from contacts between the E and F helices of two interacting subunits (Royer et al., 2001; Royer et al., 2005). One member of this group of hemoglobins, the dimeric HbI from the blood clam Scapharca inaequivalvis, has an intersubunit-interaction between the distal His and Lys 96′ either directly (unliganded state) or via an ordered water molecule (CO or O2 liganded state) (Condon and Royer, 1994; Royer, 1994) (Figure 1). This interaction potentially restricts the motions of the distal His that would facilitate ligand escape through the distal His route. As a result, the major route for ligand entry and exit in HbI might involve ligand migration through internal cavities within the protein (Chiancone et al., 1993). In the present study, internal cavities and potential ligand routes in HbI have been identified using Xe binding, organic halide binding, computational analysis by implicit ligand sampling approach and ligand rebinding kinetics in solution. The utilization of the cavities by photodissociated ligands was followed by time-resolved crystallography, which demonstrates a substantial influence of the crystal lattice on ligand migration both in differences between subunits and in the diminished exit of dissociated CO ligands from the protein matrix. Filling the cavities either by mutagenesis or bound Xe does not substantially alter the geminate rebinding of oxygen, which argues against these cavities as a major route for ligand exit. These results, thus, are consistent with the recent proposal that the distal histidine gate provides an important ligand migration route in HbI (Nienhaus et al., 2007).
Figure 1.
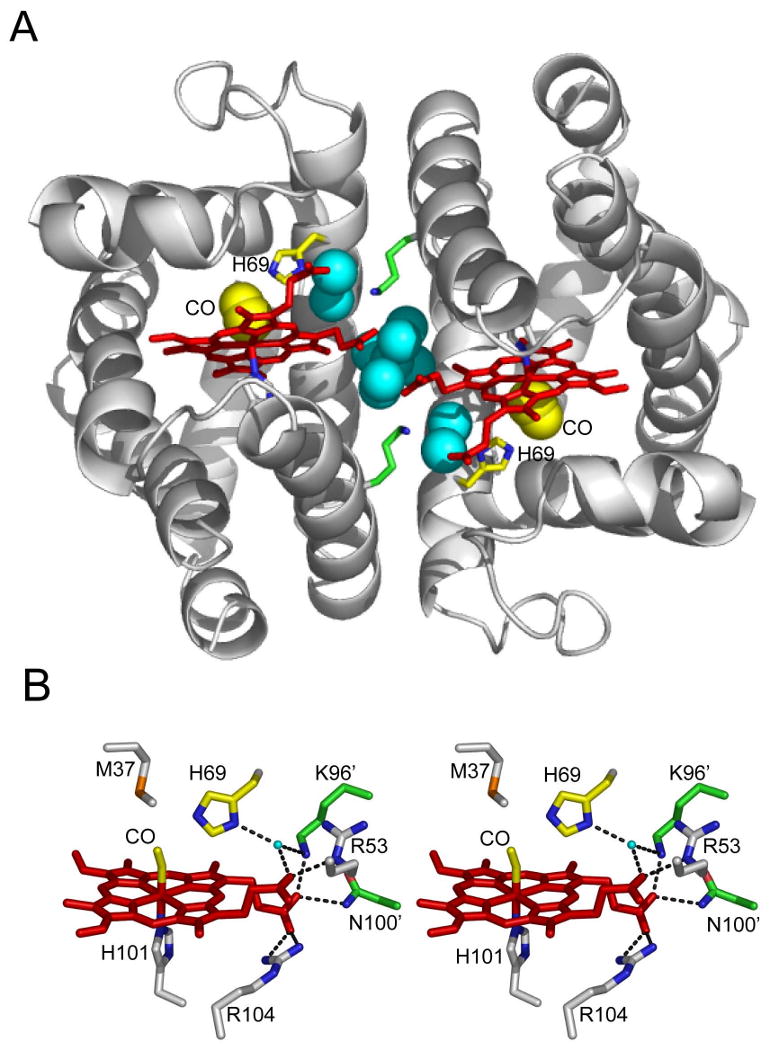
Involvement of the distal histidine in the dimeric interface of HbI, from PDB coordinates 3SDH (Royer, 1994).
(A) HbI dimer with ribbon representation of backbone (gray), heme group (red), side chains for Lys 96 (green), distal His (yellow and blue), proximal His (gray and blue), interface water molecules (blue spheres) and CO ligand (yellow spheres).
(B) Stereo stick representation of the vicinity of distal histidine. Stick resprenations are included for heme (red), CO ligand (yellow) and side chains of Met 37 (gray and orange), His 69 (yellow and blue), Arg 53 (gray and blue), Arg 104 (gray and blue) and His 101 (gray and blue) from one subunit and Lys 96 and Asn 100 from the second subunit. Also included is a small cyan ball for a water molecule in hydrogen bonding distance of His 69 and Lys 96′ (primes designate second subunit). Likely hydrogen bonds, with distances of 2.7-2.9Å, are shown as black dashes. (An additional potential hydrogen bond between the illustrated bound water and Arg 53 would involve distances of 3.25 and 3.3Å in the two subunits and is therefore not shown.) Exit of CO through a swinging histidine gate would require movement of the water molecule and Lys96′, disrupting the hydrogen bond network present in the dimeric interface.
Results
Binding of Xe and organic halides to cavities within HbI-CO
The noble gas Xe is an excellent probe of internal hydrophobic cavities, as shown in Mb (Tilton et al., 1984). Diffraction data on Xe-exposed crystals of the I25W mutant of HbI demonstrated Xe binding to Xe1 and Xe2 cavities (Nienhaus et al., 2007), but not to the Xe4 cavity that is filled by the mutant Trp 25. Here we explore locations and occupancies of Xe binding sites in wild type (WT) HbI-CO crystals (Table 1). Electron density maps at 0.97Å resolution reveal four peaks corresponding to Xe atoms in WT HbI with each site being separated from neighboring sites by a single shell of hydrophobic atoms (Figure 2C). The similarity in observed Xe sites in HbI and Mb is evident in Figure 2A and 2B. Two mutually exclusive Xe sites in HbI, termed Xe4a and Xe4b, are located near the Xe4 site in Mb, in a cavity bordered by side chains from the B, E and G helices. Xe4a and Xe4b are 7.8 and 5.2 Å away from the Xe4 site in Mb, respectively. The other two Xe sites, Xe1 and Xe2, observed also in I25W HbI-CO mutant (Nienhaus et al., 2007) are 1.8 and 3.1 Å away from equivalent Xe sites in Mb, respectively (note that the Xe1 cavity in unliganded HbI is filled by the Phe97 side chain). The occupancy of Xe sites in WT HbI-CO follows the trend Xe4a > Xe2 > Xe4b > Xe1, with largest fractional occupancy at the Xe4a site of 0.9 and low occupancies at the Xe4b and Xe1 sites of 0.1 and 0.05, respectively. The Xe occupancy in Mb follows the trend Xe1 > Xe2 > Xe3 > Xe4, with all sites having fractional occupancies greater than 0.45 and Xe1 having an occupancy of 0.94 (Tilton et al., 1984). No site analogous to the Xe3 site in Mb was found in HbI (at Xe pressures of 200-400 psi, 1.4 – 2.8 · 106 Pa).
Table 1.
Refinement statistics of the WT HbI-CO-with Xe.
| Sample | HbI-CO-Xe |
|---|---|
| PDB code | 3G46 |
| Space group | C2 |
| Cell Constants | |
| a (Å) | 92.5 |
| b (Å) | 43.5 |
| c (Å) | 82.9 |
| β (°) | 122.3 |
| Resolution range (Å) | 50 – 0.91 (1.01-0.97)* |
| Reflections | 160199 (12152) |
| Completeness (%) | 77.9 (59.3) |
| Multiplicity | 4 (2.1) |
| I/σI | 20 (4.7) |
| Rsym | 0.075 (0.189) |
| Working Set Reflections | 152165 |
| Test Set Reflections | 8034 |
| Rfactor | 0.129 |
| Rfree | 0.156 |
| Number of non-hydrogen atoms | |
| Protein atoms** | 2246 |
| Heme and ligand atoms | 90 |
| Xe atoms | 8 |
| Solvent molecules | 432 |
| Number of alternate conformations | 29 |
| Average B-factors (Å2) | |
| Main-chain atoms | 15.5 |
| Side-chain atoms | 14.3 |
| Heme atoms | 9.5 |
| CO ligands | 8.9 |
| Solvent atoms | 26.5 |
| Xe atoms | 15.1 |
| RMS Deviations from ideal value | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.76 |
| Dihedral angles (°) | 19.7 |
| Improper angles (°) | 1.34 |
| Ramachandran Plot | |
| Most Favored Regions (%) | 95.2 |
| Allowed Regions (%) | 4.8 |
Diffraction data were collected to a resolution of 0.91Å. The 0.97-0.93Å shell is 29.1% complete and that from 0.93-0.91Å is 4.8% complete. Although this is a 0.97 Å structure, the higher resolution reflections were included to improve the refinement and quality of the electron density maps.
not including alternate conformations
Figure 2.
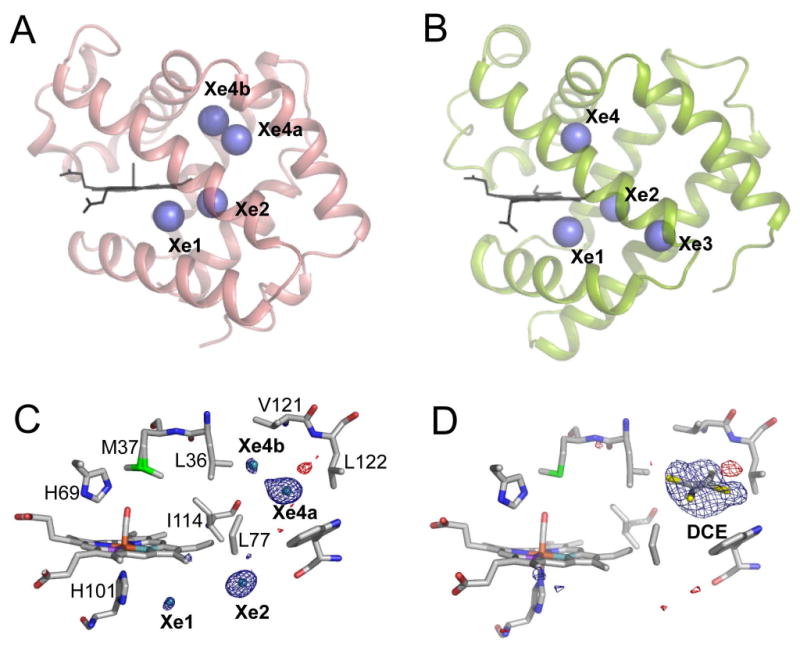
Xe and organic halide binding to HbI and Mb.
(A and B) Comparison of the Xe binding sites within HbI (A) and Mb (B; PDB file 1J52). The Xe1 and Xe2 sites from both globins are at nearly identical locations whereas Xe4a/Xe4b site in HbI is close to, but not at identical location as Xe4 in Mb. Xe3 in Mb is not present in HbI.
(C and D) A close up view of the Xe (C) and DCE (D) binding sites is superimposed upon the corresponding FoXe-FoNative or FoDCE-FoNative difference electron density map, contoured at ± 3σ. The DCE binding site is equivalent to the Xe4a binding site.
In addition to Xe binding, the HbI cavities were explored by binding of organic halides such as dichloroethane (DCE). These compounds are hydrophobic, like O2 and CO, and have been demonstrated to bind to hydrophobic pockets within proteins such as insulin (Gursky et al., 1994). DCE and other organic halides were found to bind to the Xe4 cavity within wild type HbI-CO, as evident by positive features in Foorganic-Fonative difference electron density maps (see Figure 2D, Figures S1 and S2, and Tables S1-S3, Supplemental Data). The binding of organic halides does not greatly change either the tertiary or quaternary structures (Table S4). It is unlikely that a swinging distal histidine gate in the dimeric interface of HbI could accommodate entry of such bulky compounds, suggesting a more direct route for these compounds to the Xe4 cavity within HbI crystals. Thus, alternate routes appear to exist between the solvent and the protein cavity network in crystalline HbI, but the question remains which routes are preferred and most efficient for entry and exit of small diatomic ligands such as CO and O2 in solution and the crystal.
Computational analysis of protein cavities
Cavities in HbI were also explored computationally by determining the potential of mean force; that is, the energetic favorability/probability of a ligand being present at every point in space within an ensemble of protein conformations (Cohen et al., 2006; Cohen and Schulten, 2007). This computational approach is valuable not only because it helps to visualize ligand accessible pathways and docking sites, but because it also provides numerical estimates of how likely it is that the ligand would be present at a given location compared to the ligand being present in bulk solution. Xe ligands were modeled in a geminate state with the high oxygen affinity R-state structure (HbI-CO PDB model 3SDH, but removing water molecules and the bound CO ligand). This implicit ligand sampling (ILS) approach (Cohen et al., 2006) revealed six cavities and putative Xe binding sites within R-state HbI, as shown in Figure 3. Two putative Xe sites (I-1 and I-2 in Figure 3A) were found at the subunit interface, but do not exist in dimeric HbI because of the presence of interfacial water molecules (Royer et al., 1996). A third site is located in the distal pocket and corresponds to the standard distal pocket “B site” for the photo-dissociated ligand, observed in time-resolved crystallographic experiments both in WT HbI (below) and in the M37V mutant of HbI (Knapp et al., 2006). Xe binding to this site is blocked when O2 or CO ligand is modeled bound to the heme iron. Three additional sites were found within 1-2 Å of the experimentally observed Xe binding sites Xe1, Xe2, and Xe4 (a+b) (Figure 3A), indicative of the accuracy and value of this computational approach in providing additional corroboration to our experimental results.
Figure 3.
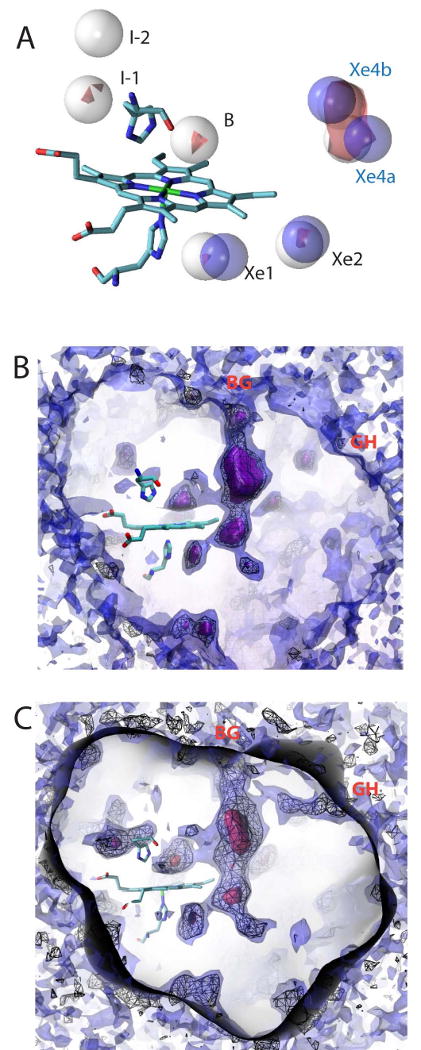
Computational analysis of the binding of Xe, CO and O2 to HbI.
(A) Predicted Xe binding sites. Six predicted Xe sites (white spheres) were located with two at the dimer interface (I1 and I2), two in or near the proximal pocket (Xe1 and Xe2), and two in or near the distal pocket (B and Xe4). Blue spheres represent crystallographically determined Xe positions for comparision. The red surface shows locations within the energy landscape map less than -5kcal/mol.
(B) The CO energy landscape map contoured at 0 kcal/mol (purple surface), 1.6 kcal/mol (black wire mesh), and 5kcal/mol (blue surface). Contour values less than 1.6 kcal/mol indicates regions where molecular CO docking to HbI is more favorable to CO than being dissolved in solution. Thus, a smaller value indicates more favorable binding.
(C) The O2 energy landscape map contoured at -3kcal/mol (red surface), 2 kcal/mole (black wire mesh), and 5kcal/mol (blue surface). A value of 0 kT is equivalent to oxygen binding within a vacuum. Contour values less than 2 kcal/mole indicates regions where molecular oxygen docking to HbI is more favorable than when oxygen is dissolved in solution. Both the CO and O2 contour maps show the presence of clear tunnel connecting the distal pocket with the proximal pocket and the bulk solvent through a major route (BG). This graphic was created with VMD (Humphrey et al., 1996). The O2 energy landscape map shows an additional tunnel between the bulk solvent and the proximal pocket through a minor route (GH).
ILS was then used to delineate cavities that are sufficiently favorable for CO and O2 docking. Figure 3B and 3C present energy contour plots with surface renderings indicating regions of the structure to which CO and O2 binds favorably, with docking sites represented in purple. The black mesh within the protein represents regions that are more favorable for CO, in terms of free energy, than the solvent. The calculated energy diagram shows a connection between the distal and proximal pockets via the Xe4 cavity network. This large cavity contains the most favorable, non-heme site for O2 and CO binding. The energy landscape also shows a connection from the cavity network to the exterior, suggesting a pathway from the bulk solvent to the distal pocket. The major (BG) pathway between the Xe4 cavity and the protein exterior is bordered by the side chains from residues Ile 25, Asn 32, Ala 35, Leu 36, Val 121, Ser 124, and Lys 125. This analysis is consistent with direct access between the Xe4 cavity and solvent. Note that a minor route (GH) between the bulk solvent and the distal pocket exists in the energy contour maps of HbI-O2 but not in the corresponding maps of HbI-CO.
Geminate ligand binding kinetics in HbI solution
Geminate phase rebinding of photolyzed O2 provides a useful probe of ligand migration. The fractional rebinding of O2 to WT and I25W was followed at Xe pressures of 100, 200 and 300 psi (6.9, 13.8, 20.7 · 105 Pa). For both proteins, the data could be best fit by two fast geminate rates and a slower rate corresponding to bimolecular rebinding. Overall geminate rebinding fraction of O2 increases from about 45% to 50% in wild-type upon addition of Xe, whereas the somewhat higher geminate fraction for I25W (∼55%) remains largely unchanged by Xe (Figure 4). Although the overall geminate fraction only changes marginally by addition of Xe, the relative proportion of the two fast geminate rates change significantly. In WT, the amplitude of the fastest component (7·107 sec-1) decreases by about half as Xe pressure increases, while the other geminate component (1.8·107 sec-1) increases by more than two-fold. In contrast, the amplitude of the fastest geminate component in I25W (2.5·108 sec-1) increases by 50%, whereas that for the other geminate component (1.9·107 sec-1) decreases. This complex reaction to Xe must reflect the alteration of available cavities for migrating O2. It appears plausible, in view of our structures of HbI (above and (Nienhaus et al., 2007)), that the decreased amplitude of the faster rate for WT could correlate directly with filling the Xe4 cavity. Despite filling this and the Xe2 cavity, the overall geminate fraction only marginally increases. In I25W, the Xe4 cavity is already filled by Trp 25, so its faster rate, whose amplitude increases rather than decreases, must result from photodissociated O2 migrating from other locations, with the most likely location being the B-site within the distal pocket. These results do indicate that the Xe binding sites are visited during ligand migration, but the lack of a large change in geminate fraction upon filling Xe4, argues against it as providing a major exit path from the heme binding site.
Figure 4.
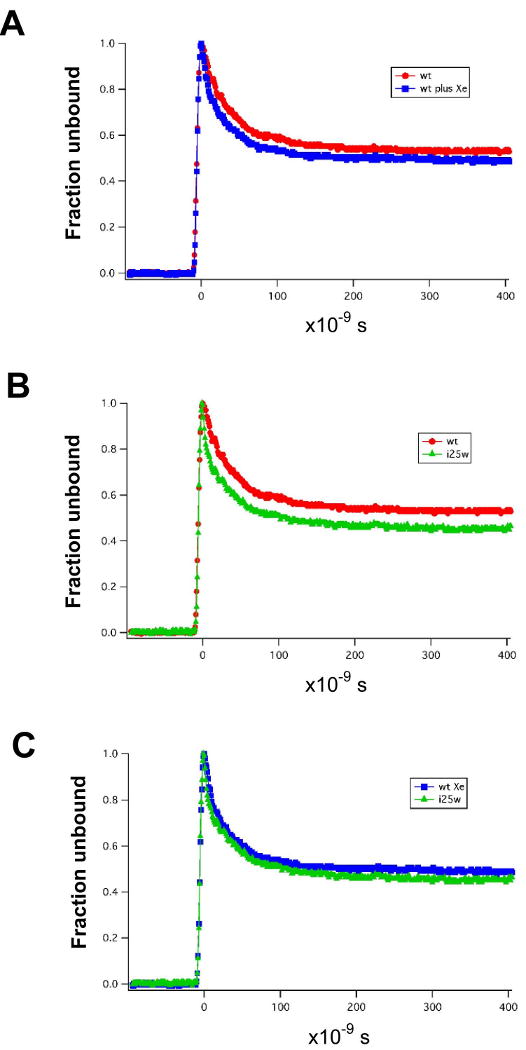
Geminate rebinding of oxygen to WT HbI and I25W in solution.
(A) Effect of Xe (300 psi, 2.07 · 106 Pa) on geminate oxygen binding to wild-type HbI. The presence of Xe has only a minor effect on either the fraction of geminate oxygen rebinding or the rate of geminate oxygen rebinding.
(B) Comparison of geminate oxygen binding to wild-type HbI and mutant I25W. Mutation of Ile 25 to Trp has a small effect, increasing the geminate fraction to around 55%.
(C) Effect of Xe (300 psi, 2.07 · 106 Pa) on geminate oxygen binding to I25W.
Probing CO migration by time-resolved crystallography
Migration of the photo-dissociated CO ligand within HbI was followed in real time by time-resolved Laue crystallography (Bourgeois et al., 2006; Schmidt et al., 2005; Schotte et al., 2003; Srajer et al., 2001; Srajer et al., 1996). Ten X-ray diffraction data sets for the WT HbI-CO photoproduct were collected between 5ns and 30μs following photo-dissociation of the ligand initiated by a 7ns laser pulse (Knapp et al., 2006). Data sets with (light) and without (dark) laser pulse were collected from the same set of crystals. Real space data for two subunits at 5ns are presented in Figure 5A and B. Difference electron density maps Folight -Fodark are shown, with positive (blue) and negative (red) features at the locations within the photoproduct (light) that have more and less electrons, respectively, than the native (dark) structure.
Figure 5.
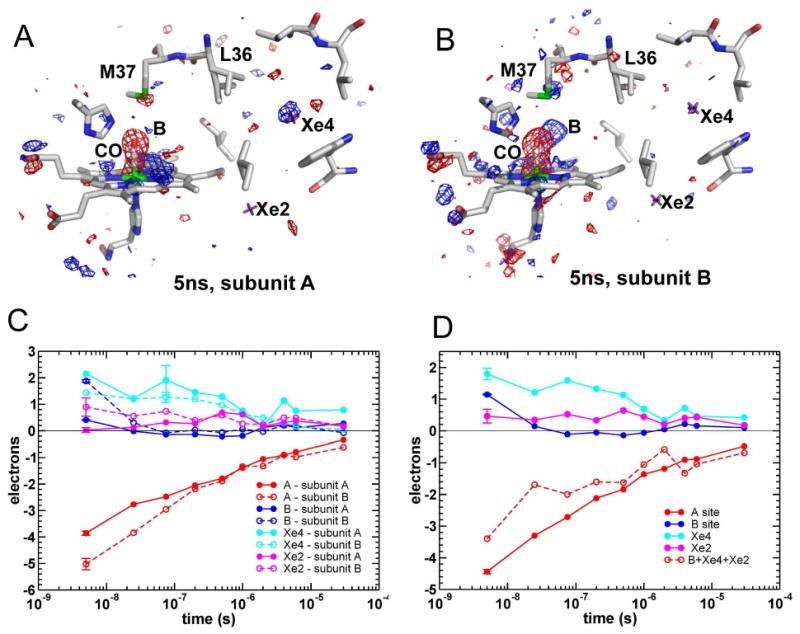
HbI difference electron density maps following CO photo-dissociation.
(A and B) Maps, contoured at +/- 3.5σ (blue and red, respectively), shown for the heme region and Xe2 and Xe4 cavities in subunits A (A) and B (B). Xe2 and X4 sites are marked by purple crosses.
(C and D) Time courses for integrated difference electron densities at ligand binding site and docking sites B, Xe2 and Xe4 are shown for subunits A and B separately (C) and averaged over the two subunits (D). The length of the error bar associated with each time course represents the absolute value of the integrated Δρ for the same volumes in a difference map calculated using two dark, HbI-CO, data sets.
As a result of ligand photo-dissociation, a large negative electron density feature appears at 5ns at the location of the bound CO in the native structure (labeled CO in Figure 5) and corresponding to the spectroscopic ligand site A. Integration of the difference electron density peak at this location shows that ∼32% of the CO is photo-dissociated (average value for A and B subunits). Positive electron density features are present at the spectroscopic ligand site B (within the distal pocket), as well as Xe4 and Xe2 sites, resulting from docked photo-dissociated ligands. Integration of electron density in the Folight -Fodark maps indicate that about 66% of photo-dissociated CO (CO*) is distributed between the ligand B and Xe4 sites. (Of the two Xe4 sites, Xe4a site is more dominant; in the following analysis, integrated electron densities at sites Xe4a and Xe4b are summed.) An additional ∼10% of CO* is localized at the Xe2 site at this time point, indicative of a rapid translocation of CO from the distal pocket throughout the remote Xe cavity network. The remaining ∼24% of photo-dissociated CO* molecules at 5ns could not be located.
Substantial differences in ligand migration are observed between the two crystallographically independent subunits of the HbI homodimer. Photolysis levels at 5ns for subunits A and B are estimated at 28% and 36%, respectively. Differences between subunits are striking when considering docking sites for photolyzed ligands. Docking of CO ligands 5ns following photolysis occurs primarily at the Xe4 site in subunit A, whereas the B-site is favored in subunit B (Figure 5). These results are consistent with the differences between the two subunits in the occupancy of alternate conformations of the side chains Leu 36 (shown in Figure 5), and Met 37, that have been observed in a 1.08 Å resolution room temperature conventional, static crystal structure of HbI-CO (unpublished data). Leu 36 forms the barrier between the distal pocket and the Xe4 site. The “in” conformation packs against the B-site potentially destabilizing ligand docking at this site whereas the “out” conformation packs against the Xe4 site, potentially destabilizing ligand docking to this site. Leu 36 occupies an “in”/”out” ratio of 60/40 in the A subunit and a 50/50 ratio in the B subunit. Met 37 packs against the ligand, both when the ligand is bound to the heme iron and when the ligand docks at the B-site. However, the Met 37 minor conformation sterically restricts ligand access to the B-site. The minor Met 37 conformation has occupancy of 38% in the A subunit but is absent in the B-subunit, based on the static 1.08 Å HbI-CO structure. This suggests that ligand docking at the B-site may be disfavored ∼75% of the time in subunit A by the combined unfavorable alternate conformations of Leu 36 (60%) and Met 37 (38%) but only ∼50% of the time in the B subunit, by the unfavorable conformation of Leu 36. The decreased B-site affinity within the A subunit may lead to increased sub-nanosecond geminate rebinding, resulting in decreased photodissociated ligand density within the distal pocket.
Decay of the negative density at the bound CO location (ligand A site) as well as time courses of positive densities at ligand sites B, Xe4 and Xe2 are shown in Figure 5D, averaged over subunits A and B. The CO population at the distal docking site B decays rapidly, with a half-life of ∼10ns. Similar rapid initial decay of negative A-site density is observed on the ns time scale, indicating a fast geminate ligand rebinding phase from the B site. The decay of positive electron densities at the Xe2 and Xe4 sites is significantly slower than the decay of the B-site density. Populations of these Xe sites are largely constant throughout the ns time domain and decay only on the μs timescale. A complete quantitative analysis of ligand migration and rebinding requires data with significantly larger number of time-points and with improved signal-to-noise ratio, over time domain extended to both shorter and longer times. The four decays shown in Figure 5D (for ligand sites A, B, Xe2, Xe4) do however indicate the presence of at least two phases and non-exponential nature of the decays. In the simplest description that assumes only two phases, the rates for the fast and slow phases are estimated as ∼7.5·107 s-1 and ∼1.9·106 s-1, with the contribution of the fast phase to ligand rebinding (ligand A site) of ∼25% (Figure 6). The rate of the fast phase seems to be somewhat faster than the geminate CO rebinding rate in solution of 1.4·107 s-1 (Chiancone et al., 1993). The sum of positive B, Xe2 and Xe4 densities is also shown in Figure 5D (dashed red line), with an inverted sign to be easily compared with the ligand rebinding (solid red line). The comparison shows that throughout the ns time domain some of the dissociated ligands are not located at these three major docking sites (missing fraction is ∼25%). Additional sites with low occupancies are possible and can contribute to the overall fast geminate rebinding phase, in addition to the geminate rebinding from the B site. On the longer μs timescale the A-site density matches (within the noise) both the amplitude and the time course of the Xe2+Xe4 density, indicating that all photo-dissociated ligands have either already rebound or are located at the Xe2 and Xe4 sites. Rapid return of the ligands from the Xe cavity system to the B site or significant exit from the protein matrix were therefore not observed. The observation of a rapid translocation of the ligand throughout the Xe cavity system (less than 5ns) but the slow rebinding from the Xe sites suggests rapid structural changes that destabilize binding at the distal pocket. Although the data does not unambiguously identify these structural changes, some density features indicate that the slow binding from the Xe sites might be caused by a rapid change in the position of the heme group (and thus the ligand binding site). The results presented here (Figures 5 and 7) and in our earlier time-resolved crystallographic study of M37V (Knapp et al., 2006) indicate a heme buckling within 5ns that includes heme atoms moving towards the distal His, allowing the side chain of this residue and that of Met 37 to sterically interfere with CO rebinding. This steric barrier is reduced by the structural changes during the microsecond time domain that accompany the allosteric transition.
Figure 6.
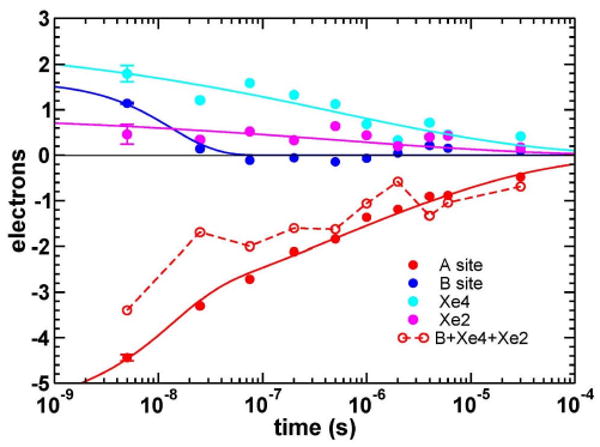
Global fit to four time courses of integrated difference electron densities corresponding to ligand sites A, B, Xe2 and Xe4.
In a simplest model, two common rates, k1 and k2, are used to fit all time courses with a fast exponential and a slower stretched exponential phase: C1exp(-k1t)+C2exp(-(k2t)β). In case of ligand rebinding (A-site), both fast and slow phases are included. The fit results in the amplitude for the fast exponential phase of 23%. For the distal docking site B only fast exponential phase is evident and amplitude of the second, slower phase was fixed to a value of 0 during the fit. For sites Xe2 and Xe4, a slow phase is predominant and amplitude of the fast phase was fixed to a value of 0 during the fit. Common rates resulting from the described global fit are k1=7.5·107 s-1 and k2=1.9·106 s-1, β=0.22.
Figure 7.
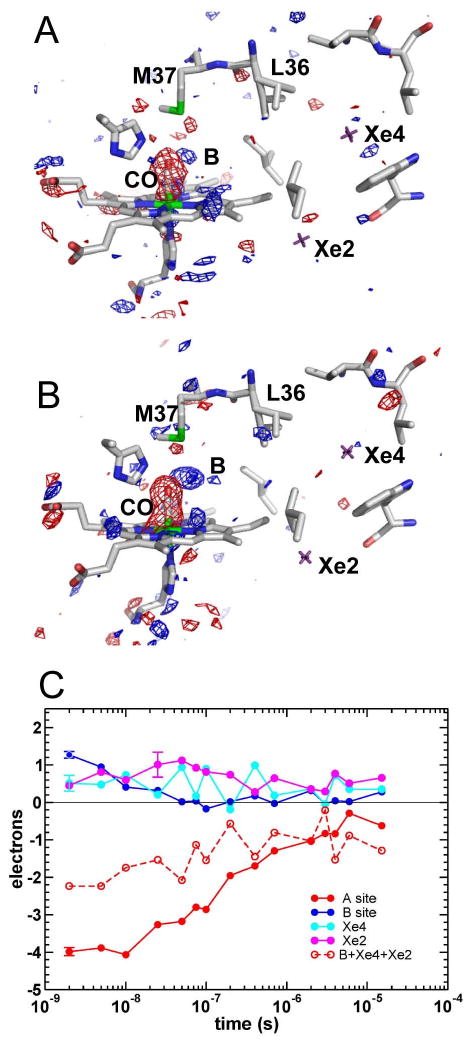
Difference electron density maps at 2ns following CO photo-dissociation on wild-type HbI with bound DCE.
(A and B) Electron density maps, contoured at +/- 3.5σ (blue and red, respectively), are shown for the heme region and Xe2 and Xe4 cavities in subunits A (A) and B (B). Xe2 and X4 sites are marked by purple crosses.
(C) Time courses for integrated difference electron densities at ligand sites A, B, Xe2 and Xe4 averaged over the two subunits. The length of the error bar associated with each time course represents the absolute value of the integrated Δρ for the same volumes in a difference map calculated using two dark, HbI-CO, data sets.
Effect of DCE bound in Xe4 cavity on ligand migration
WT HbI time-resolved crystallographic data sets were also collected with DCE bound at the Xe4 site, at 15 time delays between 2ns and 15μs following flash photolysis. Difference electron density maps, Folight-Fodark, for subunits A and B at 2ns are presented in Figure 7A and 7B, while ligand rebinding and time courses for docking sites of the photo-dissociated ligand (averaged over subunits A and B) are shown in Figure 7C. Despite the similarity of CO rebinding and B state decay between WT and WT/DCE, the utilization of the cavity network has changed by restricting the Xe4 cavity volume with bound DCE. Occupancy of Xe2 is enhanced in WT/DCE as compared to WT. Although in WT/DCE the Xe4 cavity itself is occupied by DCE some positive difference density is still detected at the Xe4 site. Comparison of the B+Xe2+Xe4 population decay with ligand rebinding (A-site), shows that roughly 50% of the photo-dissociated ligands are not accounted for at B, Xe2 and Xe4 sites on the ns time scale in WT/DCE, a fraction significantly higher than in WT (∼25%). On the μs time scale, all unbound ligands are located at the Xe2 and Xe4 sites, similar to WT. This observation implies that CO is most likely delocalized within the protein matrix; however, the possibility that CO rapidly moves to and from the solvent channels cannot be ruled out.
Discussion
As with myoglobin, substantial cavities are present in Scapharca HbI that could participate in ligand migration. Although time-resolved crystallography results show that these cavities are utilized as docking sites, ligands largely do not exit the protein matrix from these sites prior to rebinding.
The lack of ligand escape from the protein in the crystal indicates a substantial impact of the crystal lattice on ligand migration. The fraction of geminate CO rebinding for WT HbI in solution is only 5%, with most photo-dissociated ligands escaping into the solvent and rebinding bi-molecularly (Chiancone et al., 1993). In the crystal, a significant ns geminate phase is present (at least ∼25%). Even more striking is our finding that photo-dissociated ligands that do not rebind rapidly in the HbI crystal remain primarily trapped at the Xe2 and Xe4 sites until they rebind on the μs timescale. Crystal packing in Mb also influences ligand escape, with 2-5% of CO rebinding geminately on the ns time scale in solution (Henry et al., 1983; Tian et al., 1993) compared with approximately 22% rebinding geminately in the crystal (Srajer et al., 2001). However, in sharp contrast with HbI, the remaining 78% of ligands in Mb are apparently able to exit the protein before rebinding with a rate consistent with bimolecular binding (Figure 8) (Srajer et al., 2001).
Figure 8.
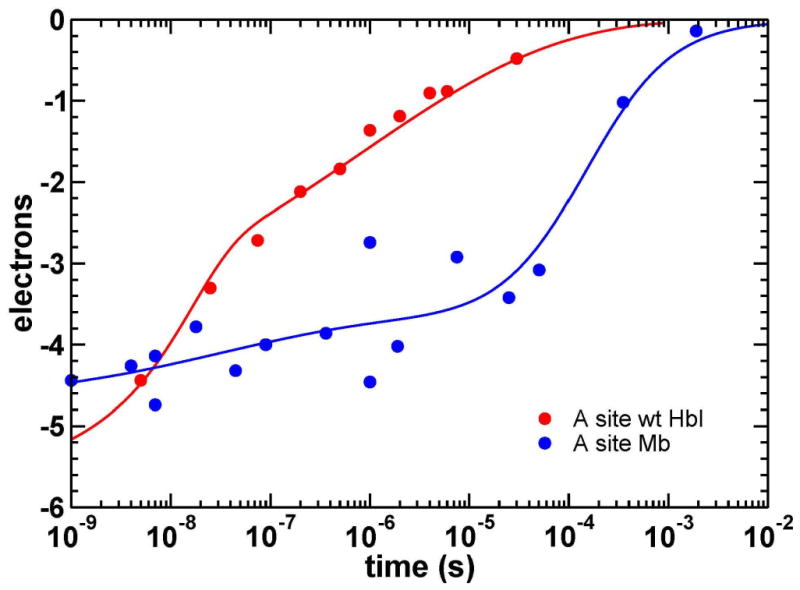
Time course for the integrated electron density at the CO binding site (A) from time-resolved crystallographic studies of Mb (Srajer et al., 2001) and wild-type HbI. In HbI (red), the rates for two rebinding phases are estimated as ∼7.5·107 s-1 and ∼1.9·106 s-1 (Figure 5), with the contribution of the fast geminate phase to ligand rebinding of at least ∼25%. Also, notice the very stretched nature of the second, slower phase. The time-points for Mb (blue) are shown with the best fit with a sum a stretched exponential (22%) and bimolecular (78%) functions (Srajer et al., 2001).
One possible explanation for the inhibition of ligand exit from the protein matrix in HbI crystals could be that lattice contacts directly block a major exit route. However, inspection of lattice interactions does not reveal significant contacts at likely exit points. The distal histidine is involved in the dimeric interface and is well away from any lattice contacts. For the alternative BG exit pathway from the Xe4 cavity, residues 25, 32, 35, 36, 121, 122, and 124 along the pathway also are not involved in such contacts. The only residue on this pathway that participates in lattice contacts is Lys 125 from the B subunit. Moreover, the ability of bulky organic halide compounds to enter the Xe4 cavity in the crystal suggests that the lattice is compatible with the use of a direct entrance to Xe4, possibly through the BG route illuminated by the computational analysis. An alternate possibility is that the lattice could impact ligand exit if the packing of molecules dampens larger movements that are required for ligand release. While exit through the BG route would not appear to require large movements, as the exit point is largely exposed to solvent in the crystal, exit through a distal His gate, proposed earlier as a primary exit route for ligands in HbI (Nienhaus et al., 2007), could require such movements because of distal histidine's participation in the dimeric interface (Figure 1). Swinging of the distal His to allow ligand exit will require displacement of an interfacial water molecule, which forms hydrogen bonds with the distal His, heme propionate and Lys 96′ from the partner subunit. These interactions are part of a hydrogen bonding network (Figure 1b), suggesting that a swinging His motion could require a concerted rearrangement of groups in the interface. One possibility is that a transient subunit reorientation is coupled with such a rearrangement that would permit ligands to exit. The tight packing of the HbI-CO lattice (43.7% solvent) could restrict such a subunit reorientation and prevent most ligands from exiting the protein before they rebind. Although subunits are able to undergo the full quaternary transition to a T-state within the crystal lattice (Knapp and Royer, 2003), it has been shown by time-resolved crystallography that this subunit rotation is slow in M37V HbI mutant, achieving less than 20% of the expected ligand-linked subunit rotation by 80μs (Knapp et al., 2006). Thus, one plausible explanation for differences in the ability of ligands to exit the protein in crystals of HbI and Mb (Figure 8) is that larger conformational transitions are required in HbI, in which the distal His is involved in a dimeric contact (Figure 1), than in the monomeric Mb.
The results presented here argue against the Xe cavities being along the primary route for the ligand escape into the solvent in HbI. Our time-resolved crystallographic analysis, revealing substantially greater rebinding of CO than for solution experiments, both during the fast (ns) geminate phase and longer (μs) time-points, indicate the absence of a readily accessible exit pathway in the crystal. Such an exit path is present in solution and is able to effectively compete with the fast geminate rebinding. As discussed above, lattice damping of required protein motion associated with a distal histidine swing could explain these findings; it is more difficult to rationalize this effect if a direct path from the Xe4 site were used for ligand exit. In solution, the addition of Xe, which fills the Xe4 cavity, has only a minor effect on the fraction of oxygen that rebinds geminately within 400ns both in WT and in I25W (Figure 4). If the Xe cavity system provided the most favorable route for ligand entry or exit, blocking of this route by Xe binding would result in a significant increase in geminate rebinding fraction which is not observed.
Additionally, the similar rebinding behavior of CO in the presence and absence of dichloroethane in the Xe4 cavity (Figures 5 and 7) argues against the Xe4 cavity being on the major exit pathway. These studies, thus, are consistent with the alternate proposal that the distal histidine gate may play a primary role in ligand transit (Nienhaus et al., 2007), but do not exclude the possibility that multiple exit routes are operative in HbI.
Also consistent with ligand escape through a distal histidine gate is the behavior of the M37V (B10) mutant. Solution experiments on M37V showed a reduced fraction of geminate rebinding of oxygen compared with wild-type, prompting its use for the first time-resolved studies on HbI (Knapp et al., 2006). Those time-resolved studies revealed that much of the photolyzed CO remains docked in the enlarged distal pocket of M37V for hundreds of nanoseconds. The reduced fraction of oxygen geminate rebinding in M37V may result because dissociated ligands are docked nearer distal His gate than they are in wild-type. This apparent advantage of M37V does not, however, extend to the behavior of CO rebinding in crystalline M37V (Knapp et al., 2006), consistent with the crystal lattice damping required motions for ligand exit. Thus, our combined experimental evidence from crystal and solution studies is consistent with the proposal (Nienhaus et al., 2007) that in dimeric HbI, as in Mb, the distal His gate is an important pathway for small diatomic ligands like O2 and CO.
Experimental Procedures
Xe binding experiments
Recombinant wild type HbI was expressed in E. coli, purified by chromatographic techniques, and crystallized as described previously (Royer, 1994; Summerford et al., 1995). HbI-CO crystals were first coated with Paratone N oil (Hampton Research) before Xe was introduced into these crystals at pressures between 200 and 400psi (1.4 and 2.8 · 106 Pa) (Hampton Research). The Xe exposed crystals were flash frozen in liquid nitrogen. Diffraction data were collected at the BioCARS 14-BM-C beamline, Advanced Photon Source (APS), Argonne National Laboratory (ANL), using an ADSC Q315 CCD detector. Both high resolution and low resolution passes were collected. Data to 0.97 Å resolution were processed with HKL2000 (Otwinowski and Minor, 1997) and CCP4 packages (CCP4, 1994). A subset of reflections was set aside to calculate a free R-factor (Brunger, 1992). The 0.82 Å resolution HbI-CO structure (unpublished) served as the initial model for refinement and fitting with SHELXL-97 (Sheldrick and Schneider, 1997) and O (Jones et al., 1991) (Table 1). The refined model included anisotropic B-factors and alternate side chains for twenty-nine side chains. The model did not include hydrogen atoms. Two models were used to refine the Xe occupancies. The first model optimized the eight Xe atoms independently. The second model optimized the Xe4a and Xe4b sites of each subunit as alternate conformations of the same site. Both models gave nearly identical values for the two Xe4 atoms in each subunit and did not differ in either the conventional or free R-factor. The second model was chosen based on the observation that the two sites were closer than the sum of the van der Waals radii of the two Xe atoms and the observation the sums of the occupancies of the two Xe4a/Xe4b pairs were approximately one. The Xe1 atoms, despite their low occupancy, were included in the model because each subunit displayed a positive peak in FoXe -Fonative difference electron density maps. Moreover, removal of the Xe1 atoms results in positive peaks at the Xe1 positions in Fo-Fc difference electron density maps and a slight increase in the free R-factor.
Crystallographic analysis of HbI-CO/organic halide complexes
Chloroform (CF), dichloroethane (DCE), dichloropropane (DCP), chloropentane (CP), chlorobenzene (CB), chloromethyl benzene (CMB), chloroethyl benzene (CEB), and chloropropyl benzene (CPB) were obtained from Sigma Aldrich. Individual HbI-CO crystals, were transferred to glass capillary tubes to which 5-10 μl aliquots of organic halide were added to the crystal. Each organic halide was allowed to equilibrate through direct liquid contact with the crystal for 2 minutes. Excess compound was removed and mother liquor was added. Capillary tubes were sealed and equilibrated overnight. Diffraction data were measured with either a Mar 345 image plate system or an R-Axis4 image plate system as described previously (Knapp et al., 2005). Data were processed as described above. Models were fitted to the observed data with O (Jones et al., 1991) and refined with CNS (Brunger et al., 1998) (Tables S1-S3).
Computational analysis of protein cavities
Implicit ligand sampling(Cohen et al., 2006) was used to determine the potential mean force (PMF) of the ligand at each position within the HbI model. PMF relates the probability of a ligand being present at each position within a given set of ensemble structures compared to its presence in either a vacuum or in bulk solvent. The PMF algorithm has been implemented as part of the VMD 1.8.4 software package (Humphrey et al., 1996) as described previously (Cohen et al., 2006). All molecular dynamic calculations were made with NAMD (Phillips et al., 2005) and the parameters used for the identical calculation made on Mb (Humphrey et al., 1996).
Kinetic analysis of geminate rebinding in HbI solution
Wild type HbI-O2 solutions were prepared as described above (Knapp et al., 2005). Samples were placed into cells pressurized with 100 to 300 psi (6.9 - 20.7 · 105 Pa) of Xe. Flash photolysis of ligands and spectroscopic analysis were done using the procedures and instrumentation described previously (Knapp et al., 2005), with and without Xe added. (Unfortunately, similar experiments with bound organic halide compounds proved impossible due to very high Fe oxidation rates in the presence of those compounds).
Time resolved crystallographic analysis of ligand migration
Crystals of native WT HbI-CO (Royer, 1994; Summerford et al., 1995) were mounted in capillary tubes with the base of the crystal being glued to a glass rod which was in turn glued to the capillary tube by epoxy. The epoxy-glued crystals are sufficiently stable to prevent elongated spot profiles when crystals are exposed to laser pulses (Knapp et al., 2004). A second batch of crystals was mounted with a polyvinyl film (Knapp et al., 2004; Rayment, 1985). The polyvinyl resin is initially dissolved in DCE, such that exposure of the crystals to this solution results in DCE translocating to the Xe4 site as the polyvinyl film forms around the crystal. Time-resolved experiments were carried out as described previously (Knapp et al., 2006) at BioCARS beamline 14-ID-B, at the APS.
Pump-probe Laue data were collected utilizing 7ns laser pulses (at 615nm) as a pump and either a single X-ray pulse (100ps duration) or a “super-pulse” (500ns duration) as a probe in the hybrid mode of the APS storage ring. All experimental conditions and data collection protocols are as described in (Knapp et al., 2006). Two time series were collected for the WT crystals, while three time series were collected for the WT/DCE crystals. All time series included a dark (no laser) frame and a series of time delays between laser and X-ray pulses. Time series 1 for WT was collected from three crystals at time delays of 5ns, 25ns, 75ns and 200ns. Time series 1 for WT/DCE was collected from four crystals at time delays of 2ns, 5ns, 25ns, 75ns, 200ns, 700ns, and 3μs. For WT/DCE crystals, additional time series 2 was also collected in the ns time domain, from four crystals at 10ns, 50ns, 100ns, and 400ns time delays. For these three time series with short time delays, a single X-ray pulse was used as a probe, with each diffraction image resulting from 75 pump/probe exposures at 0.5Hz. Data for longer laser-X-ray time delays were collected utilizing X-ray pulses of 500ns as a probe. Time series 2 for WT was collected from three crystals at time delays of 500ns, 1μs, 2μs, 4μs, 6μs and 30μs and time series 3 for WT/DCE was collected from four crystals at time delays of 2μs, 4μs, 6μs, and 15μs.
Laue data were processed with software package Precognition (Zhong Ren, www.renzresearch.com). Laue data reduction statistics is shown in Table 2. Weighted difference electron density maps (light-dark) were calculated as described earlier (Knapp et al., 2006), with phases from the published structure of HbI-CO (3SDH).
Table 2.
Data Reduction Statistics for WT HbI Time-resolved Laue Data
| Statistics for representative WT HbI crystals for Series 1 and Series 2 (20 images per dark/light data set for each crystal; 9° angular increment) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Series | Crystal | Time delay | X-ray exposure time | R(F2) (%) |
R(|F|) (%) |
Completeness (%) | ||||
| Overall 100-1.6 Å |
Last Shell 1.67-1.6 Å |
|||||||||
| 1 | 1 of 3 | dark | 100ps ×177 | 5.4 | 3.1 | 62.7 | 22.5 | |||
| 1 | 1 of 3 | 5ns | 100ps × 75 | 5.9 | 3.5 | 54.6 | 14.6 | |||
| 1 | 1 of 3 | 25ns | 100ps × 75 | 5.9 | 3.5 | 54.3 | 14.2 | |||
| 1 | 1 of 3 | 75ns | 100ps × 75 | 6.1 | 3.6 | 53.1 | 13.9 | |||
| 1 | 1 of 3 | 200ns | 100ps × 75 | 6.5 | 3.7 | 51.8 | 13.4 | |||
| 2 | 1 of 3 | dark | 500ns × 20 | 4.1 | 2.5 | 65.1 | 26.1 | |||
| 2 | 1 of 3 | 500ns | 500ns × 15 | 5.0 | 3.1 | 57.4 | 19.7 | |||
| 2 | 1 of 3 | 1μs | 500ns × 15 | 4.7 | 2.9 | 59.5 | 20.5 | |||
| 2 | 1 of 3 | 2μs | 500ns × 15 | 4.5 | 2.7 | 60.1 | 21.4 | |||
| 2 | 1 of 3 | 4μs | 500ns × 15 | 4.4 | 2.7 | 60.2 | 21.1 | |||
| 2 | 1 of 3 | 6μs | 500ns × 15 | 4.6 | 2.8 | 60.1 | 21.2 | |||
| 2 | 1 of 3 | 30μs | 500ns × 15 | 4.6 | 2.8 | 60.1 | 21.2 | |||
| Completeness of ΔF(t) WT HbI data sets (ΔF(t) merged across 3 crystals) | ||||||||||
| Resolution range (Å) | 5ns | 25ns | 75ns | 200ns | 500ns | 1μs | 2μs | 4μs | 6μs | 30μs |
| 100 – 1.60 | 70.5 | 69.1 | 68.6 | 66.1 | 71.4 | 72.9 | 73.2 | 72.4 | 72.6 | 72.5 |
| 100 – 3.20 | 85.0 | 79.8 | 80.9 | 75.3 | 75.2 | 77.6 | 78.7 | 73.4 | 74.6 | 73.3 |
| 3.20 – 2.54 | 90.4 | 89.3 | 89.7 | 88.1 | 89.9 | 90.4 | 90.7 | 90.1 | 89.9 | 90.6 |
| 2.54 – 2.22 | 88.2 | 87.2 | 86.7 | 85.8 | 89.2 | 90.1 | 89.7 | 89.8 | 90.1 | 89.6 |
| 2.22 – 2.02 | 85.7 | 85.9 | 84.8 | 83.9 | 87.2 | 88.5 | 88.4 | 88.5 | 88.8 | 88.6 |
| 2.02 – 1.87 | 73.7 | 73.8 | 73.0 | 70.5 | 76.4 | 78.5 | 78.4 | 78.5 | 78.2 | 78.6 |
| 1.87 – 1.76 | 62.2 | 60.9 | 59.5 | 57.2 | 66.1 | 67.1 | 67.7 | 67.6 | 67.9 | 67.6 |
| 1.76 – 1.67 | 49.4 | 48.8 | 47.5 | 43.6 | 53.9 | 55.9 | 56.4 | 56.6 | 56.2 | 57.2 |
| 1.67 – 1.60 | 29.6 | 27.4 | 26.5 | 24.5 | 33.6 | 35.1 | 35.4 | 35.8 | 35.2 | 34.9 |
Regions of the difference electron density maps were integrated by the program PROMSK (Schmidt et al., 2005) by summing up the density at grid points within a certain radius around a specific set of coordinates as described previously (Knapp et al., 2006). A radius of 1.2Å was used for CO A-site integration, while a radius of 1.6Å was used for Xe sites integration to capture possibly more distributed density at these sites. We note that values for electron content of difference electron density features obtained by such integration and the resulting time courses do include various sources of errors. Such errors can result, for example, from the overlap of electron density features in some regions of interest (that in effect cancel each other in the difference maps) or from the problem of choosing an appropriate radius of integration.
Supplementary Material
Acknowledgments
We thank the BioCARS staff for assistance during data collection and Yi Wang for assistance with Figure 3B. This work was supported by National Institutes of Health (NIH) Grant GM66756 (to W.E.R. and V.S.) and Grant RR05969 (to K.S.); and by the National Science Foundation Grant SCI04-38712 (to K.S.). Early work was supported by a postdoctoral fellowship to J.E.K. from the New England Affiliate of the American Heart Association (9920261T). APS is supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science (Contract W-31-109-Eng-38). Use of the BioCARS Sector 14 is supported by the National Institutes of Health, National Center for Research Resources, under grant number RR007707.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bourgeois D, Vallone B, Arcovito A, Sciara G, Schotte F, Anfinrud PA, Brunori M. Extended subnanosecond structural dynamics of myoglobin revealed by Laue crystallography. Proc Natl Acad Sci U S A. 2006;103:4924–4929. doi: 10.1073/pnas.0508880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Brunori M. Structural dynamics of myoglobin. Biophys Chem. 2000;86:221–230. doi: 10.1016/s0301-4622(00)00142-3. [DOI] [PubMed] [Google Scholar]
- Brunori M, Gibson QH. Cavities and packing defects in the structural dynamics of myoglobin. EMBO Rep. 2001;2:674–679. doi: 10.1093/embo-reports/kve159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Chiancone E, Elber R, Royer WE, Jr, Regan R, Gibson QH. Ligand binding and conformation change in the dimeric hemoglobin of the clam Scapharca inaequivalvis. J Biol Chem. 1993;268:5711–5718. [PubMed] [Google Scholar]
- Cohen J, Arkhipov A, Braun R, Schulten K. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys J. 2006;91:1844–1857. doi: 10.1529/biophysj.106.085746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J, Schulten K. O2 migration pathways are not conserved across proteins of a similar fold. Biophys J. 2007;93:3591–3600. doi: 10.1529/biophysj.107.108712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon PJ, Royer WE., Jr Crystal structure of oxygenated Scapharca dimeric hemoglobin at 1.7-A resolution. J Biol Chem. 1994;269:25259–25267. doi: 10.2210/pdb1hbi/pdb. [DOI] [PubMed] [Google Scholar]
- Gursky O, Fontano E, Bhyravbhatla B, Caspar DL. Stereospecific dihaloalkane binding in a pH-sensitive cavity in cubic insulin crystals. Proc Natl Acad Sci U S A. 1994;91:12388–12392. doi: 10.1073/pnas.91.26.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry ER, Sommer JH, Hofrichter J, Eaton WA. Geminate recombination of carbon monoxide to myoglobin. J Mol Biol. 1983;166:443–451. doi: 10.1016/s0022-2836(83)80094-1. [DOI] [PubMed] [Google Scholar]
- Huang X, Boxer SG. Discovery of new ligand binding pathways in myoglobin by random mutagenesis. Nat Struct Biol. 1994;1:226–229. doi: 10.1038/nsb0494-226. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Knapp JE, Bonham MA, Gibson QH, Nichols JC, Royer WE., Jr Residue F4 plays a key role in modulating oxygen affinity and cooperativity in Scapharca dimeric hemoglobin. Biochemistry. 2005;44:14419–14430. doi: 10.1021/bi051052+. [DOI] [PubMed] [Google Scholar]
- Knapp JE, Pahl R, Srajer V, Royer WE., Jr Allosteric action in real time: time-resolved crystallographic studies of a cooperative dimeric hemoglobin. Proc Natl Acad Sci U S A. 2006;103:7649–7654. doi: 10.1073/pnas.0509411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp JE, Royer WE., Jr Ligand-linked structural transitions in crystals of a cooperative dimeric hemoglobin. Biochemistry. 2003;42:4640–4647. doi: 10.1021/bi027136p. [DOI] [PubMed] [Google Scholar]
- Knapp JE, Srajer V, Pahl R, Royer WE., Jr Immobilization of Scapharca HbI crystals improves data quality in time-resolved crystallographic experiments. Micron. 2004;35:107–108. doi: 10.1016/j.micron.2003.10.039. [DOI] [PubMed] [Google Scholar]
- Milani M, Pesce A, Ouellet Y, Ascenzi P, Guertin M, Bolognesi M. Mycobacterium tuberculosis hemoglobin N displays a protein tunnel suited for O2 diffusion to the heme. EMBO J. 2001;20:3902–3909. doi: 10.1093/emboj/20.15.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzarelli A, Bettati S, Rivetti C, Rossi GL, Colotti G, Chiancone E. Cooperative oxygen binding to scapharca inaequivalvis hemoglobin in the crystal. J Biol Chem. 1996;271:3627–3632. doi: 10.1074/jbc.271.7.3627. [DOI] [PubMed] [Google Scholar]
- Nienhaus K, Knapp JE, Palladino P, Royer WE, Jr, Nienhaus GU. Ligand migration and binding in the dimeric hemoglobin of Scapharca inaequivalvis. Biochemistry. 2007;46:14018–14031. doi: 10.1021/bi7016798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JS, Soman J, Phillips GN., Jr Ligand pathways in myoglobin: a review of Trp cavity mutations. IUBMB Life. 2007;59:552–562. doi: 10.1080/15216540701230495. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Perutz MF, Mathews FS. An x-ray study of azide methaemoglobin. J Mol Biol. 1966;21:199–202. doi: 10.1016/0022-2836(66)90088-x. [DOI] [PubMed] [Google Scholar]
- Pesce A, Nardini M, Dewilde S, Geuens E, Yamauchi K, Ascenzi P, Riggs AF, Moens L, Bolognesi M. The 109 residue nerve tissue minihemoglobin from Cerebratulus lacteus highlights striking structural plasticity of the alpha-helical globin fold. Structure. 2002;10:725–735. doi: 10.1016/s0969-2126(02)00763-3. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I. Diffraction methods for biological macromolecules. Treatment and manipulation of crystals. Methods Enzymol. 1985;114:136–140. doi: 10.1016/0076-6879(85)14012-7. [DOI] [PubMed] [Google Scholar]
- Royer WE., Jr High-resolution crystallographic analysis of a co-operative dimeric hemoglobin. J Mol Biol. 1994;235:657–681. doi: 10.1006/jmbi.1994.1019. [DOI] [PubMed] [Google Scholar]
- Royer WE, Jr, Knapp JE, Strand K, Heaslet HA. Cooperative hemoglobins: conserved fold, diverse quaternary assemblies and allosteric mechanisms. Trends Biochem Sci. 2001;26:297–304. doi: 10.1016/s0968-0004(01)01811-4. [DOI] [PubMed] [Google Scholar]
- Royer WE, Jr, Pardanani A, Gibson QH, Peterson ES, Friedman JM. Ordered water molecules as key allosteric mediators in a cooperative dimeric hemoglobin. Proc Natl Acad Sci U S A. 1996;93:14526–14531. doi: 10.1073/pnas.93.25.14526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer WE, Jr, Zhu H, Gorr TA, Flores JF, Knapp JE. Allosteric hemoglobin assembly: diversity and similarity. J Biol Chem. 2005;280:27477–27480. doi: 10.1074/jbc.R500006200. [DOI] [PubMed] [Google Scholar]
- Salter MD, Nienhaus K, Nienhaus GU, Dewilde S, Moens L, Pesce A, Nardini M, Bolognesi M, Olson JS. The apolar channel in Cerebratulus lacteus hemoglobin is the route for O2 entry and exit. J Biol Chem. 2008;283:35689–35702. doi: 10.1074/jbc.M805727200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Nienhaus K, Pahl R, Krasselt A, Anderson S, Parak F, Nienhaus GU, Srajer V. Ligand migration pathway and protein dynamics in myoglobin: a time-resolved crystallographic study on L29W MbCO. Proc Natl Acad Sci U S A. 2005;102:11704–11709. doi: 10.1073/pnas.0504932102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotte F, Lim M, Jackson TA, Smirnov AV, Soman J, Olson JS, Phillips GN, Jr, Wulff M, Anfinrud PA. Watching a protein as it functions with 150-ps time-resolved x-ray crystallography. Science. 2003;300:1944–1947. doi: 10.1126/science.1078797. [DOI] [PubMed] [Google Scholar]
- Scott EE, Gibson QH, Olson JS. Mapping the pathways for O2 entry into and exit from myoglobin. J Biol Chem. 2001;276:5177–5188. doi: 10.1074/jbc.M008282200. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM, Schneider TR. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- Srajer V, Ren Z, Teng TY, Schmidt M, Ursby T, Bourgeois D, Pradervand C, Schildkamp W, Wulff M, Moffat K. Protein conformational relaxation and ligand migration in myoglobin: a nanosecond to millisecond molecular movie from time-resolved Laue X-ray diffraction. Biochemistry. 2001;40:13802–13815. doi: 10.1021/bi010715u. [DOI] [PubMed] [Google Scholar]
- Srajer V, Teng T, Ursby T, Pradervand C, Ren Z, Adachi S, Schildkamp W, Bourgeois D, Wulff M, Moffat K. Photolysis of the carbon monoxide complex of myoglobin: nanosecond time-resolved crystallography. Science. 1996;274:1726–1729. doi: 10.1126/science.274.5293.1726. [DOI] [PubMed] [Google Scholar]
- Summerford CM, Pardanani A, Betts AH, Poteete AR, Colotti G, Royer WE., Jr Bacterial expression of Scapharca dimeric hemoglobin: a simple model system for investigating protein cooperatively. Protein Eng. 1995;8:593–599. doi: 10.1093/protein/8.6.593. [DOI] [PubMed] [Google Scholar]
- Tetreau C, Blouquit Y, Novikov E, Quiniou E, Lavalette D. Competition with xenon elicits ligand migration and escape pathways in myoglobin. Biophys J. 2004;86:435–447. doi: 10.1016/S0006-3495(04)74120-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian WD, Sage JT, Champion PM. Investigations of ligand association and dissociation rates in the “open” and “closed” states of myoglobin. J Mol Biol. 1993;233:155–166. doi: 10.1006/jmbi.1993.1491. [DOI] [PubMed] [Google Scholar]
- Tilton RF, Jr, Kuntz ID, Jr, Petsko GA. Cavities in proteins: structure of a metmyoglobin-xenon complex solved to 1.9 A. Biochemistry. 1984;23:2849–2857. doi: 10.1021/bi00308a002. [DOI] [PubMed] [Google Scholar]
- Vojtechovsky J, Chu K, Berendzen J, Sweet RM, Schlichting I. Crystal structures of myoglobin-ligand complexes at near-atomic resolution. Biophys J. 1999;77:2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Phillips GN., Jr Crystal structures of CO-, deoxy- and met-myoglobins at various pH values. J Mol Biol. 1996;256:762–774. doi: 10.1006/jmbi.1996.0123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.