

Abstract
AIM: To clarify the significance of JC virus (JCV) T-antigen (T-Ag) expression in human gastric cancer.
METHODS: We investigated the relationship between T-Ag detected by immunohistochemistry and Epstein-Barr virus (EBV) infection, microsatellite instability (MSI), and genetic and epigenetic alterations in gastric cancers. Mutations in the p53, β-catenin, KRAS, BRAF, PIK3CA genes were analyzed by polymerase chain reaction (PCR)-single strand conformation polymorphism and DNA sequencing. Allelic losses were determined by PCR at 7 microsatellite loci. Aberrant DNA methylation was analyzed by MethyLight assay.
RESULTS: JCV T-Ag protein expression was found in 49% of 90 gastric cancer tissues. T-Ag positivity was not correlated with clinicopathological characteristics. T-Ag expression was detected in a similar percentage of EBV positive cancers (4 of 9, 44%) and EBV negative cancers (35 of 73, 48%). T-Ag expression was detected in a significantly lower percentage of MSI-H cancers (14%) than in non MSI-H cancers (55%, P = 0.005). T-Ag expression was detected in a significantly higher percentage of cancers with nuclear/cytoplasmic localization of β-catenin (15 of 21, 71%) than in cancers without (42%, P = 0.018). p53 mutations were detected in a significantly lower percentage of T-Ag positive cancers (32%) than in T-Ag negative cancers (57%, P = 0.018). T-Ag positive gastric cancers showed a significant increase in the allelic losses and aberrant methylation compared with T-Ag negative gastric cancers (P = 0.008 and P = 0.003).
CONCLUSION: The results suggest that JCV T-Ag is involved in gastric carcinogenesis through multiple mechanisms of genetic and epigenetic alterations.
Keywords: JC virus, T-antigen, Epstein-Barr virus, Microsatellite instability, Gastric cancer
INTRODUCTION
Viruses have been proposed to play an etiologic role in human cancers. JC virus (JCV) is a polyomavirus that ubiquitously infects humans worldwide, and more than 80% of the adult population carries antibodies against the virus[1]. JCV has been implicated in various types of human cancers[2-9]. It has been reported that JCV sequences are frequently present throughout the normal human gastrointestinal tract and in colorectal and gastric cancers[2-9].
JCV encodes a transforming gene, T-antigen (T-Ag), which is believed to mediate the oncogenic potential of the virus. T-Ag protein expression was specifically present in the nuclei of colon cancer cells, but not in any adjacent normal colonic epithelium[8]. Previous studies have identified multiple pathways including p53, pRb, IRS, NF1/NF2 and β-catenin, which may be dysregulated by T-Ag[4,10,11]. T-Ag can bind and inactivate p53, pRb, and the spindle assembly checkpoint protein Bub1, resulting in disruption of chromosomal integrity and cell cycle checkpoints[12,13]. T-Ag can also bind and stabilize β-catenin[4,11]. T-Ag protein expression, rather than the simple presence of JCV DNA sequences, has been significantly associated with chromosomal instability (CIN) and the methylator phenotype in colorectal cancers[8]. The possible involvement in aberrant methylation is thought to provide a secondary link to microsatellite instability (MSI) in colorectal cancers[8]. Therefore, it has been suggested that JCV is involved in colorectal cancers through multiple mechanisms of genetic and epigenetic alterations[8]. Bhattacharyya et al[14] recently reported the interplay between β-catenin and Rac1 that is initiated by T-Ag and results in stabilization of β-catenin and its presence in cell membrane ruffles. T-Ag and β-catenin synergistically activate Rac1, an event that can trigger several oncogenic factors[14].
The association between T-Ag expression and aberrant promoter methylation in colorectal cancers suggests that this viral oncogene induces the methylator phenotype[8]. Genomic methylation is a host defense mechanism that silences the transcription of transposons and retroviruses that have accumulated in the mammalian genome[8,15,16]. Methylation of host cell genes is not unique to JCV and occurs with other oncogenic viruses. It has been suggested that the key protein triggering methylation events for the polyomavirus SV40 is T-Ag, which mediates cellular transformation of cultured epithelial cells by regulating the activities of key de novo DNA methyltransferases, such as DNMT3b[8,17].
A significant correlation has been found between Epstein-Barr virus (EBV) and methylation of multiple genes in gastric cancers[18,19]. A mutually negative association between EBV and MSI has been reported in gastric cancers[20]. T-Ag protein expression was found in 9 (39%) of 23 gastric cancers, whereas no expression was observed in any of the non-cancer tissues[9]. In contrast to colorectal cancers[8], however, little is known about the significance of T-Ag expression in gastric cancers. In the current study, we investigated the relationship between T-Ag protein expression and EBV infection, MSI, and genetic and epigenetic alterations in gastric cancers.
MATERIALS AND METHODS
Tissue samples
A total of 90 paired specimens of gastric adenocarcinoma and adjacent noncarcinoma tissue were obtained from Japanese patients who had undergone surgical treatment. Informed consent was obtained from each patient. The tumor-node-metastasis (TNM) system of the American Joint Committee on Cancer and the International Union against Cancer was used for the pathologic diagnosis and classification of variables. Clinicopathological characteristics were as follows: age (69 ± 10 years), gender (59 male and 31 female), Lauren histology (44 intestinal and 46 diffuse), and pTNM stages (stage I, 23; stage II, 16; stage III, 30; stage IV, 21).
Immunohistochemistry and in situ hybridization
Immunohistochemistry using a mouse monoclonal antibody against SV40 large T-Ag (clone PAb416, 1:100 dilution; Oncogene Research Products, San Diego, CA, USA), which cross-reacts with T-Ag of JCV, was performed as described previously[8]. Immunohistochemistry with an anti-human β-catenin monoclonal antibody (BD Transduction Laboratories, San Jose, CA, USA) was done as described previously[21]. Cancer cases were categorized into the following 4 groups corresponding to immunostaining patterns of β-catenin as described previously[21]: membranous, membranous staining pattern similar to that in normal epithelium; weak, no staining or weaker staining than normal epithelium; cytoplasmic, diffuse staining in the cytoplasm as well as at the cell membrane; accumulated, strong staining in the nucleus and cytoplasm. EBV infection was analyzed by in situ hybridization for EBER-1[20].
MSI analysis
MSI was analyzed by polymerase chain reaction (PCR) using the mononucleotide (BAT26 and BAT25) and dinucleotide markers (D2S123, D5S346, and D17S250) proposed by the NCI workshop[22]. Based on the number of markers showing instability per cancer, cancers were divided into 3 groups; those with 2 or more of the 5 markers displaying instability (MSI-H), those with 1 of 5 markers displaying instability (MSI-low; MSI-L), and those with no instability (microsatellite stable; MSS).
Mutation analysis
Mutations in exons 2-9 of p53, exon 3 of β-catenin, codons 12 and 13 of KRAS, codon 600 of BRAF, exons 9 and 20 of PIK3CA genes were analyzed by PCR-single strand conformation polymorphism and DNA sequencing as described previously[23-25]. Interstitial deletions spanning exon 3 of β-catenin was analyzed as described previously[24,25].
LOH analysis
LOH was analyzed as described previously[26]. Seven sets of microsatellite loci that are linked to tumor suppressor genes were used to identify significant allelic losses in gastric cancers. DNA was amplified by PCR at microsatellite loci linked to the APC locus on 5q21 (D5S505), possible tumor suppressor/senescence gene locus on 10p15 (D10S501 and D10S602), p53 locus on 17p13 (TP53), BRCA1 locus on 17q21 (D17S855), and DCC locus on 18q21 (D18S58 and D18S61)[26]. Assessment of LOH was assigned when a tumor allele showed at least a 50% reduction in the relative intensity of 1 allele in cancer tissue compared with the matched non-cancer DNA as described previously[26].
Quantitative DNA methylation analysis by real-time PCR (MethyLight assay)
Sodium bisulfite treatment of genomic DNA and MethyLight assay were performed as described previously[25,27-30]. We analyzed six promoters: calcium channel, voltage dependent, T type a-1G subunit (CACNA1G), cellular retinoic acid binding protein 1 (CRABP1), neurogenin 1 (NEUROG1), CDKN2A (p16), RUNX3, and SOCS1. Primers, probes, and percentage of methylated reference (PMR, i.e. degree of methylation) were described previously[29,30]. The cutoff value of 4 (except for 6 in CRABP1) was based on previously validated data[28].
Statistical analysis
The results were assessed for associations with clinicopathological parameters, using the following statistical tests: Student’s t test for age, the Mann-Whitney test for depth of invasion, lymph node metastasis, and pTNM stage, and the chi-square test or Fisher’s exact test for the remaining parameters. P < 0.05 was considered significant. A P value between 0.05 and 0.10 was considered as a trend toward an association.
RESULTS
T-Ag protein expression in gastric cancers
T-Ag protein expression was found in 44 (48.9%) of 90 gastric cancers, whereas no expression was observed in adjacent normal gastric epithelial cells or stromal cells (Figure 1 and data not shown). T-Ag positivity was not correlated with clinicopathological characteristics (data not shown).
Figure 1.
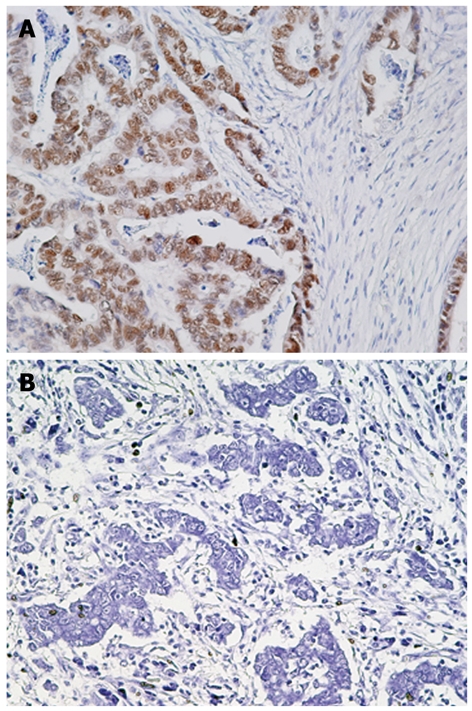
Immunohistochemistry for JCV T-Ag in gastric cancer tissues. A: Gastric adenocarcinoma positive for JCV T-Ag; B: Gastric adenocarcinoma negative for JCV T-Ag. Original magnification, × 200.
EBV infection
EBER-1 expression was found in 9 (11.0%) of 82 gastric cancers, whereas no expression was observed in any of the non-cancer tissues (Figure 2). EBV infection was detected more frequently in diffuse type (7 of 44, 16%) than intestinal type (2 of 38, 5%), although this did not reach statistical significance. T-Ag expression was detected in a similar percentage of EBV positive cancers (4 of 9, 44%) and EBV negative cancers (35 of 73, 48%). The results for T-Ag protein expression and EBV infection were summarized in a Figure 3.
Figure 2.
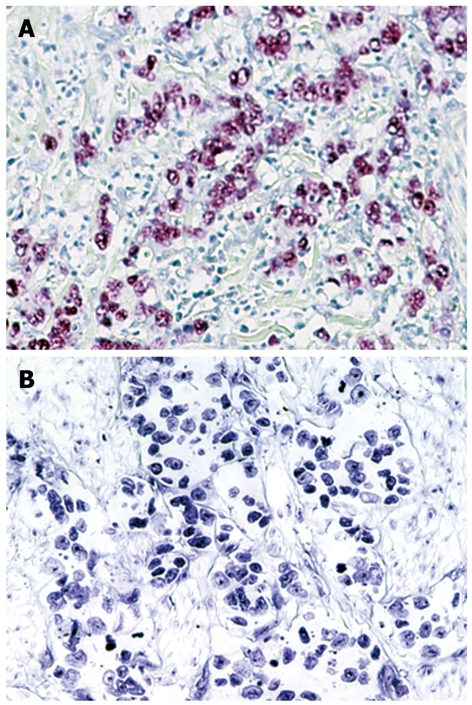
In situ hybridization for EBER-1 in gastric cancer tissues. A: Gastric adenocarcinoma positive for EBER-1; B: Gastric adenocarcinoma negative for EBER-1. Original magnification, × 200.
Figure 3.

Classification of 90 gastric cancer tissues based on T-Ag protein expression and EBV infection.
MSI status
Following the NCI criteria, a total of 90 gastric cancers was classified as follows: 14 (16%) MSI-H, 10 (11%) MSI-L, and 66 (73%) MSS (Figure 4). T-Ag expression was detected in a significantly lower percentage of MSI-H cancers (14%) than in non MSI-H cancers (55%, P = 0.005). None of the MSI-H tumors were EBV positive.
Figure 4.

MSI analysis in gastric cancer tissues. MSI was analyzed by PCR using the mononucleotide (BAT26 and BAT25) and dinucleotide markers (D2S123, D5S346, and D17S250). Results of matched normal (upper panel) and tumor (bottom panel) samples (MSI-H) are shown. The arrows mean instability positive marker. RFU: Relative fluorescent units.
Alterations of β-catenin
Figure 5 shows representative results of immunohistochemistry for β-catenin in gastric cancer tissues. In normal gastric epithelium cells, β-catenin was moderately stained in a membranous distribution (Figure 5A), whereas fibroblasts, endothelial cells, and smooth muscle cells were negative. Membranous, weak, cytoplasmic, and accumulated pattern was observed in 36 (40%), 33 (37%), 11 (12%), and 10 (11%), respectively. Neither point mutations nor interstitial deletions in exon 3 were detected (data not shown). T-Ag expression was detected in a significantly higher percentage of cancers with nuclear/cytoplasmic localization of β-catenin (15 of 21, 71%) than in cancers without (42%, P = 0.018).
Figure 5.

Immunohistochemistry for β-catenin in gastric normal (A) and cancer (B-E) tissues. A: Moderate membrane staining; B: Membrane staining pattern similar to that seen in normal epithelium; C: Weak staining; D: Diffuse staining in the cytoplasm and membrane; E: Strong staining of the nucleus and cytoplasm (Original magnification, × 200).
Mutations of the p53, KRAS, BRAF, PIK3CA genes in gastric cancer tissues
p53 mutations were detected in 40 (44%) of 90 gastric cancer tissues. p53 mutations were detected in a significantly lower percentage of T-Ag positive cancers (32%) than in T-Ag negative cancers (57%, P = 0.018). p53 mutations were detected in a significantly lower percentage of MSI-H cancers (14%) than in MSI-L cancers (60%, P = 0.028) or in MSS cancers (48%, P = 0.021). KRAS mutations were detected in only 5 (6%) of 90 gastric cancers. BRAF mutations were not detected. PIK3CA mutations were detected in 4 (4%) of 90 cancers. The exact mutations and their frequencies were described in a Table 1.
Table 1.
The exact mutations and their frequencies in gastric cancer tissues
| p53 | n | n | n | KRAS | n | BRAF | PIK3CA | n | ||
| 44% (40/90) | 6% (5/90) | 0% (0/90) | 4% (4/90) | |||||||
| G524A (R175H) | 3 | G469T (V157F) | 2 | G733A (G245S) | 2 | G35A (G12D) | 3 | A3140G (H1047R) | 3 | |
| C742T (R248W) | 2 | G743A (R248Q) | 2 | C817T (R273C) | 2 | G35T (G12V) | 2 | G1624A (E542K) | 1 | |
| G818A (R273H) | 2 | C844T (R282W) | 2 | C200G (P67R) | 1 | |||||
| G313T (G105C) | 1 | G422A (C141Y) | 1 | G461T (G154V) | 1 | |||||
| G473A (R158H) | 1 | C489G (Y163X) | 1 | G524T (R175L) | 1 | |||||
| C574T (Q192X) | 1 | A578G (H193R) | 1 | G586T (R196X) | 1 | |||||
| G587C (R196P) | 1 | A614G (Y205C) | 1 | C637T (R213X) | 1 | |||||
| G638A (R213Q) | 1 | G646A (V216M) | 1 | A659G (Y220C) | 1 | |||||
| G725T (C242F) | 1 | G731T (G244V) | 1 | G733T (G245V) | 1 | |||||
| C742G (R248G) | 1 | C817A (R273S) | 1 | T823G (C275G) | 1 | |||||
| C847T (R283C) | 1 |
Nucleotide substitution (amino acid change) is shown.
LOH analysis
Overall, 62 (69%) of 90 cancers had at least 1 LOH, with most frequent chromosomal losses observed on 17p (43%), followed by 18q (33%), 10p (27%), 5q (24%), and 17q (12%) (Figure 6A and B). Of 62 cancers with LOH, 4 (6%) were also MSI-H. The data were expressed in terms of an allelic loss index (ALI), which reflects the number of chromosomal losses per the number of chromosomal loci showing heterozygosity but not MSI in each cancer specimen. T-Ag positive gastric cancers showed a significant increase in the ALI compared with T-Ag negative gastric cancers (0.41 ± 0.12 vs 0.21 ± 0.09, P = 0.008).
Figure 6.

LOH analysis in gastric cancer tissues. A: A representative example of a tumor with allelic loss in 5q21 (D5S505) is shown. The upper and bottom panels show the intensity plots of both the normal and tumor lane, respectively, demonstrating reduced relative intensity of allele one in the tumor sample (arrow) compared to the corresponding normal sample; B: LOH frequencies based on each individual marker.
Methylation analysis
The results were summarized based on each individual marker (Figure 7A and B). The data were expressed in terms of a methylation index (MI), which reflects the number of abnormally methylated promoters per cancer specimen within the subsets of cancers. T-Ag positive gastric cancers showed a significant increase in the MI compared with T-Ag negative gastric cancers (3.01 ± 0.92 vs 1.51 ± 0.71, P = 0.003).
Figure 7.

MethyLight analysis in gastric cancer tissues. A: Results of the CRABP1 gene are shown. Bisulfite-converted DNA was used for quantitative methylation-specific PCR. The amount of hypermethylated DNA was determined by reading the midpoint of the linear portion of the S-shaped real-time curves, called the Ct point or threshold cycle. The Ct refers to the number of cycles it takes a sample to reach a specific fluorescence threshold; B: DNA methylation frequencies based on each individual marker.
DISCUSSION
In the current study, T-Ag protein expression was positive in 44 (49%) of 90 gastric cancer tissues and it was observed specifically in gastric cancer cells but not in the adjacent normal gastric epithelial cells or stromal cells. Previous study has also shown that T-Ag protein expression was found in 9 (39%) of 23 gastric cancers, whereas no expression was observed in any of the non-cancer tissues[9]. These results suggest an active role for this oncogenic protein in gastric carcinogenesis[9].
The frequencies of EBV infection (11%) and MSI-H (16%) in this study were almost consistent with those reported in previous studies[20,31,32]. None of the MSI-H cancers were EBV positive. A mutually negative association between EBV and MSI has been reported in gastric cancers[20]. T-Ag was detected in a significantly lower percentage of MSI-H cancers (14%) than in non MSI-H cancers (55%). In contrast, T-Ag expression was present in similar frequencies in MSI-H and (8/15, 53%) and MSS/MSI-L (35/85, 41%) colorectal cancers[8]. From the view point of MSI, these results suggest a differential role of T-Ag in gastric and colorectal carcinogenesis.
T-Ag expression was detected in a significantly higher percentage of cancers with nuclear/cytoplasmic localization of β-catenin than in cancers without. T-Ag interacts with β-catenin, leading to its stabilization and resultant dysregulation of the WNT-signaling pathway in gastrointestinal cancers[4]. Neither point mutations nor interstitial deletions in exon 3 were detected. Although not analyzed in this study, the frequency of APC mutations is relatively low in gastric cancers. Therefore, T-Ag may play an important role in the stabilization of β-catenin in gastric cancers.
p53 mutation was detected in a significantly lower percentage of T-Ag positive cancers (32%) than in T-Ag negative cancers (57%). T-Ag can bind and inactivate p53[12], resulting in disruption of chromosomal integrity and cell cycle checkpoints. Therefore, T-Ag expression may eliminate the selective pressure for p53 mutations in a subset of gastric cancers.
A significant association was observed between T-Ag expression and allelic losses, being consistent with that in colorectal cancers[8]. It has been previously reported that the introduction of JCV into a diploid cell line leads to the rapid induction of CIN[33]. JCV can also induce CIN in a diploid colon cancer cell line RKO, which has wild-type APC, p53, and β-catenin genes[33]. These results suggest that T-Ag play a role in genomic damage during gastric carcinogenesis.
Significant association was also observed between T-Ag expression and aberrant methylation, being consistent with that in colorectal cancers[8]. A significant correlation has been found between EBV and methylation of multiple genes in gastric cancers[8,18,19]. These results suggest that JCV T-Ag and EBV play a similar role, at least in part, during gastric carcinogenesis.
Taken together, our results suggest that JCV T-Ag is involved in gastric cancers through multiple mechanisms of genetic and epigenetic alterations[34]. Further analysis is required to determine whether there is a plausible molecular mechanism by which JCV can induce genetic and epigenetic alterations in gastric carcinogenesis.
COMMENTS
Background
Gastric cancer is one of the most common cancer and leading cause of cancer-related death in the world. Understanding the molecular biological features of gastric cancer is necessary for early diagnosis and better prognosis. The potential role of viral infection in human cancer is receiving increasing attention.
Research frontiers
A significant correlation has been found between Epstein-Barr virus (EBV) and methylation of multiple genes in gastric cancer. However, little is known about the significance of JC virus (JCV) T-antigen (T-Ag) expression in gastric cancers. In this study, the authors demonstrate that JCV T-Ag plays a key role in gastric carcinogenesis.
Innovations and breakthroughs
This is the first study to report that JCV T-Ag plays a key role in gastric carcinogenesis. Furthermore, T-Ag expression was associated with nuclear/cytoplasmic localization of β-catenin, allelic losses and aberrant DNA methylation, suggesting that JCV T-Ag is involved in gastric carcinogenesis through multiple mechanisms of genetic and epigenetic alterations.
Applications
JCV T-Ag expression could be future diagnostic and/or therapeutic targets in clinical settings. Understanding of a plausible molecular mechanism by which JCV can induce genetic and epigenetic alterations may represent a future strategy for therapeutic intervention in the treatment of patients with gastric cancer.
Terminology
JCV: JCV is a polyomavirus that ubiquitously infects humans worldwide, and more than 80% of the adult population carries antibodies against the virus. EBV: EBV is a double-stranded DNA virus and takes a linear form in the viral particles. EBV was the first virus discovered from human neoplastic cells, a Burkitt’s lymphoma cell lines. Microsatellite instability (MSI): MSI is a type of genetic instability characterized by length alterations within simple repeated microsatellite sequences.
Peer review
This paper reports the characteristics of gastric cancer with JCV T-Ag expression. The authors showed that JCV T-Ag plays a key role in gastric carcinogenesis. The study sounds interesting and the information could be useful for other gastric cancer researchers.
Footnotes
Supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Yamamoto H, Imai K and Shinomura Y) and Grants-in-Aid for Cancer Research from the Ministry of Health, Labor and Welfare of Japan (Yamamoto H)
Peer reviewers: Mark Bloomston, MD, FACS, Assistant Professor of Surgery, Division of Surgical Oncology, N924 Doan Hall, 410 W.10th Avenue, Columbus, Ohio 43082, United States; Lin Zhang, Associate Professor, Department of Pharmacology & Chemical Biology, University of Pittsburgh, UPCI Research Pavilion, Room 2.42, Hillman Cancer Center, 5117 Centre Ave, Pittsburgh, PA 15214, United States
S- Editor Tian L L- Editor O’Neill M E- Editor Zheng XM
References
- 1.Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 1992;5:49–73. doi: 10.1128/cmr.5.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laghi L, Randolph AE, Chauhan DP, Marra G, Major EO, Neel JV, Boland CR. JC virus DNA is present in the mucosa of the human colon and in colorectal cancers. Proc Natl Acad Sci USA. 1999;96:7484–7489. doi: 10.1073/pnas.96.13.7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ricciardiello L, Laghi L, Ramamirtham P, Chang CL, Chang DK, Randolph AE, Boland CR. JC virus DNA sequences are frequently present in the human upper and lower gastrointestinal tract. Gastroenterology. 2000;119:1228–1235. doi: 10.1053/gast.2000.19269. [DOI] [PubMed] [Google Scholar]
- 4.Enam S, Del Valle L, Lara C, Gan DD, Ortiz-Hidalgo C, Palazzo JP, Khalili K. Association of human polyomavirus JCV with colon cancer: evidence for interaction of viral T-antigen and beta-catenin. Cancer Res. 2002;62:7093–7101. [PubMed] [Google Scholar]
- 5.Khalili K, Del Valle L, Otte J, Weaver M, Gordon J. Human neurotropic polyomavirus, JCV, and its role in carcinogenesis. Oncogene. 2003;22:5181–5191. doi: 10.1038/sj.onc.1206559. [DOI] [PubMed] [Google Scholar]
- 6.Niv Y, Goel A, Boland CR. JC virus and colorectal cancer: a possible trigger in the chromosomal instability pathways. Curr Opin Gastroenterol. 2005;21:85–89. [PubMed] [Google Scholar]
- 7.Casini B, Borgese L, Del Nonno F, Galati G, Izzo L, Caputo M, Perrone Donnorso R, Castelli M, Risuleo G, Visca P. Presence and incidence of DNA sequences of human polyomaviruses BKV and JCV in colorectal tumor tissues. Anticancer Res. 2005;25:1079–1085. [PubMed] [Google Scholar]
- 8.Goel A, Li MS, Nagasaka T, Shin SK, Fuerst F, Ricciardiello L, Wasserman L, Boland CR. Association of JC virus T-antigen expression with the methylator phenotype in sporadic colorectal cancers. Gastroenterology. 2006;130:1950–1961. doi: 10.1053/j.gastro.2006.02.061. [DOI] [PubMed] [Google Scholar]
- 9.Shin SK, Li MS, Fuerst F, Hotchkiss E, Meyer R, Kim IT, Goel A, Boland CR. Oncogenic T-antigen of JC virus is present frequently in human gastric cancers. Cancer. 2006;107:481–488. doi: 10.1002/cncr.22028. [DOI] [PubMed] [Google Scholar]
- 10.Shollar D, Del Valle L, Khalili K, Otte J, Gordon J. JCV T-antigen interacts with the neurofibromatosis type 2 gene product in a transgenic mouse model of malignant peripheral nerve sheath tumors. Oncogene. 2004;23:5459–5467. doi: 10.1038/sj.onc.1207728. [DOI] [PubMed] [Google Scholar]
- 11.Gan DD, Khalili K. Interaction between JCV large T-antigen and beta-catenin. Oncogene. 2004;23:483–490. doi: 10.1038/sj.onc.1207018. [DOI] [PubMed] [Google Scholar]
- 12.Ludlow JW. Interactions between SV40 large-tumor antigen and the growth suppressor proteins pRB and p53. FASEB J. 1993;7:866–871. doi: 10.1096/fasebj.7.10.8344486. [DOI] [PubMed] [Google Scholar]
- 13.Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D, Freire R, Entwistle A, Golemis EA, Roberts TM, et al. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci USA. 2004;101:947–952. doi: 10.1073/pnas.0308006100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharyya R, Noch EK, Khalili K. A novel role of Rac1 GTPase in JCV T-antigen-mediated beta-catenin stabilization. Oncogene. 2007;26:7628–7636. doi: 10.1038/sj.onc.1210576. [DOI] [PubMed] [Google Scholar]
- 15.Bestor TH. The host defence function of genomic methylation patterns. Novartis Found Symp. 1998;214:187–195; discussion 195-199, 228-232. doi: 10.1002/9780470515501.ch11. [DOI] [PubMed] [Google Scholar]
- 16.Doerfler W. DNA methylation: eukaryotic defense against the transcription of foreign genes? Microb Pathog. 1992;12:1–8. doi: 10.1016/0882-4010(92)90060-2. [DOI] [PubMed] [Google Scholar]
- 17.Soejima K, Fang W, Rollins BJ. DNA methyltransferase 3b contributes to oncogenic transformation induced by SV40T antigen and activated Ras. Oncogene. 2003;22:4723–4733. doi: 10.1038/sj.onc.1206510. [DOI] [PubMed] [Google Scholar]
- 18.Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, Ro JY. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol. 2002;160:787–794. doi: 10.1016/S0002-9440(10)64901-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kusano M, Toyota M, Suzuki H, Akino K, Aoki F, Fujita M, Hosokawa M, Shinomura Y, Imai K, Tokino T. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein-Barr virus. Cancer. 2006;106:1467–1479. doi: 10.1002/cncr.21789. [DOI] [PubMed] [Google Scholar]
- 20.Chang MS, Lee HS, Kim HS, Kim SH, Choi SI, Lee BL, Kim CW, Kim YI, Yang M, Kim WH. Epstein-Barr virus and microsatellite instability in gastric carcinogenesis. J Pathol. 2003;199:447–452. doi: 10.1002/path.1302. [DOI] [PubMed] [Google Scholar]
- 21.Sasaki Y, Morimoto I, Kusano M, Hosokawa M, Itoh F, Yanagihara K, Imai K, Tokino T. Mutational analysis of the beta-catenin gene in gastric carcinomas. Tumour Biol. 2001;22:123–130. doi: 10.1159/000050606. [DOI] [PubMed] [Google Scholar]
- 22.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 23.Kahlenberg MS, Stoler DL, Basik M, Petrelli NJ, Rodriguez-Bigas M, Anderson GR. p53 tumor suppressor gene status and the degree of genomic instability in sporadic colorectal cancers. J Natl Cancer Inst. 1996;88:1665–1670. doi: 10.1093/jnci/88.22.1665. [DOI] [PubMed] [Google Scholar]
- 24.Mikami M, Nosho K, Yamamoto H, Takahashi T, Maehata T, Taniguchi H, Adachi Y, Imamura A, Fujita M, Hosokawa M, et al. Mutational analysis of beta-catenin and the RAS-RAF signalling pathway in early flat-type colorectal tumours. Eur J Cancer. 2006;42:3065–3072. doi: 10.1016/j.ejca.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 25.Nosho K, Yamamoto H, Takahashi T, Mikami M, Taniguchi H, Miyamoto N, Adachi Y, Arimura Y, Itoh F, Imai K, et al. Genetic and epigenetic profiling in early colorectal tumors and prediction of invasive potential in pT1 (early invasive) colorectal cancers. Carcinogenesis. 2007;28:1364–1370. doi: 10.1093/carcin/bgl246. [DOI] [PubMed] [Google Scholar]
- 26.Hiyama T, Tanaka S, Yoshihara M, Sasao S, Kose K, Shima H, Tuncel H, Ueno Y, Ito M, Kitadai Y, et al. Chromosomal and microsatellite instability in sporadic gastric cancer. J Gastroenterol Hepatol. 2004;19:756–760. doi: 10.1111/j.1440-1746.2004.03369.x. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi T, Nosho K, Yamamoto H, Mikami M, Taniguchi H, Miyamoto N, Adachi Y, Itoh F, Imai K, Shinomura Y. Flat-type colorectal advanced adenomas (laterally spreading tumors) have different genetic and epigenetic alterations from protruded-type advanced adenomas. Mod Pathol. 2007;20:139–147. doi: 10.1038/modpathol.3800722. [DOI] [PubMed] [Google Scholar]
- 28.Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, et al. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn. 2006;8:209–217. doi: 10.2353/jmoldx.2006.050135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 30.Kawasaki T, Ohnishi M, Suemoto Y, Kirkner GJ, Liu Z, Yamamoto H, Loda M, Fuchs CS, Ogino S. WRN promoter methylation possibly connects mucinous differentiation, microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod Pathol. 2008;21:150–158. doi: 10.1038/modpathol.3800996. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto H, Imai K, Perucho M. Gastrointestinal cancer of the microsatellite mutator phenotype pathway. J Gastroenterol. 2002;37:153–163. doi: 10.1007/s005350200015. [DOI] [PubMed] [Google Scholar]
- 32.Imai K, Yamamoto H. Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics. Carcinogenesis. 2008;29:673–680. doi: 10.1093/carcin/bgm228. [DOI] [PubMed] [Google Scholar]
- 33.Ricciardiello L, Baglioni M, Giovannini C, Pariali M, Cenacchi G, Ripalti A, Landini MP, Sawa H, Nagashima K, Frisque RJ, et al. Induction of chromosomal instability in colonic cells by the human polyomavirus JC virus. Cancer Res. 2003;63:7256–7262. [PubMed] [Google Scholar]
- 34.Ottini L, Falchetti M, Lupi R, Rizzolo P, Agnese V, Colucci G, Bazan V, Russo A. Patterns of genomic instability in gastric cancer: clinical implications and perspectives. Ann Oncol. 2006;17 Suppl 7:vii97–vii102. doi: 10.1093/annonc/mdl960. [DOI] [PubMed] [Google Scholar]