

Abstract
In prokaryotes, mono-ADP-ribose transfer enzymes represent a family of exotoxins that display activity in a variety of bacterial pathogens responsible for causing disease in plants and animals, including those affecting mankind, such as diphtheria, cholera, and whooping cough. We report here that NarE, a putative ADP-ribosylating toxin previously identified from Neisseria meningitidis, which shares structural homologies with Escherichia coli heat labile enterotoxin and toxin from Vibrio cholerae, possesses an iron-sulfur center. The recombinant protein was expressed in E. coli, and when purified at high concentration, NarE is a distinctive golden brown in color. Evidence from UV-visible spectrophotometry and EPR spectroscopy revealed characteristics consistent of an iron-binding protein. The presence of iron was determined by colorimetric method and by an atomic absorption spectrophotometer. To identify the amino acids involved in binding iron, a combination of site-directed mutagenesis and UV-visible and enzymatic assays were performed. All four cysteine residues were individually replaced by serine. Substitution of Cys67 and Cys128 into serine caused a drastic reduction in the E420/E280 ratio, suggesting that these two residues are essential for the formation of a stable coordination. This modification led to a consistent loss in ADP-ribosyltransferase activity, while decrease in NAD-glycohydrolase activity was less dramatic in these mutants, indicating that the correct assembly of the iron-binding site is essential for transferase but not hydrolase activity. This is the first observation suggesting that a member of the ADP-ribosyltransferase family contains an Fe-S cluster implicated in catalysis. This observation may unravel novel functions exerted by this class of enzymes.
INTRODUCTION
Mono-ADP-ribosylation is a covalent and enzyme-catalyzed reaction in which β-nicotinamide adenine dinucleotide (NAD+), beyond its role in energy metabolism, acts as substrate. Mono-ADP-ribosyltransferases split the N-glycosidic bond of NAD+ to yield ADP-ribose moiety, which is selectively linked to specific amino acids in their protein targets, and nicotinamide, which is simultaneously released into the medium (1). Among the several toxins used by pathogenic bacteria to target and kill their host cells, proteins that exert ADP-ribosylation activity represent a large family of dangerous and potentially lethal toxins that interfere with cellular, metabolic, and regulatory pathways (2). Many bacteria (e.g. Corinebacterium diphteriae, Vibrio cholerae, Escherichia coli, Bordetella pertussis, Salmonella enterica, Clostridium botulinum, Staphylococcus aureus, and Pseudomonas aeruginosa) that pose significant human health threats (e.g. diphtheria, cholera, whooping cough, and severe diaorrhea) utilize ADP-ribosylating toxins to block protein synthesis, to alter signal transduction, or to modify cytoskeletal functions. All the effects that these enzymes produce are mediated by ADP-ribosylation of key target proteins such as elongation factor-2 (3, 4), GTP- binding proteins (5), or actin (6). A computer-based analysis allowed us to identify NarE (Neisseria ADP-ribosylating enzyme), a previously unidentified ADP- ribosyltransferase, in strain MC58 of the Gram-negative aerobic-anaerobic facultative Neisseria meningitidis (7). The gene-encoding NarE is found in virulent strains (8) and when present is always located at the same genome locus between nmb1342 and nmb1344 (9), which code for the dihydrolipoyl acetyltransferase (E2) and the dihydrolipoyl dehydrogenase (E3), respectively. These two enzymes, together with E1, form the pyruvate dehydrogenase multienzymatic complex (PDC)2 conserved in several prokaryotic and eukaryotic organisms. NarE shares structural features with ADP-ribosylating toxins from V. cholerae and E. coli, and, like cholera toxin (10) and the related heat-labile enterotoxins (11), retains the ability to ADP-ribosylate arginine and small guanidine compounds like agmatine and to hydrolase NAD in ADP-ribose and nicotinamide in the absence of an ADP-ribose acceptor (12). Although the mechanism of host cells disruption varies considerably among toxins, toxicity is strictly dependent upon the presence of ADP-ribosyltransferase activity. Mutants of either cholera toxin or heat-labile enterotoxins lacking enzymatic activity lose toxicity, although they retain strong mucosal adjuvanticity (12, 13, 14). The importance of a solid and robust catalytic activity in the onset of the diseases caused by ADP-ribosylating toxins prompted us to elucidate the structural and catalytic characteristics of NarE. Since NarE contains a Cys-X-Cys motif, which confers iron sensitivity, an attractive hypothesis was that NarE was capable of binding iron. Here, we used a combination of biophysical methods (UV spectra and EPR) to show that NarE contains an iron-sulfur (Fe-S) center with structural features resembling those present in the rubredoxin protein family (15). Fe-S clusters are typically bound to proteins via four cysteines. Therefore, to identify the iron ligands in NarE and investigate the biochemical effects of their substitutions, we generated a series of mutants in which the four cysteines, iron putative ligands (Cys11, Cys67, Cys128, and Cys130), were individually replaced by serine. Effects of serine substitution on Fe-S cluster stability were assessed by comparing spectroscopic properties and catalytic activities of wild-type protein with molecular variants. The results reported here indicate that Cys67 and Cys128 are important for maintaining a coordinated iron center. This center is crucial for ADP-ribosyltransferase but not for NAD-glycohydrolase (NADase) activity.
EXPERIMENTAL PROCEDURES
Materials
[adenine-U-14C]NAD (274 mCi/mmol) and [carbonyl-14C]NAD (53 mCi/mmol) were purchased from Amersham Biosciences; Dowex AG1-X2, the Bradford reagent for protein quantification, and the immunoblotting detection system were purchased from Bio-Rad, whereas bovine serum albumin standard and bacterial protein extraction reagent were from Pierce. MHAB N45 filter plates were purchased from Millipore, and isopropyl-1-thio-β-d-thiogalactopyranoside was purchased from Calbiochem. SimplyBlue SafeStain was from Invitrogen, and glove boxes were purchased by VWR, whereas all other reagents used in this study were from Sigma Aldrich.
Overexpression and Purification of Recombinant NarE Protein
An overnight culture of E. coli BL21(DE3) transformed with plasmid pET21b+ carrying the narE gene in terrific broth (100 mg ml−1 ampicillin) was used to inoculate a new culture of the same medium. The narE gene was cloned in pET21b+ as a fusion construct as described previously (12), with a carboxyl-terminal His6 tag separated from the last amino acid of the protein by a linker of two amino acid residues. The cultures were grown at 37 °C with gentle shaking (180 rpm), and expression of the narE gene was induced to an A600 of 0.5–0.6 by the addition of 1 mm isopropyl-1-thio-β-d-thiogalactopyranoside. Synthesis of recombinant protein was allowed to proceed for 3 h at 25 °C. Following induction, bacteria were harvested by centrifugation for 15 min at 8,000 × g at 4 °C, and the pellet was stored at −80 °C for subsequent analysis. All purification steps were carried out either under anaerobic conditions inside a portable glove box saturated with a mixture of N2 and H2 or under aerobic conditions. Cells were thawed and resuspended in a bacterial protein extraction reagent using a ratio of 10 ml for 3 g of pellet in the presence of 0.1 mm MgCl2, 100 units of DNase I, and 1 mg/ml of chicken egg lysozyme. The mixture, supplemented with 1 mm phenylmethylsulfonyl fluoride, was stirred for 40 min at room temperature. Debris and membranes were pelleted by centrifugation at 16,000 × g for 30 min and then discarded. The supernatant was loaded onto an immobilized metal affinity chromatography column using a nickel-nitriloacetic acid matrix (Ni-NTA; Pharmacia; 1.5 ml matrix/liter of culture volume). The column was equilibrated with buffer A (50 mm sodium phosphate buffer, pH 7.4, and 300 mm NaCl) containing 10 mm imidazole. The column was initially washed with 10 bed volumes of buffer A containing 10 mm imidazole and 20 bed volumes of buffer A containing 50 mm imidazole. Bound proteins were eluted with buffer A containing 250 mm imidazole. Fractions containing NarE were identified using SDS-PAGE and were pooled and stored at 4 °C for subsequent analysis.
Electrophoresis and Immunoblot Analysis
Protein samples were resolved by SDS-PAGE using the NuPAGE gel system (Invitrogen) with MES as running buffer. Gels were stained with Coomassie Blue. Separated proteins were electrophoretically transferred onto nitrocellulose using the iBlot apparatus (Invitrogen). Immunoblotting was carried out with primary polyclonal α-NarE antiserum generated in mouse against recombinant NarE (1:10,000 dilution). A secondary rabbit α-mouse horseradish peroxidase-conjugated antibody (Jackson ImmunoResearch Laboratories) was added at a dilution of 1:10,000. Bound antibodies were visualized using an ECL immunoblotting detection system (Bio-Rad) according to the manufacturer's instructions. Molecular masses were estimated from calibration standards included on each gel.
ADP-ribosyltransferase Assays
ADP-ribosyltransferase activity was measured using standard assay (16) in a final volume of 0.3 ml, containing 50 mm potassium phosphate, pH 7.5, 20 mm agmatine as ADP-ribose acceptor and 0.1 mm [adenine-U-14C]NAD (0.05 μCi). After incubation at 30 °C, duplicate samples (100 μl) were applied to 1 ml columns of Dowex AG1-X2. [adenine-14C]ADP-ribosylagmatine was eluted for liquid scintillation counting with 5 ml of H2O. Alternatively, ADP-ribosyltransferase activity was assayed using a filter plate-based assay (17). Assays were performed as above in 0.3 ml total volume with 0.6 mg of poly-arginine replacing agmatine as an ADP-ribose acceptor. After incubation at 30 °C, samples were precipitated with 0.3 ml of 50% (w/v) trichloroacetic acid, left for 30 min on ice, and applied in quadruplicate (100 μl) under vacuum conditions to each well of a mixed cellulose esters filter plate. The unreacted [adenine-U-14C]NAD was washed out with 10 volumes of 5% (w/v) trichloroacetic acid, and the incorporated radioactivity was measured in a Packard Top counter.
NAD-glycohydrolase Assay
An analog assay based on nicotinamide release was employed to measure the NADase activity of NarE (18). This assay was carried out in 50 mm potassium phosphate, pH 7.5, 0.1 mm [carbonyl-14C]NAD (0.05 μCi) replacing [adenine-U-14C]NAD in a total volume of 0.3 ml. Samples (100 μl) after incubation at 30 °C were applied to a 1-ml column of Dowex AG1-X2 and 14C-nicotinamide eluted with 5 ml of H2O for liquid scintillation counting.
Iron Content Determination
Concentration of wild-type NarE iron content was determined colorimetrically using ferrozine under reducing conditions as described (19). To completely release the complexed iron, samples were incubated 2 h at 60 °C in 2.25% KMnO4 and 0.6 n HCl. After a second reducing step at room temperature for 30 min, absorbances of samples were measured at 562 nm in a Cary 50 UV-visible spectrophotometer. The iron content was also measured by an atomic absorption spectrophotometer with a Varian 820-MS ICP mass spectrometer.
Site-directed Mutagenesis
To mutate amino acid residues of NarE, a PCR-based method was used. Oligonucleotides indicated below carrying a single nucleotide mismatch in the underlined positions were used to generate the mutants: C11S, AGAGGCATTAGTTCCCACAAGATAG; C67S, TCTATATGACGGATCTTATATATCTACA; C128S, AGCTGAAGATTCTGGCTGGCTGTATTCCTGAAG; and C130S, AGCTGAAGATTGTGGCTCTATTCCTGG. After mutagenesis and DNA sequencing, all of the generated mutants were cloned into pET21b+, expressed, and purified following the procedure described previously.
UV-visible and EPR Measurements
Ultraviolet-visible spectroscopy of purified recombinant NarE was carried out at room temperature using a Lambda 2 Ultraspec spectrophotometer. Continuous wave X-band (9.4 GHz) EPR spectra were obtained using a spectrometer Bruker E500 Elexsys Series using the Bruker ER4122 SHQE cavity and an Oxford helium continuous flow cryostat (ESR900) system. EPR signals of the Fe-S center at X-band consist of three parts, one for each of the Kramers doublets. If the magnetic field is not too large compared with the energy separation between the Kramers doublets, each of these three parts may be described by a set of three effective g values. The lowest Kramers doublet gives rise to a signal starting at approximately geff = 9. The second Kramers doublet consists of a complex pattern centered near geff = 4.3. The third signal arising from the highest Kramers doublet is similar to that arising from the lowest and is not usually observed independently of it (20–24). All these contributions are rationalized using an isotropic S = 5/2 spin Hamiltonian of the form,
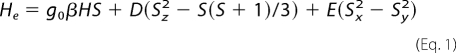 |
where D and E are the axial and rhombic zero-field splitting parameters, respectively. Samples of NarE suitable for EPR spectroscopy were prepared inside the glove box and frozen immediately in liquid nitrogen after elution.
Protein Assay
Protein content was determined by the Bradford protein assay kit (Bio-Rad) according to manufacturer's instructions using bovine serum albumin for standardization.
RESULTS
Production and Purification of Recombinant NarE
The His6-tagged recombinant NarE was overexpressed in E. coli BL21(DE3) using a T7 RNA polymerase-driven system. The protein used in this study was purified to apparent homogeneity by a single chromatographic step on an immobilized metal ion chromatography (Ni-NTA) column. Fig. 1A shows an SDS-PAGE analysis of the purified NarE protein, which gave an essentially pure band running slightly above the predicted molecular mass for NarE of 17 kDa. MALDI-TOF mass spectrometry gave a more precise mass of 17,269 Da (Fig. 1B). Both results were consistent with the theoretical mass for His-tagged NarE (17,269 Da). Western immunoblotting (Fig. 1A, lane 3) and peptide mass analysis (data not shown) were also performed to confirm the identity of the purified protein.
FIGURE 1.

Expression, purification, and NarE identification. SDS-PAGE and Western immunoblot analysis of NarE purification (A). Soluble fraction of cell-free extract prepared from E. coli BL21(DE3) carrying pET21b+ after induction with isopropyl-1-thio-β-d-thiogalactopyranoside (lane 1) and Ni-NTA eluate (5 μg protein) (lane 2). Proteins were resolved on a 4–12% NuPAGE gel and stained with Coomassie Blue. The purified protein (1 μg) was further identified by Western immunoblot (lane 3) as described under “Experimental Procedures.” Molecular mass markers (M) are indicated. MALDI-TOF mass spectrometry of purified NarE (B). The estimated mass of 17,262 Da is consistent with the theoretical mass of His-tagged NarE (17,269 Da). Data presented are from one representative experiment. a.i. arbitrary intensity.
NarE Contains an Iron-Sulfur Center
The high solubility of the protein allowed a highly concentrated sample of NarE to be used. Interestingly, when concentrations of ≥7 mg/ml were reached, the protein solution appeared to be golden brown in color, whereas the protein solution at lower concentrations was colorless. A UV absorption spectrum of the protein revealed an increase in absorption at 340 nm and ∼420 nm, in addition to the normal peak at 280 nm caused by side chains of aromatic amino acid residues (Fig. 2A) and with an E420/E280 ratio of 2.3 × 10−2. To maximize purification of the active enzyme, the protein was purified under anaerobic conditions, as described under “Experimental Procedures,” and analyzed soon after purification or anaerobically sealed in vials and kept at 4 °C for subsequent analysis. Absorption peaks in the 340–420 nm range can result from a variety of common biological chromophores ranging from carotenes to phorphyrins, but these features are also typical for many proteins containing Fe-S clusters. The iron content of NarE preparation was determined by colorimetric assay and by an atomic absorption spectrophotometer. The results of the two assays are very similar and converge to a ratio <1 iron atom per monomer that is lower than it would be expected for an intact rubredoxin-like cluster. Due to the high level production of recombinant protein only incomplete iron-sulfur cluster formation was achieved leading to a substechiometric ratio. Furthermore, we have not performed all the procedures in strictly anaerobic conditions. Another observation that further supports the presence of iron came from the strong inhibition of transferase activity by the addition of high specific iron chelator O-phenanthroline to the assay mixture (Fig. 2B). In contrast, NADase activity measured in the presence of the iron chelator was less dramatically affected (Fig. 2C).
FIGURE 2.

UV-visible absorption spectrum of NarE and dose-response curve of ADP-ribosyltransferase and NAD-glycohydrolase activities in the presence of iron-chelating agent. The UV-visible spectrum of anaerobically purified NarE (6.5 mg/ml) was recorded in 50 mm sodium phosphate buffer, pH 7.4, 300 mm NaCl, and 250 mm imidazole (A). Shown are data representative of one experiment of three performed with different protein preparations. B, the ADP-ribosyltransferase activity of anaerobically purified NarE was assayed with a recently developed filter-based assay (17). Affinity-purified NarE was incubated for 18 h at 30 °C with 0.6 mg of poly-arginine as acceptor molecule and 0.1 mm [adenine-U-14C]NAD (0.05 μCi) in a final volume of 0.3 ml with (gray bars) or without (white bars) 10 mm of O-phenanthroline and increasing concentration of NarE, as specified in the B. After trichloroacetic acid precipitation, samples (100 μl) were applied under vacuum condition in each well of a mixed cellulose ester filter plate and washed with 10 volumes of 5% trichloroacetic acid. The incorporated radioactivity was measured in a Packard Top counter. C, to test NADase activity, the same enzyme following a similar method was used. Enzyme activity was assayed at 30 °C for 18 h with 0.1 mm [carbonyl-14C]NAD in a final volume of 0.3 ml in 50 mm potassium phosphate buffer, pH 7.5, with (gray bars) or without (white bars) 10 mm O-phenanthroline and increasing concentrations of purified NarE as specified in C. At the end of incubation, 100-μl samples were applied to AG1-X2 columns and 14C-nicotinamide eluted by water and counted. The values presented represent the means ± S.D. of three independent assays. Each activity measurement was run in duplicate and analyzed with four replicates.
Spectroscopic Characterization of the Iron-Sulfur Cluster in NarE
EPR spectroscopy is a well established technique to confirm the presence of iron-sulfur centers in proteins and assign them to specific cluster type. To this purpose, low temperature EPR spectroscopy was performed. The use of UV-visible spectrophotometry analysis paired with EPR spectroscopy assures that no structural change occurs at the iron site, lowering the temperature. EPR spectra were recorded using a fresh sample of the anaerobically purified NarE that had been frozen in glass needles in liquid N2 soon after purification. The native state of the protein shows a complex resonance with g values equal to 4.4, 4.3, and 4.2, as reported in Fig. 3a. This is indicative of a high spin ferric iron, as it occurs in similar cases such as rubredoxin, where the ferric iron is bound to four sulfur atoms in a rhombic site (20). EPR spectra of oxidized rubredoxin display a number of features in the g = 4.0–4.8 range arising from the ±3/2 Kramers doublet of a S = 5/2 ground spin multiplet and a g ∼ 9.5 signal from the lowest doublet of this multiplet. In our case, the resonances centered around g = 4.3 are clearly evident, but the contribution at g ∼ 9 is too low to be detected in the native state. The NarE protein was also analyzed in the presence of 5 mm K3Fe(CN)6 and 10 mm FeCl3. FeCl3 strongly increases the intensity of the pattern centered around g = 4.3 and a signal at g = 9.3 becomes evident, as shown in Fig. 3b. In particular with FeCl3, a better outlined series of signals, but very low in intensity, comes out at g = 9.3, 7.3, and 5.7 in addition to the pattern also visible in the native state at g = 4.4, 4.3, and 4.2. This result shows that the agent acts at the metal site of the protein. In contrast, the addition of 5 mm K3Fe(CN)6 does not induce any changes at the iron site of the protein (Fig. 3c), keeping the intensity of the signal at g = 4.3 unchanged. A new broad peak appears due to the presence of the low spin contribution of the Fe(III)-inorganic salt (25), which remains free in solution, as shown in Fig. 3c.
FIGURE 3.

EPR spectra of the affinity-purified NarE. EPR spectrum of 0.1 mm NarE in 50 mm potassium phosphate buffer, pH 7.4, 250 mm imidazole, and 300 mm NaCl (a). NarE spectra were also recorded in the same buffer in the presence of 10 mm FeCl3 (b) and 5 mm K3Fe(CN)6 (c). Similar results were observed for three independent experiments. mT, millitesla.
Identification of Iron-Sulfur Ligands and Role of Cluster in Catalysis
A variety of amino acids, including histidine, aspartate, and arginine (26), have the ability to coordinate iron. However, cysteine residues, by far the most common ligands, usually hold a critical role in iron coordination. Distribution of cysteine ligands in Fe-S proteins is very heterogeneous, and sometimes this family lacks a unique consensus pattern. NarE contains four cysteines (Cys11, Cys67, Cys128, and Cys130) not involved in disulfide bridges (7) and a Cys-X-Cys iron-sensing motif at C-terminal that is present in well characterized Fe-S proteins like AFT1 (27) and the IscA/SufA family reviewed in Ref. 28. Therefore, to probe the role of the four cysteines in cluster assembly, we constructed a panel of site-directed mutant forms of the protein in which each cysteine was individually changed to serine. The mutated enzymes were produced in E. coli BL21(DE3) under conditions identical to the wild-type enzyme and purified as described under “Experimental Procedures” (Fig. 4A). All the mutant forms were highly soluble, expressed at levels comparable to wild-type NarE, and recognized by α-NarE antibodies (Fig. 4B). C67S and C128S mutants were colorless upon purification, even at concentration >7 mg/ml, which may reflect reduced iron content. The presence of iron in these forms is reinforced by EPR spectroscopy that showed a weak but clear paramagnetic signal (data not shown). Moreover, UV-visible spectroscopy analyses of C67S and C128S mutants provided a spectrum in which absorption signals at 320 and 420 nm were highly reduced with respect to the wild-type protein (Fig. 5A), suggesting an altered cluster assembly. In contrast, spectroscopic analyses of C11S and C130S mutants showed spectra overlapping with WT NarE (Fig. 5B). To assess the functional relevance of the Fe-S cluster in NarE, transferase activity of the four mutant proteins was tested. Quantification of enzyme activity as a function of protein concentration revealed a 20-fold reduction in the ADP-ribosyltransferase specific activity of C67S and C128S proteins compared with WT (Fig. 6A). By contrast, C11S and C130S mutants maintained transferase activity at a comparable level to wild-type protein (Fig. 6A). We have also measured the E420/E280 ratio in the four generated mutants and compared with the WT protein. While this value was drastically reduced (0.72 and 0.99 × 10−2) in the mutants lacking transferase activity it remained at a level (2.8 and 2.2 × 10−2) comparable with WT protein (2.3 × 10−2) in the mutants with unaltered activity. Like most of the ADP-ribosyltransferases, NarE, in addition to its ability to transfer ADP-ribose to small guanidine compounds, possesses the ability to hydrolyze NAD in the absence of a specific substrate. Therefore, we compared NADase activity in WT and NarE mutants to determine whether mutations that alter cluster organization completely inactivate the enzyme or affect only its ADP-ribosyltransferase activity. Although C67S and C128S had not detectable ADP-ribosyltransferase activity, they still possessed NADase activity, which was reduced by ∼45 and 39%, respectively (Fig. 6B). The presence of O-phenanthroline, an iron-chelator, inhibits ∼40 and 45% the transferase activity of C11S and C130S (Fig. 7), thus reinforcing the hypothesis that the substitution at Cys11 and Cys130 with Ser allows the formation of a coordinate and stable Fe-S cluster. Overall, these results are in line with a previous experiment (Fig. 6) supporting the view that the transferase activity is blocked or highly reduced without a coordinated cluster, but the ability to hydrolyze NAD is only weakly affected. Coupled with spectroscopic results, these observations strongly suggest that Cys67 and Cys128 are involved in ligating iron in a correct coordination, which is crucial for NarE transferase but play a less important role for NADase activity.
FIGURE 4.

Purification profile of Cys → Ser mutants. A, the four single Cys → Ser mutants in which Cys11 (lane 1), Cys67 (lane 2), Cys128 (lane 3), and Cys130 (lane 4) were changed to serine and overexpressed in E. coli BL21(DE3). Purification was carried out after induction of NarE expression with isopropyl-1-thio-β-d-thiogalactopyranoside by metal affinity chromatography and elution using 250 mm imidazole as specified under “Experimental Procedures.” Equal amounts (1 μg) of the Ni-NTA purified mutants were visualized by SDS-PAGE and Coomassie Blue staining. Positions of molecular weight standards (M) are indicated. B, shown is an immunoblot of 0.1 μg of purified protein using α-NarE polyclonal antibodies (1:10,000 dilution). Lane positions are as above. Data presented are from one representative experiment.
FIGURE 5.

Spectroscopic characterization of wild-type and mutated NarE. Wild-type NarE and mutants were purified by metal affinity chromatography and eluted with 50 mm sodium phosphate buffer, pH 7.4, and 300 mm NaCl containing 250 mm imidazole. Panel A represents the spectra of wild-type protein (6.5 mg/ml) and the C67S (7.7 mg/ml) and C128S (7.8 mg/ml) mutants. Panel B represents wild-type protein with C11S (7 mg/ml) and C130S (7.2 mg/ml) mutants. Spectra were recorded under aerobic conditions early after purification.
FIGURE 6.

Synthesis of ADP-ribosylagmatine and nicotinamide release by NarE and site-directed mutants. Affinity-purified proteins were incubated for 18 h at 30 °C in 50 mm potassium phosphate buffer with 0.1 mm [adenine-U-14C]NAD (0.05 μCi) and 20 mm agmatine as ADP-ribose acceptor (A) or with 0.1 mm [carbonyl-14C]NAD (0.05 μCi) (B) in a final volume of 0.3 ml. ADP-ribosylagmatine or 14C-nicotinamide were purified by AG1-X2 and quantified by scintillation counting. Data are expressed as ADP-ribosylagmatine formed or nicotinamide released per mg of mutant proteins. Data presented are the means ± S.D. of three independent assays, with each activity measurement performed in duplicate.
FIGURE 7.

Effect of O-phenanthroline on the ADP-ribosyltransferase activity of NarE mutants. NarE mutants were tested for their ability to transfer ADP-ribose moiety and release nicotinamide with (white bars) and without (black bars) 10 mm O-phenanthroline. ADP-ribosyltransferase activity was measured for 18 h at 30 °C under the same conditions of previous experiments and as described under “Experimental Procedures.” Data are expressed as ADP-ribosylagmatine formed per mg of purified protein. Error bars represent the S.D. of three independent experiments.
DISCUSSION
Our study shows that the ADP-ribosyltransferase of N. meningitidis, NarE, heterologously expressed in E. coli and purified as a soluble His-tagged protein contains an iron-sulfur cluster. This is the first report describing an iron center in a member of the ADP-ribosyltransferase family. Analyses of NarE obtained by UV-visible and EPR spectrometry are consistent with the presence of a single Fe(S→Cys)4 structure similar to that present in rubredoxin, the simplest family of iron-sulfur proteins. Furthermore, using site-directed mutagenesis, cysteine residues have been shown to hold a critical role in iron binding and in transferase activity. Single substitution of the four Cys in turn to Ser led to different spectroscopic and biochemical properties. In rubredoxin, Cys → Ser mutations allow formation of FeSO3 center (21) in which Ser can substitute the sulfydryl group of cysteine ligand. We argue that the catalytically active C11S and C130S NarE mutants, which show similarity with wild-type protein in spectroscopic behavior (Fig. 5B), may carry a stable Fe(S→Cys)3(O-Ser) structure with Ser replacing Cys as a ligand without altering the cluster. In contrast, in catalytically inactive mutants, the drastic loss of signal intensity suggested the formation of unstable Fe(S→Cys)3 (OH) in which Ser67 and Ser128 side chains are not liganded. The strong influence of Cys67 and Cys128 substitution in transferase activity indicates that these two residues are essential for iron coordination, and therefore, Ser cannot structurally replace them. In contrast, these two mutants exhibited only modest reduction of NADase activity. So far, we do not have a clear answer for explaining the differences between the two activities. Remarkably, the presence of transferase activity is strictly associated with the pathogenetic effects, whereas the biological significance of NADase activity is still unknown for this class of enzymes. The individual properties of the four cysteines may reflect surface (Cys11 and Cys130) or internal (Cys67 and Cys128) positions, but structural data are needed to address this question. Proteins with ferric iron bound to four sulfur atoms in a rhombic site, like rubredoxin are usually of low molecular weight and water soluble and are primarily associated with redox chemistry (28, 29). Beyond their traditional role as cofactors participating in electron transfer in catalytic mechanisms, numbers and functions of iron-binding proteins have lately grown exponentially. Multiple functions include binding and activation of substrate, dimer formation, iron storage, regulation of gene expression, disulfide reduction, and sulfur donor (28). Clusters are invariably integrated into proteins via a multistep process involving several proteins (30) and can modulate the structure and stability of a protein. When involved in protein structure function, clusters provide some level of stability, enhancing the protein solubility. In our case the Cys → Ser mutants maintained a high level of solubility. In small polypeptides, the presence of Fe-S structure can influence the oligomerization state. Analysis of the crystal structure of E. coli SufA (31) has revealed that the conserved Cys-Gly-Cys sequence is positioned in close proximity to the dimer interface, thereby forcing the protein to assume a dimeric conformation. The arrangement of these cysteines together with Glu118 in SufA dimers allows iron coordination (31). In NarE, a biglutamic motif (Glu133-Glu134) comes within close proximity of the Cys-X-Cys motif, which may contribute to iron coordination and dimer formation. PDC is a multimeric complex with several copies of the different components, and NarE is inserted in PDC and co-transcribed with the E3 component of lipoamide dehydrogenase (12). Modulation of the PDC complex related to a loss in transferase activity could also be explained in terms of redox function of the iron center. E3 contains a structural FAD prosthetic group that undergoes oxidation/reduction and can accept and give e−, which in turn may control the redox state of the cluster modulating ADP-ribosyltranferase activity. An intriguing possibility is that NarE through cluster may sense the metabolic state of the cell and influence it by ADP-ribosylating E3, which possesses several arginines potential amino acid targets. Interestingly, in humans, pyruvate dehydrogenase kinase and phosphatase interacting with PDC are responsible for regulation of its activity by a reversible phosphorylation/dephosphorylation mechanism. Like phosphorylation, ADP-ribosylation is a covalent and reversible post-translational modification used by cells to modulate protein functions. It has been recently shown that a reduced metabolic response leads to an increased bactericidal drug resistance (32). NarE could be part of a more complex mechanism that, due to its influence on metabolic responses might increase Neisseria drug resistance. Another common role for clusters is their requirement for enzymatic activity, as reported for DNA repair enzymes like MutY (33) and XPD (34). We have shown that changing Cys67 and Cys128 to serine leads to a drastic reduction in transferase (Fig. 6A) but not NADase (Fig. 6B) activity, thereby supporting the hypothesis that correct iron coordination is required for transferase activity. Although crystal structure confirmation is needed, Cys128 of the Cys-X-Cys iron-sensing motif is located at the C terminus in close proximity to the biglutamic motif Glu118-Lys119-Glu120 essential for enzymatic activity that we have identified by site-directed mutagenesis.3 The novel observation presented in this report, that a member of the ADP-ribosyltransferase family contains a Fe-S center, suggests that NarE may exert a different role compared with the other ADP-ribosylating enzymes usually involved in the onset of infectious diseases, thus opening new horizons of investigation. Our findings imply that this structure may well function in some form of regulatory role. Furthermore, iron is necessary for survival of Neisseria into the host and is implicated in the regulation of the expression of a number of Neisseria genes involved not only in metabolism but also in virulence (35). Therefore, the ability of NarE to bind iron through its cluster could have a key role in Neisseria pathogenesis and virulence.
Acknowledgments
We thank Giorgio Corsi for precious artwork; Elisabetta Soldaini for helpful discussion, valuable advice, and assistance; and Marta Castagnini and Maria Scarselli for technical support. We also acknowledge Prof. Rod Merrill from the University of Guelph (Canada) for allowing us to perform some important experiments in the laboratory. We further acknowledge Antonietta Maiorino for assistance in editing and Nathalie Norais for technical support in mass spectrometry analysis.
This work was supported by Novartis Vaccines & Diagnostics.
F. Di Marcello, D. Veggi, M. Del Vecchio, M. Scarselli, R. Rappuoli, M. Pizza, and E. Balducci, unpublished data.
- PDC
- pyruvate dehydrogenase complex
- MES
- 4-morpholineethanesulfonic acid
- WT
- wild-type
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight
- Ni-NTA
- nickel-nitriloacetic acid matrix.
REFERENCES
- 1.Ueda K., Hayaishi O. (1985) Annu. Rev. Biochem. 54, 73–100 [DOI] [PubMed] [Google Scholar]
- 2.Moss J., Vaughan M. (1988) Adv. Enzymol. Relat. Areas Mol. Biol. 61, 303–379 [DOI] [PubMed] [Google Scholar]
- 3.Honjo T., Nishizuka Y., Hayaishi O. (1968) J. Biol. Chem. 243, 3553–3555 [PubMed] [Google Scholar]
- 4.Iglewski B. H., Kabat D. (1975) Proc. Natl. Acad. Sci. U.S.A. 72, 2284–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Locht C., Keith J. M. (1986) Science 232, 1258–1264 [DOI] [PubMed] [Google Scholar]
- 6.Aktories K., Wegner A. (1989) J. Cell Biol. 109, 1385–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masignani V., Balducci E., Serruto D., Veggi D., Aricò B., Comanducci M., Pizza M., Rappuoli R. (2004) Int. J. Med. Microbiol. 293, 471–478 [DOI] [PubMed] [Google Scholar]
- 8.Pizza M., Scarlato V., Masignani V., Giuliani M. M., Aricò B., Comanducci M., Jennings G. T., Baldi L., Bartolini E., Capecchi B., Galeotti C. L., Luzzi E., Manetti R., Marchetti E., Mora M., Nuti S., Ratti G., Santini L., Savino S., Scarselli M., Storni E., Zuo P., Broeker M., Hundt E., Knapp B., Blair E., Mason T., Tettelin H., Hood D. W., Jeffries A. C., Saunders N. J., Granoff D. M., Venter J. C., Moxon E. R., Grandi G., Rappuoli R. (2000) Science 287, 1816–1820 [DOI] [PubMed] [Google Scholar]
- 9.Ala' Aldeen D. A., Westphal A. H., De Kok A., Weston V., Atta M. S., Baldwin T. J., Bartley J., Borriello S. P. (1996) J. Med. Microbiol. 45, 419–432 [DOI] [PubMed] [Google Scholar]
- 10.Moss J., Vaughan M. (1977) J. Biol. Chem. 252, 2455–2457 [PubMed] [Google Scholar]
- 11.Gill D. M., Woolkalis M. (1985) CIBA Found. Symp. 112, 57–73 [DOI] [PubMed] [Google Scholar]
- 12.Masignani V., Balducci E., Di Marcello F., Savino S., Serruto D., Veggi D., Bambini S., Scarselli M., Aricò B., Comanducci M., Adu-Bobie J., Giuliani M. M., Rappuoli R., Pizza M. (2003) Mol. Microbiol. 50, 1055–1067 [DOI] [PubMed] [Google Scholar]
- 13.de Haan L., Verweij W. R., Feil I. K., Lijnema T. H., Hol W. G., Agsteribbe E., Wilschut J. (1996) Infect. Immun. 64, 5413–5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pizza M., Fontana M. R., Giuliani M. M., Domenighini M., Magagnoli C., Giannelli V., Nucci D., Hol W., Manetti R., Rappuoli R. (1994) J. Exp. Med. 180, 2147–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C. J., Lin Y. H., Huang Y. C., Liu M. Y. (2006) Biochem. Biophys. Res. Commun. 349, 79–90 [DOI] [PubMed] [Google Scholar]
- 16.Moss J., Stanley S. J. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 4809–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balducci E. (2005) Anal. Biochem. 344, 278–280 [DOI] [PubMed] [Google Scholar]
- 18.Moss J., Manganiello V. C., Vaughan M. (1976) Proc. Natl. Acad. Sci. U.S.A. 73, 4424–4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fish W. W. (1988) Methods Enzymol. 158, 357–364 [DOI] [PubMed] [Google Scholar]
- 20.Peisach J., Blumberg W. E., Lode E. T., Coon M. J. (1971) J. Biol. Chem. 246, 5877–5881 [PubMed] [Google Scholar]
- 21.Meyer J., Gaillard J., Lutz M. (1995) Biochem. Biophys. Res. Commun. 212, 827–833 [DOI] [PubMed] [Google Scholar]
- 22.Lode E. T., Coon M. J. (1971) J. Biol. Chem. 246, 791–802 [PubMed] [Google Scholar]
- 23.Bachmayer H., Piette L. H., Yasunobu K. T., Whiteley H. R. (1967) Proc. Natl. Acad. Sci. U.S.A. 57, 122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoo S. J., Meyer J., Achim C., Peterson J., Hendrich M. P., Münck E. (2000) J. Biol. Inorg. Chem. 5, 475–487 [DOI] [PubMed] [Google Scholar]
- 25.Duelund L., Toftlund H. (2000) Spectrochim. Acta. A Mol. Biomol. Spectrosc. 56A, 331–340 [DOI] [PubMed] [Google Scholar]
- 26.Berkovitch F., Nicolet Y., Wan J. T., Jarrett J. T., Drennan C. L. (2004) Science 303, 76–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rutherford J. C., Jaron S., Ray E., Brown P. O., Winge D. R. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14322–14327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson D. C., Dean D. R., Smith A. D., Johnson M. K. (2005) Annu. Rev. Biochem. 74, 247–281 [DOI] [PubMed] [Google Scholar]
- 29.Beinert H., Holm R. H., Münck E. (1997) Science 277, 653–659 [DOI] [PubMed] [Google Scholar]
- 30.Antoine R., Locht C. (1994) J. Biol. Chem. 269, 6450–6457 [PubMed] [Google Scholar]
- 31.Wada K., Hasegawa Y., Gong Z., Minami Y., Fukuyama K., Takahashi Y. (2005) FEBS Lett. 579, 6543–6548 [DOI] [PubMed] [Google Scholar]
- 32.Kohanski M. A., Dwyer D. J., Hayete B., Lawrence C. A., Collins J. J. (2007) Cell 130, 797–810 [DOI] [PubMed] [Google Scholar]
- 33.Porello S. L., Cannon M. J., David S. S. (1998) Biochemistry 37, 6465–6475 [DOI] [PubMed] [Google Scholar]
- 34.Rudolf J., Makrantoni V., Ingledew W. J., Stark M. J., White M. F. (2006) Mol. Cell 23, 801–808 [DOI] [PubMed] [Google Scholar]
- 35.Perkins-Balding D., Ratliff-Griffin M., Stojiljkovic I. (2004) Microbiol. Mol. Biol. Rev. 68, 154–171 [DOI] [PMC free article] [PubMed] [Google Scholar]