

Abstract
Purpose
Colon cancer is a major cause of cancer-deaths. Dietary factors contribute substantially to the risk of this malignancy. Western style diets promote development of azoxymethane (AOM)-induced colon cancer. While we showed that epidermal growth factor receptors (EGFR) controlled AOM tumorigenesis in standard fat conditions, the role of EGFR in tumor promotion by high dietary fat has not been examined.
Experimental Design
A/JxC57BL6/J mice with wild type Egfr (Egfrwt) or loss-of-function waved-2 Egfr (Egfrwa2) received AOM followed by standard (std 5% fat) or Western style (20% fat) diet. As F1 mice were resistant to AOM, we treated mice with AOM followed by one cycle of inflammation-inducing dextran sulfate sodium (DSS) to induce tumorigenesis. Mice were sacrificed 12 wks after DSS. Tumors were graded for histology and assessed for EGFR ligands and proto-oncogenes by immunostaining, Western blotting and real time PCR.
Results
Egfrwt mice gained significantly more weight and had exaggerated insulin resistance compared to Egfrwa2 mice on high fat diet. Dietary fat promoted tumor (71.2% vs. 36.7%, p<0.05) and cancer incidence (43.9% vs. 16.7%, p<0.05) only in Egfrwt mice. The lipid-rich diet also significantly increased tumor and cancer multiplicity only in Egfrwt mice. In tumors, dietary fat and Egfrwt up-regulated TGF‐α, amphiregulin, CTNNB1, MYC, and CCND1, whereas PTGS2 was only increased in Egfrwt mice and further up-regulated by dietary fat. Notably, dietary fat increased TGF-α in normal colon.
Conclusions
EGFR is required for dietary fat-induced weight gain and tumor promotion. EGFR-dependent increases in receptor ligands and PTGS2 likely drive diet-related tumor promotion.
Keywords: epidermal growth factor receptor, CTNNB1, MYC, CCND1, experimental colon cancer
Introduction
Colon cancer is the second leading cause of cancer-related deaths in males and females in the United States (1). Germ line mutations, such as those occurring in familial adenomatous polyposis (FAP) syndrome and hereditary non-polyposis colon cancer (HNPCC), cause hereditary forms of colon cancer. Environmental factors especially dietary constituents, however, are believed to play major roles in sporadic forms of this malignancy (2). The 20-fold differences in world-wide colon cancer incidence rates, and rapidly changing incidence in immigrant populations support environmental exposure as a causal factor for colon cancer (3). Historically, for example, colon cancer rates were low in Japan, but within two generations the incidence of colon cancer among Japanese Americans approached rates for Caucasian Americans (4). Diets rich in animal fat and red meat, and relatively deficient in fiber and micronutrients, have been implicated in this increased risk in the industrialized Western world (2).
Experimental animal models have been widely used to study the role of dietary factors in colonic carcinogenesis. Azoxymethane (AOM) is a mutagen that methylates guanine bases resulting in activating mutations in K-ras and CTNNBI that encodes β-catenin. The AOM model mimics many features of sporadic human colon cancer, including promotion by dietary fat (5). Using this model, we showed that epidermal growth factor receptor (EGFR) plays an important role in colonic tumorigenesis (6, 7). To assess the role of EGFR in tumor promotion by dietary fat, we examined mice with wild type Egfr (Egfrwt) and waved-2 (Egfrwa2) since Egfr null mice are not viable (8). Egfrwa2 possesses a naturally occurring hypomorphic mutation in the kinase domain that abrogates 90% of kinase activity in vitro (9). This mutation has been shown to attenuate intestinal tumorigenesis in Apc mutant Min mice, a model of FAP syndrome (10). We compared AOM-induced tumorigenesis in Egfrwt and Egfrwa2 mice fed standard rodent chow (5% fat), or a Western style high fat diet (20% fat). A modification of this lipid-rich diet, which mimicked a Westernized diet high in animal fat and low in calcium and vitamin D, has been shown to induce spontaneous colonic tumors in mice during long-term feeding (11). As these mice were resistant to AOM alone, we modified the protocol to include AOM followed by dextran sulfate sodium (AOM/DSS). DSS is a non mutagenic agent that arrests crypt cell proliferation, leading to colonic crypt shortening and eventual mucosal ulcerations and inflammation (12). Many strains of mice resistant to AOM alone are susceptible to the pro-inflammatory and tumor-promoting effects of AOM/DSS (13). While EGFR contributes to AOM tumorigenesis, the role of this receptor in AOM/DSS tumor promotion by dietary fat has not been examined. There are other potential tumor-promoting factors modulated by dietary fat that might drive tumor promotion independent of EGFR signals. These include increases in colonic luminal secondary bile acids and circulating insulin-like growth factors (14-16).
To begin to elucidate potential EGFR effectors that might mediate tumor promotion by dietary fat, we examined several proto-oncogenes, including CTNNB1, MYC, CCND1 (cyclin D1) and PTGS2 (cyclooxygenase-2) that are known to play important roles in colonic tumorigenesis. MYC, CCND1 and PTGS2 are transcriptional targets of CTNNB1 (17-21). Recent studies have shown that dietary fat enhances expression of these proto-oncogenes in colonic carcinogenesis (22-24). Furthermore, EGFR regulates tyrosine phosphorylation and nuclear localization of CTNNB1 as well as MYC expression (25, 26). We have shown, moreover, that EGFR regulates CCND1 and PTGS2 levels in the AOM model under standard dietary fat conditions (6, 7). In the current study we demonstrate that CTNNB1, MYC and CCND1 up-regulations by dietary fat are amplified by EGFR signals. In contrast, diet-related increases in PTGS2 require EGFR signals. To identify potential up-stream effectors of EGFR induced by dietary fat, we also examined the influence of diet on TGF-α and amphiregulin, two EGFR ligands that are increased in colonic tumorigenesis (6, 7).
Materials and Methods
Materials
C57BL6/J Egfrwt/wa2 mice were interbred with A/J Egfrwt/wa2 mice to generate the F1 hybrid C57BL6/J × A/J experimental group. Formulated high fat diet was based on Western style diet that contained 20% fat as described (11). A standard fat diet was also formulated that contained 5% fat with the additional calories provided by cornstarch. Harlan Teklad laboratories (Madison, WI) prepared these diets, and also supplied AIN-76A rodent chow. The specific dietary components are provided in Table 1S in the Supplemental data section. Azoxymethane was obtained from Midwest Research (Kansas City, MO), the NCI Chemical Carcinogen Reference Standard Repository. Superfrost Plus slides were purchased from Fisher Scientific (Pittsburgh, PA). Polyclonal antibodies to CCND1 (SC-718) and monoclonal antibodies to MYC (SC-40, clone 9E10) and VEGF (SC-7269) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-CTNNB1 antibodies (#610153) were obtained from BD Pharmingen (Palo Alto, CA). Rabbit polyclonal anti-PTGS2 antibodies (#160106) were purchased from Cayman Chemicals (Ann Arbor, MI). Monoclonal β-actin antibodies were purchased from Sigma-Aldrich (St. Louis, MO). DNeasy kit (#69504) and RNeasy lipid extraction kit (#78404) and HotStarTaq™ DNA polymerase were obtained from Qiagen (Valencia, CA). FokI restriction enzyme was purchased from New England Biolabs (Beverly, MA). RNAlater™ RNA storage solution, and DNA-free™ DNase-I kit were purchased from Ambion (Austin, TX). TRIzol® RNA/DNA/Protein isolation reagent was obtained from Gibco BRL (Gaithersburg, MD). RiboGreen® reagent for RNA quantitation was purchased from Molecular Probes (Eugene, OR). Custom PCR primers were obtained from Integrated DNA Technologies, Inc. (Coralville, IA). Other PCR reagents, including Moloney murine leukemia virus reverse transcriptase, random hexamers, and SYBR Green were purchased from Applied Biosystems (Foster City, CA). SuperScript III Platinum Two-Step qRT-PCR kit was obtained from Invitrogen (Carlsbad, California). Electrophoretic grade acrylamide, bisacrylamide, Tris, SDS, prestained molecular weight markers and RC-DC protein assay were from Bio-Rad Laboratories (Richmond, CA). Kodak (Rochester, NY) supplied the X-OMAT AR film. PVDF membranes (Immobilon-P) were purchased from Millipore Inc. (Bedford, MA). Unless otherwise noted, all other reagents were of the highest quality available and were obtained from Sigma-Aldrich.
Methods
Egfr Genotyping
The Egfrwa2 point mutation is a T to G transversion (valine to glycine) that creates a recognition site for the restriction enzyme Fok I (GGATG). To genotype this locus, we PCR amplified genomic sequences and digested products with Fok that were separated on 2% agarose containing 100 bp DNA markers (Jackson Laboratories protocol). Primers are in intron 19 and exon 20 of mouse Egfr, respectively, and amplify 326 bp fragments for Egfrwt and Egfrwa2 (27). Fok I cuts the Egfrwa2 sequence (GGATG), but not the Egfrwt sequence (TGATG) to generate a doublet of 166 and 160 bp.
Experimental Animal Protocol
Mice were treated with AOM (7.5 mg i.p./kg body wt) or saline (AOM vehicle) weekly × 6 wks and maintained on AIN-76A diet. Two wks after the last AOM treatment animals were started on standard (Std) or high fat diets. The high fat diet is based on a diet formulation that approximates dietary amounts consumed in Western diets with increased animal fat and lower levels of vitamin D3 and calcium (11). The diet compositions are shown in Supplemental Data Table 1S. Chow was replaced weekly and remaining chow weighed to estimate food intake. Animals were weighed weekly.
As animals in the first cohort sacrificed did not develop tumors, we modified the protocol by giving AOM/DSS. Mice were switched to AIN-76A chow for 2 wks and then treated with AOM (7.5 mg/kg body weight) weekly × 2 wks. One wk after the 2nd AOM injection mice received 2.5% DSS in the drinking water for 5 days. Control animals received i.p. saline (AOM vehicle) and were provided tap water (DSS control) for drinking. Two wks after completing DSS or vehicle, animals were re-started on standard or high fat diets. Twelve wks after DSS administration, mice were anesthetized and colons excised. Perirenal and mesenteric fat were collected to estimate visceral fat stores. Colons were cleared of feces and opened longitudinally. Tumors were harvested and fixed in 10% buffered formalin and embedded in paraffin. A small portion of tumors was flash frozen in liquid nitrogen for RNA and proteins. Tumors were classified according to histological grade by an expert GI pathologist (JH) following consensus criteria (28). A 1 cm left colonic segment (distal margin 1 cm above the anus) that was cleared of any tumor was scraped and the mucosa flash frozen for protein or RNA. The remaining colons were fixed flat in 10% formalin for immunostaining, or in 70% ethanol to preserve proteins for Western blotting.
Blood glucose and serum insulin levels
Blood samples from non-fasted mice were obtained at the time of sacrifice and serum separated from clotted blood. Glucose levels were measured using an Abbott Laboratory blood glucose monitoring system. Insulin levels were measured by EIA using an insulin assay with a standard insulin curve from 0-6.9 ng/ml following the manufacturer's directions (Alpco, Salem NH).
Real-time PCR
Frozen colonic mucosa or tumors were thawed and RNA extracted using RNeasy Lipid Tissue Mini Kit. Samples were homogenized with a Polytron and loaded onto an RNA-binding spin column, washed, digested with DNase I and eluted in 30 μl of elution buffer. RNA samples were tested by Agilent chip for RNA purity and quantified by Ribogreen. RNA (250 ng) was reverse transcribed into cDNA using SuperScript III Platinum Two-Step qRT-PCR kit in 20 μL total volume. Incubation conditions were 25°C for 10 min, 42°C for 50 min, and 85°C for 5 min. Samples were then incubated with RNase H at 37°C for 20 min. The resulting first-strand complementary DNA (cDNA) was used as template for quantitative PCR in triplicate using SYBR Green QPCR Master Mix kit. Oligonucleotide PCR primer pairs were designed to cross intron-exon boundaries from published mouse sequences in the GenBank database using Primer3 (29). The TGF-α primers were: forward 5′-TGGGCACTTGTTGAAGTGAG-3′ and reverse 5′-TGCTAGCGCTGGGTATCC-3′. The amphiregulin primers were: forward 5′-GCTATT-GGCATCGGCATC-3′ and reverse: 5′-ACAGTCCCGTTTTCTTGTCG-3′. Reverse transcribed cDNA (1 μL of 1:8 dilution) and primers were mixed with SYBR Green dye I master mixture in 25 μl. Reactants were initially heated to 95°C for 5 min followed by 40 cycles: denaturation at 95°C for 10 sec, and then combined annealing and extension step at 60°C for 30 sec. The last cycle was followed by a 7 min extension at 72°C, and thermal denaturing profile to identify the Tm. PCR amplification was verified by melting curve and electrophoretic analysis of the PCR products on 3% agarose gel. Negative controls (no reverse transcriptase and no template) yielded no products. The data were analyzed using the comparative ΔΔCt method, and mRNA abundance normalized to β-actin mRNA and expressed as fold-control (30).
Immunohistochemistry
Five-micron sections of formalin-fixed paraffin-embedded colonic tissue (normal colons or tumors) were cut and mounted on Vectabond-coated Superfrost Plus slides. The slides were heated to 60°C for 1 hr, deparaffinized by three washes of 5 min each in xylene, hydrated in a graded series of ethanol washes and rinsed with distilled water. Epitope retrievals were achieved by microwave heating for 15 min in 0.01 M citrate buffer, pH 6 (CTNNB1), or in a steamer with Tris-EDTA buffer, pH 9 (CCND1). The antigen retrieval step was omitted for MYC staining. Frozen sections were used for PTGS2 staining and the peroxidase-blocking step was omitted. Following epitope retrieval, sections were washed three times for 2 min each in Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBST). The endogenous peroxidase activity was quenched by incubation for 15 min in methanol/H2O2 solution (0.5%) protected from light. Sections were washed three times in TBST for 2 min each and nonspecific binding saturated using Protein Block (Dako, Carpinteria, CA) for 20 min. The sections were incubated with primary antibody for 24 hrs at room temperature (1:150 dilution for CTNNB1; 1:25 dilution for MYC; 1:50 dilution for CCND1; 1:100 dilution for PTGS2). After three TBST washes, the slides were incubated at room temperature for 30 min with 1:200 dilution of biotinylated secondary antibodies. Antigen-antibody complexes were detected using an HRP labeled DAKO EnVision™+ System (DAKO LSAB™+ System), and 3,3′-diaminobenzidine as substrate. After washing with distilled water, the slides were counterstained with Gill's III hematoxylin, rinsed with water, dehydrated in ethanol and cleared with xylene. Tumors of comparable stage were used for immunostaining comparisons. For negative controls, primary antibodies were omitted or sections were incubated with isotype matched non-immune antibodies. Control sections showed no specific staining.
Western Blotting
Proteins were extracted in SDS-containing Laemmli buffer, quantified by RC-DC protein assay and subjected to Western blotting as described (31). Briefly, proteins were separated by SDS-PAGE on 4-20% resolving polyacrylamide gradient gels and electroblotted to PVDF membranes. Blots were incubated overnight at 4°C with specific primary antibodies followed by 1 hr incubation with appropriate peroxidase-coupled secondary antibodies that were detected by enhanced chemiluminescence using X-OMAT film. Xerograms were digitized using an Epson scanner (San Jose, CA) and band intensity quantified using UN-SCAN-IT gel 5.3 software (Silk Scientific, Orem UT). Protein expression levels in tumors were expressed as fold of control colonic mucosa (means ± SD), matched for diet and Egfr genotype. Separate aliquots were probed for β-actin to assess loading and expression levels normalized to β-actin levels. Protein lysates from tumors and colonic mucosa with equal protein abundance as assessed by RC-DC assays also showed comparable Western blotting β-actin levels. Tumors of comparable stage were used for Western blotting comparisons.
Statistical Methods
Continuous data (glucose, insulin, weight, and fat ratio) were summarized as mean ± SD, and compared between groups using Student's t-test. Analyses for all values summarized in Table 1 were log-transformed. Differences in Western blotting protein expression were compared by unpaired Student's t-test. Real time PCR samples were run in triplicate, and Ct values were averaged. Untransformed Ct values were compared between groups using saturated ANOVA models with genotype, diet, and tissue type (tumor or normal mucosa) effects and their interactions (30). Relative abundance, expressed as 2-ΔΔCt, was calculated by exponentiating the estimated differences in Ct between individual groups. Tumor incidence was defined as the proportion of mice with at least one tumor. Tumor multiplicity was defined as the average number of tumors in a given group. Nonparametric trend test was used to test for trends in tumor and cancer multiplicity across Egfrwt/wt, Egfrwt/wa2, and Egfrwa2/wa2 genotypes. Since in general Egfrwa2 behaves as a recessive allele (10), Egfrwt/wt and Egfrwt/wa2 genotypes were combined in subsequent analyses. Tumor incidence was compared between groups using logistic regression. Tumor multiplicity was compared between groups using negative binomial regression (32). Estimates and p-values reported in Table 2 are based on the corresponding saturated regression models with genotype, diet, and genotype × diet interaction. All statistical analyses were performed using SAS v. 9.1 or Stata v. 10, and p-values < 0.05 were considered statistically significant.
Table 1.
Effects of Egfr genotype and diet on glucose, insulin, body weight, and visceral fat/body weight
| Egfr genotype | Number | Diet | Glucose (mg/100 ml) | Insulin (ng/ml) | Body wt g | Visceral fat/body wt |
|---|---|---|---|---|---|---|
| Egfrwt/ | 14 | StdF1 | 121±11 | 2.3±0.8 | 35±3 | 0.05±0.03 |
| 13 | HF2 | 242±233 | 5.2±1.64 | 45±65 | 0.09±0.016 | |
| Egfrwa2 | 7 | StdF | 117±16 | 2.2±1.9 | 28±3 | 0.03±0.02 |
| 7 | HF | 117±5 | 3.9±1.77 | 30±3 | 0.06±0.018 |
Indicated parameters were measured at the time of sacrifice.
StdF, standard fat (5%);
HF, high fat (20%).
p<0.05 compared to Egfrwt on StdF diet.
p<0.05 compared to Egfrwt on StdF diet.
p<0.05 compared to Egfrwt on StdF diet.
p<0.05 compared to Egfrwt on StdF diet.
p<0.05 compared to Egfrwa2 on StdF diet.
p<0.05 compared to Egfrwa2 on StdF diet.
Table 2.
Effects of Egfr genotype and diet on tumor incidence and multiplicity
| Genotype | Diet | Mice total | Tumor Incidence | Tumor multiplicity | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 152 | Mice w/Tumor |
Tumor Incidence (%) |
Mice w/Cancer |
Cancer Incidence (%) |
Total Tumors |
Ave N tumors/mouse |
Total Cancers |
Ave N Cancers/mouse |
||
| Egfrwt/ | StdF1 | 60 | 22 | 36.7 | 10 | 16.7 | 53 | 0.9 | 18 | 0.3 |
| HF2 | 66 | 47 | 71.23 | 29 | 43.94 | 133 | 2.05 | 62 | 0.96 | |
| Egfrwa2 | StdF | 11 | 5 | 45.4 | 1 | 9.1 | 8 | 0.7 | 1 | 0.09 |
| HF | 15 | 7 | 46.7 | 2 | 13.3 | 9 | 0.6 | 2 | 0.13 | |
StdF, standard fat (5%),
HF, high fat (20%);
p<0.002 compared to StdF conditions (based on logistic regression models for incidence, and negative binomial regression models for multiplicity; each model contained genotype, diet and genotype × diet interaction).
p<0.002 compared to StdF conditions (based on logistic regression models for incidence, and negative binomial regression models for multiplicity; each model contained genotype, diet and genotype × diet interaction).
p<0.002 compared to StdF conditions (based on logistic regression models for incidence, and negative binomial regression models for multiplicity; each model contained genotype, diet and genotype × diet interaction).
p<0.002 compared to StdF conditions (based on logistic regression models for incidence, and negative binomial regression models for multiplicity; each model contained genotype, diet and genotype × diet interaction).
Results
Effects of EGFR signals and dietary fat on colonic tumorigenesis
We studied F1 progeny derived from interbreeding Egfrwt/wa2 C57BL6/J and Egfrwt/wa2 A/J mice for these experiments to provide an A/J background for increased AOM susceptibility and a C57BL6/J background for greater hybrid vigor since A/J Egfrwa2/wa2 mice tolerated AOM poorly. We controlled for hybrid genetic background by using F1 littermates for the experimental groups. Mice were treated with 6 weekly injections of AOM or saline and begun on experimental diets 2 wks after the last AOM injection. The high fat diet is based on a formulation that approximates dietary amounts consumed in Western diets with increased animal fat and lower levels of vitamin D3 and calcium (11). Growth rates were comparable in mice homozygous and heterozygous for Egfrwt. We, therefore, combined these groups for growth analyses. Compared to mice fed the standard fat diet, Egfrwt mice, but not in Egfrwa2 mice, gained significantly more weight on the high fat diet (Fig. 1). Chow consumption was increased but comparable in Egfrwt and Egfrwa2 mice on the high fat diet. F1 mice, however, were resistant to AOM as no aberrant crypt foci (ACF), microadenomas or tumors developed up to one year after carcinogen treatment in the first 50 mice sacrificed regardless of genotype or diet. Colons were prepared as Swiss rolls and multiple sections extensively examined. Presumably, this reflected the relative AOM resistance of the C57BL6/J parental strain.
Fig. 1. Egfrwa2 mutation prevents dietary fat-induced weight gain.

Following AOM treatment mice were started on standard (Std, 5% fat) or high fat diet (20% fat) and weighed weekly. Shown are monthly average weights for the indicated genotype and diet normalized to the first month weight. Within 4 months of diet initiation weights were stable. Saline-treated control groups, matched for genotype and diet, gained slightly more weight than AOM treated animals, but then closely paralleled AOM treated groups for the remainder of the study with no significant differences. *p<0.05 compared to age-matched Egfrwt animals on standard fat diet.
In order to enhance tumorigenesis, the remaining AOM-treated mice received a modified protocol involving AOM/DSS administration (13). The AOM/DSS treatment protocol is summarized in supplementary data Fig. 1S. Five staggered cohorts of mice initially treated with AOM were available for AOM/DSS treatment. We ensured that the one year interval between AOM treatment and AOM/DSS protocol was identical for each of the groups. Mice were switched to AIN-76A chow for 2 wks and then treated with AOM (7.5 mg/kg body weight) weekly × 2 wks to prevent confounding effects of AOM and experimental diets. One wk after the 2nd AOM injection mice received 2.5% DSS in the drinking water for 5 days. Control animals received i.p. saline (AOM vehicle) and were provided tap water (DSS control) for drinking. The AOM and DSS treatments were well tolerated with no unexpected deaths. DSS induced mild clinical colitis, as manifested by ∼5% weight loss and loose stools that were positive for occult blood. Two wks after completing DSS or vehicle, animals were re-started on standard or high fat diets to prevent confounding DSS inflammation with effects of experimental diets. Twelve wks after DSS administration, mice were sacrificed.
The high fat diet increased visceral fat in both genotypes, but weight gain was greater in the Egfrwt group. Serum insulin was increased in both Egfrwa2 and Egfrwt mice, but levels were higher in the Egfrwt group and blood glucose was only elevated in the Egfrwt group, suggesting greater insulin resistance in the latter group (Table 1). There were no tumors in the dietary control groups treated with saline and given only water (no DSS). We examined the effects of Egfr genotype on tumorigenesis. As summarized in Table 2S (supplemental data), tumor incidence was 0.57 in Egfrwt/wt group, 0.52 in the Egfrwt/wa2 group and 0.46 in the Egfrwa2/wa2 group (p=0.62, Fisher's exact test). Cancer incidences were 0.35, 0.27 and 0.12 (p=0.08, Fisher's exact test), respectively. Tumor multiplicities in these groups were 1.7, 1.3 and 0.7, and cancer multiplicities were 0.8, 0.5 and 0.1, respectively. These decreases in tumor and cancer multiplicities across genotypes Egfrwt/wt > Egfrwt/wa2 > Egfrwa2/wa2 were statistically significant by nonparametric trend test (p=0.05 and p=0.01 for tumor and cancer multiplicity, respectively). Since the Wa2 mutation functions as a recessive allele (10), we compared the effects of wild type Egfr [Egfrwt/ = Egfrwt/wt + Egfrwt/wa2] to Egfrwa2/wa2 on tumorigenesis. Cancer incidence was significantly higher in the combined Egfrwt/ group compared to the Egfrwa2/wa2 group (31% vs. 11.5%, p=0.05, Fisher's exact test). Tumor incidence was also higher in the Egfrwt/ group compared to the Egfrwa2/Wa2 group (55% vs. 46%), although the difference was not statistically significant (p=0.52). Tumor multiplicity (1.5 vs. 0.7) and cancer multiplicity (0.6 vs. 0.1) were also significantly higher in Egfrwt/ groups compared to the Egfrwa2/wa2 group (p=0.02 and p=0.01, respectively; negative binomial regression). Thus, homozygous Egfrwa2 mutations inhibited tumor progression to cancers, with significantly lower cancer incidence and cancer multiplicity compared to Egfrwt/ mice.
We next examined the interaction of Egfr genotype and diet as summarized in Table 2. A high fat diet significantly increased tumor incidence from 36.7% to 71.2% (p<0.001) and cancer incidence from 16.7% to 43.9% (p=0.002, logistic regression) in the Egfrwt group. As also shown in Table 2, high dietary fat significantly increased tumor multiplicity from 0.9 to 2.0 (p=0.001) and cancer multiplicity from 0.3 to 0.9 in the Egfrwt group (p=0.002). In contrast, tumor incidence and tumor multiplicity were comparable in Egfrwa2/wa2 mice fed standard vs. high fat diet (Table 2). Although the interaction between diet and genotype did not reach statistical significance in these regression models, (p=0.11 and p=0.15 for tumor incidence and multiplicity), models fitted separately within each genotype confirmed highly significant increases in tumor and cancer incidence and multiplicity induced by the high fat diet in the Egfrwt group (p<0.002 in all 4 models), but not in Egfrwa2/wa2 mice. Additionally, the relatively small sample size in the Egfrwa2/wa2 group potentially limited our ability to detect a diet × genotype interaction. Thus, these results suggest that dietary fat significantly increased tumor incidence and promoted tumor progression only in Egfrwt animals.
Effects of EGFR signals and dietary fat on proto-oncogene effector signals
To begin to uncover EGFR-dependent pathways that mediate effects of dietary fat on tumor promotion, we examined expression levels of several proto-oncogenes implicated in colonic carcinogenesis. As assessed by Western blotting, CTNNB1 was significantly up-regulated in tumors compared to controls in Egfrwt animals. Dietary fat further increased CTNNB1 expression levels in tumors (Fig. 2, upper panel). Significant increases in tumor CTNNB1 were also observed in Egfrwa2 mice on high dietary fat. Note that fold-increases in CTNNB1 in tumors were higher Egfrwa2 mice compared to Egfrwt mice since the normalizing control mucosal levels were lower in the Egfrwa2 mice. CTNNB1 levels, however, were higher in tumors from Egfrwt compared to Egfrwa2 mice. We immunostained tumors and found that CTNNB1 was expressed predominantly in colonocytes (Fig. 2, upper panel). In agreement with Western blotting results, CTNNB1 staining levels were higher in tumors from Egfrwt animals compared to Egfrwa2 animals on a standard fat diet. Dietary fat further increased tumor CTNNB1 staining levels in Egfrwt and Egfrwa2 animals.
Fig. 2. CTNNB1 and MYC expression levels in colonic tumors are controlled by Egfr genotype and diet.
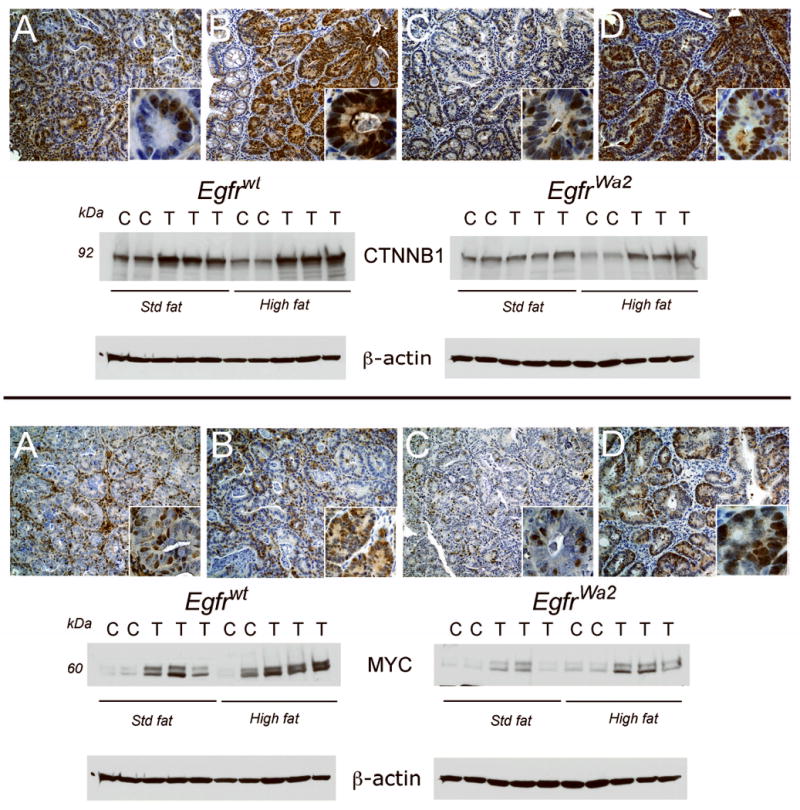
Colonic tumors were immunostained and Western blotted as described in “Materials and Methods”. Shown are representative tumors from each group. Upper panel: CTNNB1 IHC. A. Egfrwt, std fat; B. Egfrwt, high fat; C. Egfrwa2, std fat; D. Egfrwa2, high fat. Images are 20× and insets 100×. CTNNB1 Western blot. Proteins from colonic tumors (T) and control colons (N) from animals on standard fat (Std fat) or high fat diets were Western blotted for CTNNB1. Densitometry units were expressed as fold-control matched for Egfr genotype and diet. In Egfrwt animals, CTNNB1 levels were significantly higher in tumors compared to control under both Std fat (1.4±0.1-fold, p<0.05) and high fat conditions (1.6±-0.2 fold, p<0.05). In animals with Egfrwa2 on a high fat diet, CTNNB1 was 3.8±0.8-fold higher in tumors compared to control (p<0.05). Lower panel: MYC IHC. A. Egfrwt, std fat; B. Egfrwt, high fat; C. Egfrwa2, std fat; D. Egfrwa2, high fat. Images are 20× and insets 100×. MYC Western blot. In Egfrwt animals under std fat and high fat conditions, MYC in tumors was 8.7±1.9-fold (p<0.05) and 2.5±0.5 fold of control (p<0.05), respectively. In tumors from Egfrwa2 animals, MYC was significantly increased 3.8±0.8-fold control (p<0.05) in high fat conditions. Note that CTNNB1 and MYC levels were controlled by Egfr genotype and dietary fat. Under high fat conditions CTNNB1 and MYC were expressed predominantly in malignant epithelial cells with both cytoplasmic and nuclear distributions. Fold-increases in CTNNB1 and MYC were higher in tumors from Egfrwa2 mice on high fat compared to Egfrwt mice since the normalizing control mucosal levels were lower. Expression levels of these proto-oncogenes, however, were higher in tumors from Egfrwt mice compared to Egfrwa2 mice.
We next examined MYC expression. As in the case of CTNNB1, MYC tumor levels were higher in Egfrwt animals compared Egfrwa2 animals under standard fat conditions (Fig. 2, lower panel). High dietary fat increased MYC expression in tumors regardless of Egfr genotype and levels were greater in tumors from Egfrwt compared to Egfrwa2 animals. As assessed by immunostaining, MYC expression appeared to be relatively restricted to colonocytes in tumors from Egfrwa2 animals. This suggests that MYC expression might be more dependent on EGFR signals in stromal cells compared to epithelial cells. In Egfrwt animals, MYC was expressed in stromal cells and malignant colonocytes in low fat conditions, whereas under high fat conditions MYC was predominantly in colonocytes (Fig 2, lower panel compare inset A with inset B). Thus, dietary fat increased CTNNB1 and MYC in tumors regardless of Egfr genotype and the presence of Egfrwt enhanced these increases (Fig. 2). Furthermore, Egfr genotype modulated the effects of dietary fat on cell-specific MYC expression.
CCND1 was also increased in tumors compared to control mucosa in Egfrwt and Egfrwa2 mice, with the highest levels occurring in Egfrwt mice under high fat conditions (Fig. 3, upper panel). As assessed by immunostaining, CCND1 was predominantly nuclear and localized to epithelial cells in agreement with AOM studies (6, 7).
Fig. 3. CCND1 and PTGS2 expression levels in colonic tumors are controlled by Egfr genotype and diet.
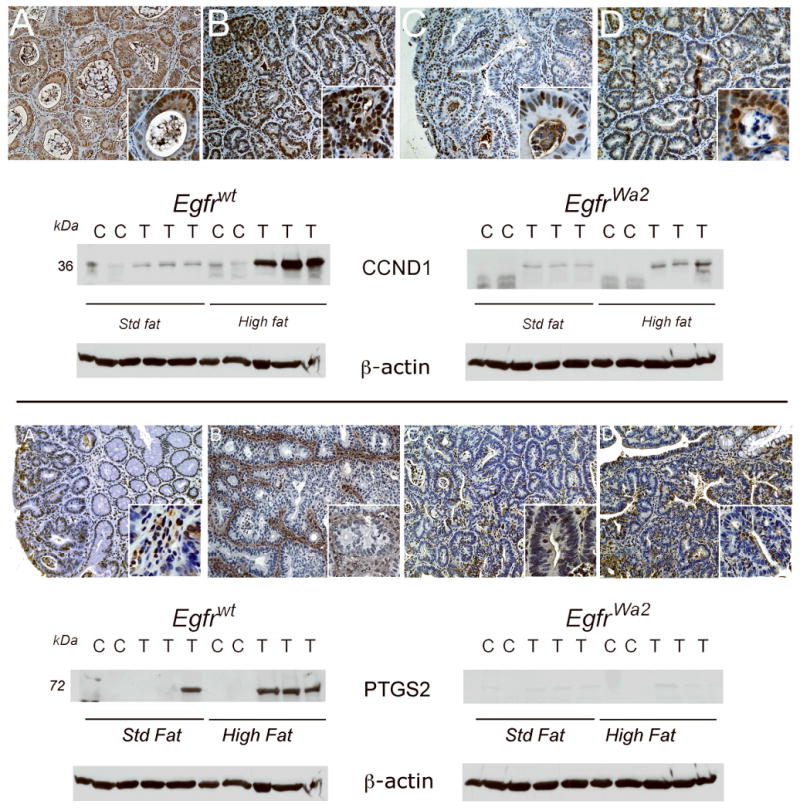
Colonic tumors were immunostained and Western blotted as described in “Materials and Methods”. Shown are representative tumors from each group. Upper panel: CCND1 IHC. A. Egfrwt, std fat; B. Egfrwt, high fat; C. Egfrwa2, std fat; D. Egfrwa2, high fat. Images are 20× and insets 100×. CCND1 Western blot. Proteins from colonic tumors (T) and control colons (N) from animals on standard fat (Std fat) or high fat diets were Western blotted for CCND1. Densitometry units were expressed as fold-control matched for Egfr genotype and diet. CCND1 levels were significantly higher in tumors compared to controls under high fat conditions in both Egfrwt animals (5.5±-1.4 fold control, p<0.05) and Egfrwa2 animals (10.0±0.5.4-fold control, p<0.05). Note that while fold-increase in tumor CCND1 was higher in Egfrwa2 mice on high fat compared to Egfrwt mice since the normalizing control mucosal levels was lower, CCND1 expression levels were much higher in tumors from Egfrwt mice. Lower panel: PTGS2 IHC. A. Egfrwt, std fat; B. Egfrwt, high fat; C. Egfrwa2, std fat; D. Egfrwa2, high fat. Images are 20× and insets 100×. PTGS2 was increased in Egfrwt animals on a high fat diet and was predominantly expressed in stromal cells (in lower panel, compare B to A). PTGS2 Western blot. In Egfrwt animals, PTGS2 levels were significantly higher in tumors compared to control under high fat conditions (21.8±-2.8 fold, p<0.005).
In contrast to CTNNB1, MYC and CCND1, PTGS2 was almost undetectable in tumors from Egfrwa2 mice fed standard or high dietary fat as assessed by Western blotting (Fig. 3, lower panel). PTGS2 up-regulation required Egfrwt and was strongly influenced by dietary fat. PTGS2 was increased in 7/8 tumors from Egfrwt animals on high fat diet, compared to only 1/7 tumors from Egfrwt animals on a standard fat diet (Table 3S in supplemental data, p<0.05). In agreement with Western blotting results, PTGS2 staining levels were greater in tumors from Egfrwt animals on high dietary fat compared to a standard fat diet (Fig. 3, lower panel, compare B to A). PTGS2 was expressed predominantly in tumor stromal cells, with lower levels in malignant colonocytes. Dietary fat also increased tumor VEGF and was higher in Egfrwt mice (data not shown).
Effects of EGFR signals and dietary fat on EGFR ligand expression
Up-regulated EGFR signals can be driven by gene amplification, activating mutations and increased ligand or receptor abundance. In colonic carcinogenesis increases in ligand abundance are very important. The effect of dietary fat on these ligands, however, has not been examined. As shown in Table 3, in normal mucosa there was a significant interaction between diet and genotype in regulating TGF-α expression (p=0.01): high fat diet significantly increased TGF-α expression in the EGFRwt/ mice (2-ΔΔCt =2.8, p=0.009), but not in the EGFRwt2/wt2 group (2-ΔΔCt=0.96, p=0.89). Diet had no significant effect on amphiregulin levels in normal mucosa regardless of genotype (data not shown). TGF-α and amphiregulin transcripts were significantly increased in tumors compared to normal colonic mucosa matched for Egfr genotype and diet. Increases ranged from 4.9 – 46 fold of normal mucosa (Table 3). In tumors, there was a significant genotype × diet interaction for TGF-α (p=0.045): high fat diet increased tumor TGF-α levels both in EGFRwt/ mice (2-ΔΔCt =10.0, p<0.0001) and EGFRwa2/wa2 mice (2-ΔΔCt =3.6, p=0.001), but the increase in the EGFRwt/ mice was much greater. Thus, there appears to be an important interaction between diet and Egfr genotype that regulates TGF-α abundance. In contrast, the increase in tumor amphiregulin due to high fat diet was smaller across genotypes (2-ΔΔCt =1.9, p=0.05) and there was not a diet × genotype interaction.
Table 3.
Effects of diet and genotype on EGFR ligands in normal mucosa and tumors
| Normal Mucosa TGF-α | Tumor TGF-α | Tumor vs. Normal TGF-α | Tumor vs. Normal AREG | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype | Diet | 2-ΔΔCt | p-value | 2-ΔΔCt | p-value | 2-ΔΔCt | p-value | 2-ΔΔCt | p-value |
| Egfr wt/ | StdF1 | 1 | - | 1 | - | 12.9 | <0.0001 | 8.8 | <0.0001 |
| HF2 | 2.8 | 0.009 | 10.0 | <0.0001 | 46.1 | <0.0001 | 29.9 | <0.0001 | |
| Egfrwa2 | StdF | 1 | - | 1 | - | 4.9 | <0.0001 | 11.8 | <0.0001 |
| HF | 0.9 | 0.9 | 3.6 | 0.001 | 18.0 | <0.0001 | 25.5 | <0.0001 | |
StdF, standard fat (5%),
HF, high fat (20%); Egfr transcript levels were measured by real-time PCR as described in “Materials and Methods”. Fold-changes (2-ΔΔCt) comparing TGF‐α expression between diets in each genotype in normal mucosa and tumors (N=6 in each group). The estimates were obtained by exponentiating the estimated differences in Ct levels based on separate 2-way ANOVA models within tissue type, diet, genotype and their interactions (30). Fold-changes (2-ΔΔCt) comparing TGF-α expression and amphiregulin (AREG) expression in tumor versus normal mucosa in each genotype and diet group were obtained by exponentiating the estimated differences in Ct levels based on a 3-way ANOVA model with diet, genotype, tissue type (tumor vs. normal), and their interactions (30). P-values were obtained post-hoc based on the saturated model.
Discussion
Diet is believed to play a key role in sporadic colonic tumorigenesis. Western style dietary fat has been shown to up-regulate several key proto-oncogenes in experimental colonic tumorigenesis including CTNNB1, CCND1 and PTGS2 that are regulated by multiple signaling pathways (23, 33). While growing lines of evidence from human and experimental animal studies support an etiologic role for EGFR in colonic carcinogenesis, dietary fat could potentially circumvent the need for this receptor. In the current report we demonstrate that this growth factor receptor is required for promotion of AOM/DSS-induced colonic tumors by a Western style diet. In Egfrwt mice a Western style high fat diet significantly increased weight gain and visceral fat as well as blood glucose and insulin levels. These metabolic derangements were accompanied by increased colonic tumor burden and tumor progression compared to a standard fat diet. In contrast, increased dietary fat did not enhance weight gain, or tumor promotion in Egfrwa2 mice. When data from the dietary groups were aggregated to assess the contribution of Egfr genotype to tumorigenesis, we found that cancer incidence and multiplicity were significantly higher in Egfrwt animals compared to Egfrwa2 mice. High dietary fat strongly promoted tumor development, increasing both tumor and cancer incidence in Egfrwt but not Egfrwa2 animals.
Luminal factors, including secondary bile acids have been implicated in diet-induced tumor promotion (14, 34). High fat diets increase colonic excretion of secondary bile acids that can activate EGFR in colorectal cancer cells (14, 35). In prior studies, we showed that dietary supplementation with cholic acid, the predominant primary bile acid, enhanced tumorigenesis in the AOM model (36). Systemic factors such as circulating insulin and insulin like growth factors are also increased by high fat diets and linked to an elevated risk of colon cancer (16). In this regard, blood sugars and serum insulin levels were higher in Egfrwt compared to Egfrwa2 mice on the high fat diet, indicating that EGFR contributes to hyperglycemia and insulin resistance in this model.
CTNNB1 is an integral part of the cytoskeleton as well as an important transcription factor in colonic tumorigenesis. CTNNB1 is up-regulated and activated in most colon cancers, and controls several key tumor-promoting genes including MYC, CCND1 and PTGS2 (17, 20, 37). Prior studies demonstrated that EGFR is an up-stream regulator of CTNNB1, inducing CTNNB1 deacetylation and nuclear localization in colon cancer cells (26). Other studies have demonstrated that Western style diets also increased CTNNB1 in premalignant colonic mucosa (23). In the current study we demonstrated that both dietary fat and EGFR controlled CTNNB1 expression in tumors. Thus, EGFR signals and dietary fat control CTNNB1 expression in premalignant and malignant colonocytes.
The proto-oncogene MYC is regulated by CTNNB1 and EGFR (17, 26). MYC was required for adenoma formation in the Apc mutant Min mouse (38). In prior studies we showed that MYC was increased in both AOM and AOM/DSS models of experimental colonic tumorigenesis (7, 39). In the current study dietary fat and EGFR controlled MYC expression. MYC levels were highest in tumors from Egfrwt animals on a high fat diet, the group with the greatest tumor burden. In Egfrwt mice, dietary fat appeared to differentially increase MYC in tumor epithelial cells compared to stromal cells. The cell context specificity of this diet-induced and EGFR-dependent effect will require further study.
The proto-oncogene CCND1 controls G1 to S cell cycle progression and is increased in human and experimental models of colon cancer (19, 31). We showed that CCND1 is controlled by EGFR under standard fat conditions in AOM colonic tumorigenesis (6, 7). In experimental colon cancer high dietary fat up-regulated colonic mucosal CCND1 (23, 40). Whether this increase required EGFR signals, however, has not been addressed. In the current study we demonstrated that EGFR and dietary fat controlled CCND1 expression in the AOM/DSS model. Dietary fat enhanced tumor CCND1 expression more in Egfrwt than in Egfrwa2 mice. In mutant mice, while dietary fat increased CTNNB1, MYC and CCND1 it failed to enhance tumorigenesis. In this regard, threshold levels for Apc (and presumably β-catenin) and CCND1 have been reported for adenoma formation in the Apc mutant Min mouse (41, 42). In addition to lower amplitudes of these proto-oncogenes, reduced tumorigenesis in Egfrwa2 mice might reflect insufficiency of other tumor-promoting signals, such as PTGS2.
The proto-oncogene PTGS2 is the rate-limiting enzyme for prostanoid biosynthesis. PTGS2 is up-regulated in human and experimental models of colon cancer (21, 31). Pharmacologic or genetic inhibition of PTGS2 inhibited experimental tumorigenesis, demonstrating its critical role in intestinal neoplasia (43, 44). Western style dietary fat has been shown to increase PTGS2 in AOM tumorigenesis (33). In prior AOM rat studies, we showed that activated Ras controlled PTGS2 expression (31). In the current study we showed that dietary fat strongly enhanced PTGS2 expression in tumors from Egfrwt but not Egfrwa2 mice. These results indicate that PTGS2 is tightly controlled by EGFR. PTGS2 was predominantly expressed in stromal cells in agreement with findings in Apc mutant Min mice and AOM/DSS-treated Egfrwt mice (45, 46). Prior studies have shown that activated K-Ras and CTNNB1 are both required to induce PTGS2 in colon cancer cells (20). EGFR is an up-stream activator of K-Ras, which is known to stabilize PTGS2 mRNA (47). Thus, our studies suggest that the pathway involving EGFR, K-Ras and PTGS2 plays a key role in tumor promotion by Western diet.
Since EGFR signals in colonic carcinogenesis are frequently driven by up-regulated ligands for this receptor, we measured TGF-α and amphiregulin transcript abundance. In prior studies we observed increases in these ligands in the AOM model (6, 7). Our finding that transcript levels of TGF-α and amphiregulin in tumors were controlled by both dietary fat and Egfr genotype explains in part tumor promotion by dietary fat. It is intriguing that increased dietary lipids up-regulated TGF-α expression even in normal colonic mucosa (without carcinogen induction). While the mechanisms by which dietary fat enhances EGFR ligand expression will require further study, it is known that insulin-like growth factors and secondary bile acids that are increased by dietary fat can enhance EGFR ligand release (48, 49). Thus, the diet-related risk of colon cancer could derive in part by a generalized “field effect” reflected by increases in EGFR ligands that expand mutant colonic crypt stem cells.
The potential tumor-promoting roles of secondary bile acids and metabolic derangements induced by Western style diets are also incompletely understood. High fat diets increase colonic secondary bile acids and also predispose to metabolic syndromes with increased insulin resistance and up-regulated IGF1 that can transactivate EGFR (15, 16). Egfrwt mice on a high fat diet had elevated blood glucose and increased serum insulin levels consistent with a metabolic syndrome. Further studies will be required to determine whether EGFR enhances tumor promotion by dietary fat via systemic effects on metabolism in addition to local receptor signals in the colon. Selective deletion of Egfr from colonocytes using floxed Egfr mice could be used to dissect colonocyte vs. systemic EGFR effects. Dietary interventions, moreover, with nutrient constituents that reduce EGFR and/or PTGS2 levels might provide novel chemopreventive strategies to inhibit the increased risk of colon cancer associated with obesity or diabetes.
Supplementary Material
Acknowledgments
These studies were funded in part by the following grants: P30DK42086 [Digestive Diseases Research Core Center], and CA036745 (M.B.), as well as the Samuel Freedman Research Laboratories for Gastrointestinal Cancer Research. The Department of Pathology Research Core Facilities of the University of Chicago provided additional funding.
The abbreviations used are
- AOM
azoxymethane
- ACF
aberrant crypt foci
- DSS
dextran sulfate sodium
- EGFR
epidermal growth factor receptor
- Egfrwt
wild type Egfr
- Egfrwa2
Waved-2 Egfr
- CTNNB1
β-catenin
- MYC
mammalian homologue of avian myelocytomatosis virus (c-Myc)
- CCND1
cyclin D1
- PTGS2
prostaglandin-endoperoxide synthase 2 (cyclooxygenase-2)
Footnotes
Statement of Translational Relevance: Colon cancer is a leading cause of cancer-related deaths in the United States. Western style diet is strongly linked to sporadic colon cancer. EGFR is also implicated in the genesis of colon cancer and development of effective receptor inhibitors suggest future strategies to prevent this disease. We previously demonstrated that EGFR regulates colonic tumorigenesis in the azoxymethane (AOM) model of colon cancer. In the current study we asked if EGFR controls tumor promotion by Western style diet. Dietary fat increases colonic secondary bile acids, enhances circulating IGF-1 levels and alters the enteric microbiome that might promote tumorigenesis by EGFR-independent mechanisms. We used a genetic approach with wild type and Egfr loss-of-function Waved-2 mutant mice to address this question. We studied a Western style diet that mimics the dietary fat composition of a large proportion of Americans. We demonstrated that EGFR was required for tumor promotion by dietary fat in the azoxymethane/dextran sulfate sodium model of colon cancer. EGFR was also required for this diet to up-regulate PTGS2. In addition, we demonstrated that dietary fat increased TGF-α transcripts in normal colonic mucosa, reflecting a “field effect” that might contribute to the increased risk of colon cancer in obesity. These findings have important implications for chemoprevention strategies that target EGFR and PTGS2. There are several naturally occurring dietary substances with such dual inhibitory activities, including curcumin, green tea and fish oil.
References
- 1.Rim SH, Seeff L, Ahmed F, King JB, Coughlin SS. Colorectal cancer incidence in the United States, 1999-2004 : an updated analysis of data from the National Program of Cancer Registries and the Surveillance, Epidemiology, and End Results Program. Cancer. 2009 doi: 10.1002/cncr.24216. [DOI] [PubMed] [Google Scholar]
- 2.Lipkin M, Reddy B, Newmark H, Lamprecht SA. Dietary factors in human colorectal cancer. Annu Rev Nutr. 1999;19:545–86. doi: 10.1146/annurev.nutr.19.1.545. [DOI] [PubMed] [Google Scholar]
- 3.Parkin DM, Muir CS, Whelan SL. Cancer incidence in five continents. Lyon: International agency for research on cancer scientific publications; 1992. [Google Scholar]
- 4.Haenszel W, Kurihara M. Studies of Japanese migrants. I. Mortality from cancer and other diseases among Japanese in the United States. J Natl Cancer Inst. 1968;40:43–68. [PubMed] [Google Scholar]
- 5.Rao CV, Hirose Y, Indranie C, Reddy BS. Modulation of experimental colon tumorigenesis by types and amounts of dietary fatty acids. Cancer Res. 2001;61:1927–33. [PubMed] [Google Scholar]
- 6.Fichera A, Little N, Jagadeeswaran S, et al. EGFR signaling is required for microadenoma formation in the mouse azoxymethane model of colonic carcinogenesis. Cancer Res. 2007;67:827–35. doi: 10.1158/0008-5472.CAN-05-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dougherty U, Sehdev A, Cerda S, et al. EGFR controls flat dysplastic ACF development and colon cancer progression in the rat azoxymethane model. Clin Cancer Res. 2008;14:2253–62. doi: 10.1158/1078-0432.CCR-07-4926. [DOI] [PubMed] [Google Scholar]
- 8.Threadgill DW, Dlugosz AA, Hansen LA, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–4. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 9.Fowler KJ, Walker F, Alexander W, et al. A mutation in the epidermal growth factor receptor in waved-2 mice has a profound effect on receptor biochemistry that results in impaired lactation. Proc Natl Acad Sci U S A. 1995;92:1465–9. doi: 10.1073/pnas.92.5.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts RB, Min L, Washington MK, et al. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2002;99:1521–6. doi: 10.1073/pnas.032678499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newmark HL, Yang K, Lipkin M, et al. A Western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis. 2001;22:1871–5. doi: 10.1093/carcin/22.11.1871. [DOI] [PubMed] [Google Scholar]
- 12.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–49. [PubMed] [Google Scholar]
- 13.Suzuki R, Kohno H, Sugie S, Nakagama H, Tanaka T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis. 2006;27:162–9. doi: 10.1093/carcin/bgi205. [DOI] [PubMed] [Google Scholar]
- 14.Narisawa T, Reddy BS, Weisburger JH. Effect of bile acids and dietary fat on large bowel carcinogenesis in animal models. Gastroenterol Jpn. 1978;13:206–12. doi: 10.1007/BF02773665. [DOI] [PubMed] [Google Scholar]
- 15.McGarr SE, Ridlon JM, Hylemon PB. Diet, anaerobic bacterial metabolism, and colon cancer: a review of the literature. Journal of clinical gastroenterology. 2005;39:98–109. [PubMed] [Google Scholar]
- 16.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–28. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 17.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 18.Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the ®-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96:5522–7. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartkova J, Lukas J, Strauss M, Bartek J. The PRAD-1/cyclin D1 oncogene product accumulates aberrantly in a subset of colorectal carcinomas. Int J Cancer. 1994;58:568–73. doi: 10.1002/ijc.2910580420. [DOI] [PubMed] [Google Scholar]
- 20.Araki Y, Okamura S, Hussain SP, et al. Regulation of cyclooxygenase-2 expression by the Wnt and ras pathways. Cancer Res. 2003;63:728–34. [PubMed] [Google Scholar]
- 21.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–8. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 22.Delage B, Bairras C, Buaud B, Pallet V, Cassand P. A high-fat diet generates alterations in nuclear receptor expression: prevention by vitamin A and links with cyclooxygenase-2 and beta-catenin. Int J Cancer. 2005;116:839–46. doi: 10.1002/ijc.21108. [DOI] [PubMed] [Google Scholar]
- 23.Fujise T, Iwakiri R, Kakimoto T, et al. Long-term feeding of various fat diets modulates azoxymethane-induced colon carcinogenesis through Wnt/beta-catenin signaling in rats. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1150–6. doi: 10.1152/ajpgi.00269.2006. [DOI] [PubMed] [Google Scholar]
- 24.Wang R, Dashwood WM, Lohr CV, et al. beta-catenin is strongly elevated in rat colonic epithelium following short-term intermittent treatment with 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and a high-fat diet. Cancer Sci. 2008;99:1754–9. doi: 10.1111/j.1349-7006.2008.00887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoschuetzky H, Aberle H, Kemler R. Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127:1375–80. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem. 2008;283:12686–90. doi: 10.1074/jbc.C700185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiter JL, Threadgill DW, Eley GD, et al. Comparative genomic sequence analysis and isolation of human and mouse alternative EGFR transcripts encoding truncated receptor isoforms. Genomics. 2001;71:1–20. doi: 10.1006/geno.2000.6341. [DOI] [PubMed] [Google Scholar]
- 28.Boivin GP, Washington K, Yang K, et al. Pathology of mouse models of intestinal cancer: Consensus report and recommendations. Gastroenterology. 2003;124:762–77. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 29.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols. Totowa, NJ: Humana Press; 2000. pp. 365–86. [DOI] [PubMed] [Google Scholar]
- 30.Yuan JS, Reed A, Chen F, Stewart CN., Jr Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85. doi: 10.1186/1471-2105-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bissonnette M, Khare S, von Lintig FC, et al. Mutational and nonmutational activation of p21ras in rat colonic azoxymethane-induced tumors: effects on mitogen-activated protein kinase, cyclooxygenase-2, and cyclin D1. Cancer Res. 2000;60:4602–9. [PubMed] [Google Scholar]
- 32.Drinkwater NR, Klotz JH. Statistical methods for the analysis of tumor multiplicity data. Cancer Res. 1981;41:113–9. [PubMed] [Google Scholar]
- 33.Singh J, Hamid R, Reddy BS. Dietary fat and colon cancer: modulation of cyclooxygenase-2 by types and amount of dietary fat during the postinitiation stage of colon carcinogenesis. Cancer Res. 1997;57:3465–70. [PubMed] [Google Scholar]
- 34.Reddy BS, Mangat S, Sheinfil A, Weisburger JH, Wynder EL. Effect of type and amount of dietary fat and 1,2-dimethylhydrazine on biliary bile acids, fecal bile acids, and neutral sterols in rats. Cancer Res. 1977;37:2132–7. [PubMed] [Google Scholar]
- 35.Im E, Martinez JD. Ursodeoxycholic acid (UDCA) can inhibit deoxycholic acid (DCA)-induced apoptosis via modulation of EGFR/Raf-1/ERK signaling in human colon cancer cells. J Nutr. 2004;134:483–6. doi: 10.1093/jn/134.2.483. [DOI] [PubMed] [Google Scholar]
- 36.Earnest DL, Holubec H, Wali RK, et al. Chemoprevention of azoxymethane-induced colonic carcinogenesis by supplemental dietary ursodeoxycholic acid. Cancer Res. 1994;54:5071–4. [PubMed] [Google Scholar]
- 37.Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 38.Sansom OJ, Meniel VS, Muncan V, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007 doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 39.Fichera A, Little N, Dougherty U, et al. A Vitamin D Analogue Inhibits Colonic Carcinogenesis in the AOM/DSS Model. J Surg Res. 2007;142:239–45. doi: 10.1016/j.jss.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 40.Yang K, Kurihara N, Fan K, et al. Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res. 2008;68:7803–10. doi: 10.1158/0008-5472.CAN-08-1209. [DOI] [PubMed] [Google Scholar]
- 41.Li Q, Ishikawa TO, Oshima M, Taketo MM. The threshold level of adenomatous polyposis coli protein for mouse intestinal tumorigenesis. Cancer Res. 2005;65:8622–7. doi: 10.1158/0008-5472.CAN-05-2145. [DOI] [PubMed] [Google Scholar]
- 42.Hulit J, Wang C, Li Z, et al. Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Mol Cell Biol. 2004;24:7598–611. doi: 10.1128/MCB.24.17.7598-7611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oshima M, Dinchuk JE, Kargman SL, et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–9. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 44.Reddy BS, Hirose Y, Lubet R, et al. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res. 2000;60:293–7. [PubMed] [Google Scholar]
- 45.Fukata M, Chen A, Klepper A, et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology. 2006;131:862–77. doi: 10.1053/j.gastro.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sonoshita M, Takaku K, Oshima M, Sugihara K, Taketo MM. Cyclooxygenase-2 expression in fibroblasts and endothelial cells of intestinal polyps. Cancer Res. 2002;62:6846–9. [PubMed] [Google Scholar]
- 47.Sheng H, Shao J, Dubois RN. K-Ras-mediated increase in cyclooxygenase 2 mRNA stability involves activation of the protein kinase B1. Cancer Res. 2001;61:2670–5. [PubMed] [Google Scholar]
- 48.El-Shewy HM, Kelly FL, Barki-Harrington L, Luttrell LM. Ectodomain shedding-dependent transactivation of epidermal growth factor receptors in response to insulin-like growth factor type I. Mol Endocrinol. 2004;18:2727–39. doi: 10.1210/me.2004-0174. [DOI] [PubMed] [Google Scholar]
- 49.Merchant NB, Rogers CM, Trivedi B, Morrow J, Coffey RJ. Ligand-dependent activation of the epidermal growth factor receptor by secondary bile acids in polarizing colon cancer cells. Surgery. 2005;138:415–21. doi: 10.1016/j.surg.2005.06.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.