

Abstract
Mammalian voltage-dependent potassium (Kv) channels regulate the excitability of nerve and muscle cells. Kv12.2 features the longest S5-P loop among all known mammalian Kv channels with the most N-linked glycosylation sites (three sites). Despite its unique structural features, Kv12.2 is not well characterized. Because glycosylation plays important roles in the folding, trafficking, and function of various Kv channels, we focused on the N-glycosylation of Kv12.2. We show that Kv12.2 is N-glycosylated in Chinese hamster ovary (CHO) cells and in cultured neurons as well as in the mouse brain. As an effect of N-glycosylation on the function of Kv12.2, we demonstrate that removal of sugar chains causes a depolarizing shift in the steady-state activation without a significant reduction in current amplitude. Unlike the previously reported shift for Shaker-type Kv channels, this shift does not appear to be due to negatively charged sialic acid residues in the sugar chains. We next examined the trafficking in CHO cells to address whether the unglycosylated Kv12.2 channels are utilized in vivo. Although double mutants, retaining only one glycosylation site, are trafficked to the surface of CHO cells irrespective of the position of the glycosylated site, unglycosylated channels are not trafficked to the cell surface. Furthermore, we could not detect unglycosylated channels in the mouse brain. Our data suggest that only glycosylated Kv12.2 channels show proper voltage dependence and are utilized in vivo.
introduction
Mammalian voltage-dependent potassium (Kv)3 channels are multisubunit membrane proteins that regulate the excitability of nerve and muscle cells (1). The channels are composed of four α-subunits, with each α-subunit containing six membrane-spanning segments (S1-S6) (Fig. 1A). The S1-S4 helices function as the voltage sensor, whereas the S5-S6 helices together with a pore helix between them tetramerize to form a central ion-conducting pore. Mammalian Kv channels constitute a large superfamily and are categorized into groups termed Kv1.x ∼ Kv12.x according to their primary sequences (2, 3). Kv12.2, also known as ether-a-go-go-like 2 (Elk2) or KCNH3, belongs to the ether-a-go-go (EAG) family, which comprises the ether-a-go-go (Eag, Kv10.x), ether-a-go-go-related gene (Erg, Kv11.x), and ether-a-go-go like (Elk, Kv12.x) subfamilies (4, 5). In mouse, rat, and human, Kv12.2 mRNA is specifically expressed in the brain (6–8). In the human brain expression levels of Kv12.2 mRNA are high in the cerebral cortex, hippocampus, amygdala, caudate, and nucleus accumbens (9). In these parts of the brain the level of Kv12.2 mRNA was found to be as high as 15∼30% that of ribosomal protein RS9, a housekeeping gene (9). Unlike Kv5.x, Kv6.x, Kv8.x, and Kv9.x, which function as modifiers for other Kv channels (10), Kv12.2 can produce a functional channel on its own when heterologously expressed (6–8, 11). In addition, human Kv12.2 may be implicated in epilepsy (12). These findings suggest that Kv12.2 is likely to play important roles in the mammalian brain. Among all known mammalian Kv channels, Kv12.2 has the longest extracellular S5-P loop located between S5 and the pore helix (Fig. 1A, right panel) and the largest number of (i.e. three) N-glycosylation consensus sequences in the S5-P loop (Fig. 1B). Despite these findings and unique structural features, Kv12.2 has not yet been well characterized at the protein level (10).
FIGURE 1.

Topological model and amino acid sequence of S5-P loops of various voltage-dependent potassium channels. A, the proposed topology of the Kv1.x and Kv12.2 monomers is shown. Kv channels have six transmembrane segments (S1-S6). Helix S4 contains positively charged residues, indicated by +. The predicted N-linked glycosylation sites are indicated by Ψ. Kv1.x channels have an N-terminal tetramerization domain (T1), and single N-glycosylation site between helix S1 and S2 with the exception of Kv1.6. Kv12.2 channels have an N-terminal Per-Arnt-Sim (PAS) domain and a C-terminal cyclic nucleotide biding domain (CNBD). Kv12.2 has three N-glycosylation sites in a particularly long extracellular S5-P loop between helix S5 and the pore helix (PH). B, shown are amino acid sequences of the predicted S5-P loops of the mouse Kv, fruit fly (Drosophila melanogaster) Elk, and zebrafish (Danio rerio) Elk1 channels. The names in parentheses refer to the subfamilies containing the corresponding mouse Kv channels. Elk, Eag, and Erg are subfamilies of the EAG family. White letters in black boxes indicate the predicted N-linked glycosylation consensus sequence Asn-Xaa-(Ser/Thr) in which Xaa can be any amino acid except proline. The numbers in parentheses above the sequences indicate the positions of the residues in Kv12.2.
Many Kv channels undergo N-linked glycosylation, which begins with the co-translational addition of a core glycan to a lumenal-exposed asparagine residue that is part of a consensus sequence. As the proteins progress through the Golgi apparatus, enzymes modify the sugar chains, resulting in high mannose, hybrid, or complex oligosaccharides (13, 14). N-Glycosylation generally promotes proper folding, increases trafficking and stability, and modifies the function of Kv channels (15–20), but the positions and roles of N-glycosylation vary among Kv channels. With the exception of Kv1.6, Shaker type Kv1.x channels have a single glycosylation consensus sequence in a loop located between helices S1 and S2 (Fig. 1A, left panel), and in some Kv1.x channels removal of the N-linked glycans results in a positive shift of the activation curve (15, 19). It has been suggested that this effect is due to negatively charged sialic acid residues in the sugar chains. In contrast, despite the fact that EAG-type Kv channels have one or more N-glycosylation sites adjacent to the pore region (Fig. 1B), only a few studies have addressed the role of N-glycosylation in the functional regulation of these channels (18). The analysis of Kv12.2 provides insights into the role of N-glycans in the function of channels of the EAG family.
Regulation of trafficking to the cell surface, where Kv channels are generally expressed, is another important role of the N-glycosylation of these channels (21, 22). The effect of glycosylation on trafficking varies between Kv channels. Glycosylation of Kv1.1 does not affect the trafficking of the channel (16, 23). It has been suggested that both glycosylated and unglycosylated Kv1.1 could be utilized in vivo and that differences in the degree of glycosylation increase the functional diversity of the channels, possibly modifying cell excitability (24). In contrast, the human Erg (HERG or Kv11.1) channel requires glycosylation for proper cell surface expression (20, 25). Mutations that prevent glycosylation of the HERG protein cause a cardiac disorder known as long QT syndrome type 2 (20). These data suggest isoform-specific differences in the sensitivity of potassium channel trafficking to N-linked glycosylation.
Here we present the evidence that Kv12.2 channels are expressed and N-glycosylated in the mouse brain and that N-glycosylation is essential for proper function of EGFP-Kv12.2 expressed in Chinese hamster ovary (CHO) cells. Furthermore, by a systematic mutational analysis of the three glycosylation sites of Kv12.2, our study provides insight into how glycosylation regulates the trafficking of Kv channels.
EXPERIMENTAL PROCEDURES
Cloning and Plasmid Construction
Kv12.2 cDNA was isolated by PCR from a mouse brain library using Prime STAR GXL DNA polymerase (Takara Bio Inc., Otsu, Japan) and specific primers (5′-primer, 5′-GCTAGCGCCACCATGCCGGCCATGCGGGGGCTCCTG-3′; 3′-primer, 5′-TTGCGGCCGCTCAGACCCCTGTGCCTTCTTCCTGGGT-3′). A NheI- Kozak sequence was introduced at the 5′ end of the PCR product and a NotI restriction site at the 3′ end, and the construct was cloned into the pGEM T-easy cloning vector (Promega). With the exception of two silent mutations (G1161A and G2682A), our mouse Kv12.2 cDNA sequence is identical to GenBankTM accession number NM_010601.3 but differs slightly from GenBankTM accession number AF109143 (8). The NheI-NotI fragment from the Kv12.2 cloning vector was ligated into digested pcDNA3.1(+) vector (Invitrogen). For the EGFP-Kv12.2 construct, EGFP was isolated by PCR from the pEGFP C2 vector (Clontech) and ligated to the 5′ end of Kv12.2 in the pcDNA3.1(+) vector. EGFP-Kv12.2 substitution mutants were generated from EGFP-Kv12.2 with the QuikChange II site-directed mutagenesis kit (Stratagene, Birmingham, AL) according to the manufacturer's instructions. To avoid undesired mutations, after point mutations were introduced into the EGFP-Kv12.2 construct, the fragments were sequenced and ligated into confirmed vectors. For expression of EGFP-Kv12.2 in primary cultured neurons, the EGFP-Kv12.2 fragment was cloned into the pCA mammalian expression vector under the chicken β-actin promoter. The pCA-based EGFP-Kv12.2 vector resulted in higher protein expression in cultured neurons than the pcDNA3.1-based vector.
CHO Cell Culture and Transfection
CHO K1 cells were grown in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% heat-inactivated fetal bovine serum (BioWhittaker), 50 units/ml penicillin, and 50 μg/ml streptomycin at 37 °C in a humidified 5% CO2 atmosphere. CHO-K1 cells in a 35-mm Petri dish were transfected with a mixture containing 1.5 μg of the EGFP-Kv12.2 expression plasmid and 5 μl of Lipofectamine (Invitrogen) according to the manufacturer's instructions. For transfection of CHO-K1 cells in 60-mm Petri dishes, twice as much EGFP-Kv12.2 expression plasmid and Lipofectamine were used.
Patch Clamp Recordings and Data Analysis
Whole-cell Kv12.2 currents were recorded 36–48 h after transfection at 25 °C. Cells were voltage-clamped with an EPC10 amplifier (HEKA Elektronik, Lambrecht, Germany). Patch pipette resistances were 2–3.5 megaohms. The external solution contained 10 mm HEPES-NaOH (pH 7.4), 112 mm NaCl, 40 mm KCl, 1.5 mm CaCl2, 1 mm MgCl2, and 10 mm glucose. The internal pipette solution contained 10 mm HEPES-KOH (pH 7.3), 10 mm NaCl, 130 mm potassium gluconate, 1.3 mm CaCl2, 2 mm MgCl2, 10 mm EGTA, and 1 mm ATP. To generate the steady-state activation curve, an established tail current protocol was used (6, 8, 11). Inward tail currents were elicited by voltage steps at −120 mV after conditioning pulses from −80 to 80 or 100 mV for 3 s. The small inward tail current for the conditioning pulse of −80 mV was subtracted from all measured currents. The peak amplitudes of the tail currents were normalized and plotted against the conditioning potential. The data points were fitted with a Boltzmann function of the form g = gmax/1 + exp(−(Vm − V1/2)/k), where gmax is the maximum conductance, V1/2 is the voltage of half-maximal activation, Vm is the applied membrane potential of the conditioning pulse, and k is the slope factor. Data are usually given as the mean value ± the S.E. The channel activity was evaluated from the average peak amplitude of tail currents at −100 mV after a conditioning potential of 120 mV for 500 ms.
Primary Hippocampal Cell Culture
E18 rat hippocampi dissected in Hanks' balanced salt solution without calcium and magnesium were treated with 0.25% trypsin for 15 min at 37 °C and dispersed with a constricted Pasteur pipette (20 times) to produce a homogeneous suspension. The isolated neurons were washed with Hanks' balanced salt solution without calcium and magnesium and subjected to electroporation using the Nucleofector system (Amaxa Inc., Gaithersburg, MD). Briefly, 2∼3 × 106 dissociated neurons were spun down at 100 × g for 3 min at 4 °C, resuspended in 100 μl of rat neuron Nucleofector solution kept at room temperature, combined with 3 μg of plasmid DNA, transferred into a cuvette, and electroporated using program O-03 of the Amaxa system. Transfected neurons (200,000 ∼ 400,000 per 60-mm dish) were plated in tissue culture dishes coated with poly-l-lysine (Sigma) in neurobasal medium (Invitrogen) containing 1/50 volume of B27 (Invitrogen) and 1 mm GlutaMAX (Invitrogen). The neurons were maintained at 37 °C in a humidified 5% CO2 atmosphere for 14 days and used for immunoblot analysis.
Antibody against the Very C Terminus of Kv12.2
Antibody against the Kv12.2 protein was raised using as antigen a peptide corresponding to its very C terminus (residues 1081–1095). The synthetic peptide was conjugated to keyhole limpet hemocyanin and injected into rabbits to generate polyclonal antibodies according to standard protocol. The antibody was affinity-purified from the rabbit serum and used for immunoblotting (anti-Kv12.2(1081)). The anti-Kv12.2(787) antibody was raised against a peptide corresponding to C-terminal residues 787–800 using the same method described for anti-Kv12.2(1081).
Immunoblot Analysis
48 h after transfection, CHO cells were washed with phosphate-buffered saline (PBS; 8.1 mm Na2HPO4, 1.5 mm KH2PO4, 137 mm NaCl, 2.7 mm KCl; Takara Bio, Inc.) and harvested. To solubilize the Kv12.2 protein, cell pellets were suspended in solubilization buffer (1% n-dodecyl-β-d-maltopyranoside (Dojindo Laboratories, Kumamoto, Japan) in 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA) supplemented with 1/100 volume of protease inhibitor mixture (Nacalai Tesque, Inc., Kyoto, Japan) and 1 mm phenylmethylsulfonyl fluoride and incubated for 30 min on ice. After removing cell debris by centrifugation at 20,000 × g for 30 min, the supernatant was resolved by SDS-PAGE (7.5% gel). The lanes were transferred to a polyvinylidene difluoride membrane (Bio-Rad), which was incubated with anti-Kv12.2 antibody at 2.5 μg/ml for about 12 h at 4 °C. The antibody was detected by horseradish peroxidase-conjugated secondary antibody (Promega) and visualized using ECL Advance (Amersham Biosciences). The bands were quantified using an LAS-3000 image analyzer (FUJI FILM).
At 14 days in vitro hippocampal neurons were washed with PBS, harvested, and treated like CHO cells except that for solubilization of neurons radioimmune precipitation assay buffer was used (1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS in 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA) supplemented with 1/100 volume of protease inhibitor mixture (Nacalai Tesque) and 1 mm phenylmethylsulfonyl fluoride.
For the detection of native Kv12.2, crude membrane fractions were prepared from freshly dissected whole brains of mice of different ages (embryonic 17 day, postnatal day 5 (PD5), P13, or adult (P210)). Crude membrane fractions prepared from livers of adult mice were used as control. Samples were homogenized in homogenization buffer (0.32 m sucrose, 10 mm HEPES-NaOH (pH 7.0), 0.1 mm CaCl2, 1 mm MgCl2, 1/100 volume of protease inhibitor mixture). The homogenate was centrifuged at 1500 × g for 10 min to remove cell debris, and the supernatant was centrifuged at 13,500 × g for 10 min. The pellet was resuspended in homogenization buffer and stored at −80 °C. Protein content was determined using the DC Protein Assay system (Bio-Rad) with bovine serum albumin as standard. 20 μg of protein was added to reducing SDS sample buffer, boiled for 3 min at 95 °C, and analyzed by immunoblotting as described above.
Enzymatic Deglycosylation
n-Dodecyl-β-d-maltopyranoside or radioimmune precipitation assay buffer-solubilized proteins from transfected cells or crude membrane fractions from mouse tissues were denatured by boiling for 3 min at 95 °C in buffer containing 1% β-mercaptoethanol and 0.5% SDS. The denatured samples were treated with 50 units/μl Endo H (New England Biolabs) or 50 units/μl PNGase F (New England Biolabs) for 1 h at 37 °C. The reaction was stopped by adding an equal volume of 2× SDS sample buffer and boiling the samples for 3 min at 95 °C.
Tunicamycin Treatment
8–10 h after transfection with the EGFP-Kv12.2 construct, CHO cells were treated with 50 μg/ml tunicamycin (Sigma) or an equal volume of dimethyl sulfoxide (DMSO) as control. CHO cells were cultured for an additional ∼36 h and analyzed by immunoblot or patch clamp experiments.
PNGase F or Neuraminidase Treatment of Live CHO Cells
For PNGase F treatment of live CHO cells, EGFP-Kv12.2-transfected cells in a 35-mm tissue culture dish were washed 3 times with PBS containing 1 mm MgCl2 and 1 mm CaCl2 (PBS-MC) and incubated with 1 ml of PBS-MC containing 0, 500, 1000, 2500, or 5000 units of PNGase F for 1 h at 37 °C. The removal of sugar chains was monitored by immunoblotting. Cells treated with 1000 units of PNGase F were used for patch clamp recordings within 1 h after treatment. Neuraminidase treatment was performed as described for PNGase F using 0, 50, or 100 units in 1 ml of PBS-MC for immunoblot experiments and 50 units for patch clamp experiments.
Fluorescence Microscopy and Immunoelectron Microscopy
For confocal immunofluorescence microscopy, CHO cells were cultured in eight-well culture slides (Nunc, Naperville, IL). 36–48 h after transfection fluorescence images were acquired at room temperature with an FV1000 confocal laser-scanning microscope (Olympus, Tokyo, Japan).
For immunoelectron microscopy, CHO cells were fixed 48 h after transfection for 2 h at room temperature with 2% paraformaldehyde (Merck) in 0.1 m phosphate buffer (PB) at pH 7.5 containing 20 μm (p-amidinophenyl)methanesulfonyl fluoride hydrochloride. After washing with 100 mm PB, cells were incubated for 15 min in 20% Blockace (Dainippon Pharmaceutical, Osaka, Japan) in 100 mm PB containing 20 μm (p-amidinophenyl)methanesulfonyl fluoride hydrochloride and 0.005% saponin (Merck). Cells were then incubated overnight at 4 °C with anti-green fluorescent protein polyclonal antibody (Abcam, Cambridge, MA) diluted 1:1000 in dilution buffer (100 mm PB containing 5% Blockace and 0.05% saponin (Merck)). After washing with washing buffer (100 mm PB containing 0.05% saponin (Merck)), cells were incubated for 2 h at room temperature with 1-nm gold-conjugated goat anti-rabbit IgG (Nanogold; Nanoprobes, Inc., Stony Brook, NY) diluted 1:100 in dilution buffer. After washing with washing buffer, cells were postfixed for 15 min with 1% glutaraldehyde in PB, and gold labels were enhanced by a 6-min incubation with a silver developer (HQ silver, Nanoprobes) in the dark. Cells were postfixed again with 0.5% OsO4 in PB for 90 min at 4 °C, stained with 4% uranyl acetate for 30 min at room temperature, and dehydrated by passage through a graded series of ethanol (50, 70, 90, and 100%). After embedding in Epon 812 (Nacalai Tesque), ultrathin sections (80 nm thickness) were cut with an LKB Ultratome (GE Healthcare ), transferred to specimen grids coated with polyvinyl formal (Nissin EM Co., Tokyo, Japan), stained with uranyl acetate and lead citrate, and inspected under an electron microscope (JEM-1013EX, JEOL, Tokyo, Japan) (26).
RESULTS
EGFP-Kv12.2 Forms Functional Channels
We cloned the mouse Kv12.2 gene (see “Experimental Procedures”) and examined the activity of its product by recording whole-cell currents in transiently transfected CHO cells using the patch clamp technique. To identify transfected cells, EGFP was co-transfected with wild-type Kv12.2, but this approach did not allow us to reliably measure currents from EGFP-positive cell (data not shown). To improve the reproducibility of our measurements, we fused EGFP to the N terminus of Kv12.2. When cells were transfected with EGFP-Kv12.2, almost all EGFP-positive cells showed voltage-dependent transient outward currents and characteristic tail currents (Fig. 2), which were absent in mock-transfected cells. The currents were similar to those of untagged Kv12.2 channels which were previously reported (6, 8, 11). We, therefore, concluded that EGFP-Kv12.2 could be used for further characterization of the channel.
FIGURE 2.
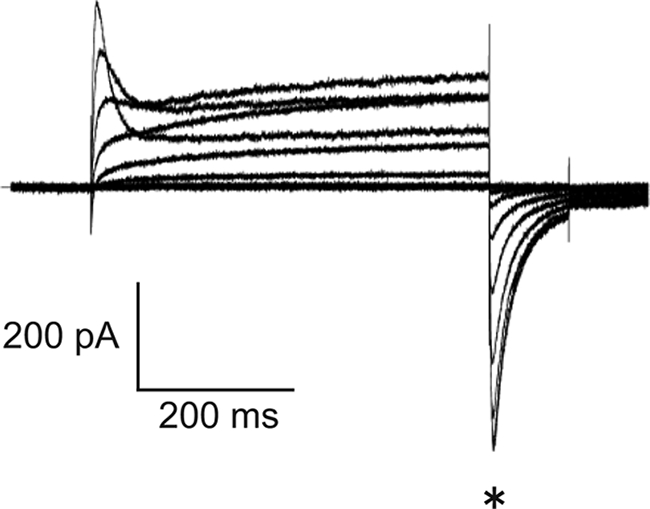
Voltage-dependent current of EGFP-Kv12.2. Representative currents were measured from CHO cells transfected with EGFP-Kv12.2. In this experiment, EGFP-Kv12.2 outward currents were elicited by voltage pulses from −80 to 80 mV for 500 ms from a holding potential of −80 mV. Tail currents were elicited by repolarizing the membrane to −120 mV at the end of the 500-ms pulses. The asterisk indicates the tail currents.
Electrophoretic Mobility of Recombinant EGFP-Kv12.2 and Native Kv12.2
To characterize the biochemical properties of Kv12.2, we performed an immunoblot analysis. We raised a polyclonal antibody against the very C terminus of mouse Kv12.2 (anti-Kv12.2(1081), see “Experimental Procedures”) and used it to detect EGFP-Kv12.2 expressed in CHO cells. The anti-Kv12.2(1081) antibody recognized three prominent bands that were absent in mock-transfected CHO cells (Fig. 3A). The bands at molecular masses of 172 and 167 kDa were strong, whereas the band at the highest molecular mass (188 kDa) was faint. These molecular masses are bigger than the calculated molecular mass of the EGFP-Kv12.2 protein of 145 kDa, which includes 27 kDa for EGFP and the linker. The antibody did not cross-react with the closely related Kv12.3 channel expressed in CHO cells (supplemental Fig. S1), and another antibody (anti-Kv12.2(787), see “Experimental Procedures”) produced a similar staining pattern on immunoblots (supplemental Fig. S1). We, therefore, conclude that the anti-Kv12.2(1081) antibody specifically recognizes Kv12.2 and that all three bands detected by anti-Kv12.2(1081) represent EGFP-Kv12.2.
FIGURE 3.

Electrophoretic mobility of Kv12.2 channels. Cell lysates or crude membranes were analyzed by SDS-PAGE and immunoblotting. A, n-dodecyl-β-d-maltopyranoside-solubilized lysates were prepared from CHO cells transfected with EGFP-Kv12.2 and mock-transfected cells. B, radioimmune precipitation assay buffer-solubilized lysates were prepared from hippocampal neurons transfected with EGFP-Kv12.2 and mock-transfected neurons. C, crude membranes were prepared from adult mouse brain and liver tissue at postnatal day 210. 20 μg of protein was loaded. D, equal amounts of crude membranes were analyzed at different developmental stages: embryonic day 17 (E17), postnatal day 5 (P5) and 13 (P13), and adult (P210). The molecular masses estimated from the protein standards are shown to the right of the lanes.
Immunoblot analysis of EGFP-Kv12.2 expressed in cultured hippocampal neurons revealed only two bands (Fig. 3B). Their apparent molecular masses of 186 and 170 kDa suggested that they correspond to the upper (188 kDa) and middle (172 kDa) band seen in immunoblots of CHO cells expressing EGFP-Kv12.2.
When immunoblots of crude membranes from mouse brain were analyzed, the anti-Kv12.2(1081) antibody recognized two bands with apparent molecular masses of 157 and 142 kDa that were not detected in immunoblots of mouse liver membranes (Fig. 3C). These bands would correspond to the upper band (188 and 186 kDa) and the middle band (172 and 170 kDa) of EGFP-Kv12.2 expressed in heterologous systems (CHO cells and cultured neurons, respectively). The lower apparent molecular masses of the native channels are due to the lack of the 27-kDa EGFP. The faint bands at 200 and 120 kDa are likely due to nonspecific binding because these bands were not detected by the anti-Kv12.2(787) antibody. To examine temporal changes in the expression level and the biochemical properties of native Kv12.2 channels in the developing brain, equal amounts of membranes from whole brains of mice at four ages (embryonic day 17 (E17), postnatal day 5 (P5), P13, and P210 (adult)) were analyzed (Fig. 3D). The intensity of the predominant 157-kDa band increased steadily in the course of brain development, with the greatest change occurring between postnatal day 5 and P13. In contrast, the intensity of the 142-kDa band peaked at P13. The detection of multiple bands in these results suggested that Kv12.2 carries co- and/or post-translational modifications both in vitro and in vivo.
Kv12.2 Is N-Glycosylated
Among the various potential modifications of Kv12.2, we decided to focus on N-glycosylation because Kv12.2 contains three N-linked glycosylation consensus sequences in the unique S5-P loop (Fig. 1B). To investigate the N-glycosylation of Kv12.2, we performed immunoblot analysis of detergent-solubilized EGFP-Kv12.2 or crude mouse brain membrane preparations after removal of either high mannose-type oligosaccharides by Endo H or all types of oligosaccharides by PNGase F.
When EGFP-Kv12.2 expressed in CHO cells was treated with Endo H, the 172-kDa band disappeared, presumably shifting the protein to the 167-kDa band, which increased in intensity (Fig. 4A, white arrowhead). The 188-kDa band was not affected by Endo H treatment (Fig. 4A, black arrow). This result indicates that the 172-kDa band contains proteins carrying high mannose-type oligosaccharides. We will refer to this protein as the immature form of EGFP-Kv12.2. Treatment of recombinant EGFP-Kv12.2 with PNGase F, which can remove both high mannose and complex oligosaccharides, caused the 188-kDa band to completely disappear, and the intensity of the 172-kDa band was significantly reduced with a concomitant increase in the intensity of the 167-kDa band (Fig. 4A). This result suggests that the 188-kDa band contains proteins carrying complex oligosaccharides, and we will refer to this protein as the mature form of EGFP-Kv12.2. The 167-kDa band did not shift even after PNGase F treatment (Fig. 4A, black arrowhead), indicating that it represents unglycosylated EGFP-Kv12.2. These results demonstrate that EGFP-Kv12.2 expressed in CHO cells is N-glycosylated.
FIGURE 4.

Enzymatic deglycosylation of Kv12.2 channels. Cell lysates or crude membranes were denatured and treated with endoglycosidases (Endo H or PNGase F) or buffer alone (−). A, lysate of CHO cells transfected with EGFP-Kv12.2 is shown. B, lysate of neurons transfected with EGFP-Kv12.2 is shown. C, crude membranes prepared from P13 mouse brain are shown. Arrows indicate the mature form of EGFP-Kv12.2 or native Kv12.2, which carries complex oligosaccharides. Open arrowheads indicate the immature form of EGFP-Kv12.2 or native Kv12.2, which carries high mannose oligosaccharides. Filled arrowheads indicate the unglycosylated or deglycosylated form of EGFP-Kv12.2 or native Kv12.2.
In primary cultured neurons, similar band shifts were observed when detergent-solubilized EGFP-Kv12.2 was treated with endoglycosidases (Fig. 4B). The experiments revealed that in cultured neurons the 186-kDa band represents the mature channel carrying complex oligosaccharides, and the 170-kDa band represents the immature channel carrying high mannose oligosaccharides. The lack of a 165-kDa band, which would correspond to unglycosylated EGFP-Kv12.2 (Fig. 4B, black arrowhead) indicates that only a negligible fraction of EGFP-Kv12.2 in cultured neurons exists in an unglycosylated form.
The 157-kDa band seen with native Kv12.2 from mouse brain was resistant to Endo H but sensitive to PNGase F, whereas the 142-kDa band was sensitive to both enzymes (Fig. 4C). Like recombinant EGFP-Kv12.2 in cultured neurons, native Kv12.2 appears to exist in the mouse brain in a mature form carrying complex oligosaccharides (157-kDa band) and an immature form carrying high-mannose oligosaccharides (142-kDa band) with an undetectable level of unglycosylated protein.
From these results we conclude that Kv12.2 is N-glycosylated both in vivo as well as in heterologous expression systems. The bands representing deglycosylated Kv12.2 after PNGase F treatment were diffuse (Fig. 4). This may be due to α1,3-linked fucose residues in the sugar chains, which render N-glycans resistant to cleavage by PNGase F (27). It is also possible that mature Kv12.2 undergoes another post-translational modification that affects the electrophoretic mobility. The ratio of mature to immature and unglycosylated Kv12.2 was higher in mouse brain and cultured neurons than in CHO cells. The inefficient maturation of EGFP-Kv12.2 in CHO cells was not due to the fused EGFP, because the analysis of Kv12.2 without EGFP expressed in CHO cells yielded similar results (supplemental Fig. S2). The observed molecular masses of deglycosylated EGFP-Kv12.2 and deglycosylated Kv12.2 from mouse brain are bigger than the calculated molecular mass of the core protein by ∼20 kDa. The difference between the apparent and calculated molecular masses may indicate that Kv12.2 carries post-translational modifications other than N-linked glycosylation or may be due to a slightly aberrant mobility of Kv12.2 in SDS-PAGE gels, which is not uncommon for membrane proteins.
Glycosylation Is Not Required for Kv12.2 Channel Function
N-Linked glycosylation may play an important role in channel function itself and/or in defining channel characteristics because the N-glycosylation sites of Kv12.2 are located in the long extracellular S5-P loop near the pore region (Fig. 1A, right panel). To determine whether glycosylation affects channel function itself, we measured the tail currents of CHO cells expressing wild-type EGFP-Kv12.2 after enzymatic removal of the sugar chains with PNGase F. Because only channels in the plasma membrane are measured, this experimental design eliminated any potential effects related to protein trafficking. We first treated live CHO cells expressing EGFP-Kv12.2 with increasing concentrations of PNGase F and evaluated its efficiency (Fig. 5A). Although PNGase F could affect high mannose-type oligosaccharides of detergent-solubilized EGFP-Kv12.2 (Fig. 4A), treatment of the cells with 500 units/ml PNGase F only affected complex sugar chains of EGFP-Kv12.2 (Fig. 5A, arrow), and even higher enzyme concentrations did not remove high mannose-type oligosaccharides (Fig. 5A, white arrowhead). This result shows that PNGase F can remove sugar chains of EGFP-Kv12.2 on live cells and suggests that only the mature form of Kv12.2 carrying complex oligosaccharides is expressed on the cell surface. When we measured the EGFP-Kv12.2 tail currents after removal of the sugar chains by 1000 units/ml PNGase F, the current amplitude was not significantly reduced as compared with the controls (Fig. 5B). This result indicates that sugar chains are not necessary for the Kv12.2 channel function itself.
FIGURE 5.

Effect of glycosylation on the current amplitude and steady-state activation of EGFP-Kv12.2 expressed in CHO cells. A, an immunoblot of lysate from CHO cells transfected with EGFP-Kv12.2 and treated with PNGase F is shown. CHO cells transfected with wild-type EGFP-Kv12.2 were incubated at 37 °C for 1 h with 0, 500, 1000, 2500, or 5000 units/ml PNGase F in PBS supplemented with 1 mm MgCl2 and 1 mm CaCl2. The arrow indicates the mature form of EGFP-Kv12.2, the open arrowhead indicates the immature form, and the filled arrowhead indicates unglycosylated EGFP-Kv12.2. B and C, CHO cells transfected with EGFP-Kv12.2 were treated with 1000 units/ml or without PNGase F (filled circles or open circles in Fig. 5C, respectively). B, shown are average current amplitudes of the treated cell. The numbers in parentheses indicate the number of experiments. C, the activation curve obtained using the tail-current protocol is shown. For PNGase F-treated cells, the half-maximal activation voltage (V1/2) was 46.5 ± 1.3 mV, and the slope factor (k) was 29.9 ± 0.7 mV (n = 5, open circles); for control cells V1/2 was 15.1 ± 2.5 mV, and k was 23.7 ± 1.7 mV (n = 5, filled circles). Error bars correspond to the S.E. of the mean.
Deglycosylation Causes a Depolarizing Shift in the Voltage-dependent Activation of Kv12.2
We next focused on the role of N-glycosylation in the channel characteristics. To examine the effect of glycosylation on the voltage dependence of Kv12.2, we determined the steady-state activation of EGFP-Kv12.2 before and after PNGase F treatment of live CHO cells expressing EGFP-Kv12.2 (Fig. 5C). The steady-state activation was evaluated according to an established tail-current protocol (see the legend to Fig. 5C and Refs. 6, 8, and 11 for details). The membrane potential was stepped from a holding potential to conditioning potentials between −80 and 80 mV. Tail currents were elicited during a subsequent repolarizing step, where the inactivated channels recover quickly to the open state as in Fig. 2. Therefore, the tail current reflects the number of open channels at a conditioning potential. The normalized peak amplitudes of the tail currents were plotted against the conditioning potentials (Fig. 5C). The half-maximal activation voltage of glycosylated EGFP-Kv12.2 was 15.1 ± 2.5 mV (n = 5, Fig. 5C, filled circles), but that of deglycosylated EGFP-Kv12.2 was 46.5 ± 1.3 mV (n = 5, Fig. 5C, open circles). Thus, removal of sugar chains by PNGase F treatment caused a depolarizing shift in the voltage-dependent activation by ∼30 mV.
The Deglycosylation-induced Shift in Voltage-dependent Activation Is Not Due to the Loss of Sialic Acid Residues
Previous studies have reported that N-glycosylation can affect voltage-dependent activation of potassium channels and sodium channels via negatively charged sialic acids. The removal of sialic acids causes a depolarizing shift in the activation voltage by ∼10–20 mV (28–31). The effect of sialic acids on the channels was attributed to the electrostatic influence of these negatively charged residues, which alters the effective electric field detected by the voltage sensors of the channels. We speculated that the effect of N-glycans on Kv12.2 (Fig. 5C) is also caused by sialic acid residues in the sugar chains. To examine this possibility, we treated cells expressing wild-type EGFP-Kv12.2 with neuraminidase (sialidase) and analyzed them by immunoblotting and patch clamp recording. Immunoblots demonstrated that neuraminidase caused a band shift of 4 kDa (Fig. 6A). This result shows that EGFP-Kv12.2 expressed in CHO cells carries sialic acids and that these can be removed by neuraminidase treatment of live CHO cells. The steady-state activation of the desialylated EGFP-Kv12.2 channel was measured by the same tail-current protocol used in Fig. 5C (Fig. 6B). The half-maximal activation voltage after neuraminidase treatment was 12.3 ± 2.8 mV (n = 4, Fig. 6B, open circles), similar to that measured with untreated cells (Fig. 6B, filled circles, same data as in Fig. 5C). This result suggests that the deglycosylation-induced shift in the steady-state activation curve of the EGFP-Kv12.2 channel is due to a mechanism that is different from the electrostatic influence of the sialic acid residues.
FIGURE 6.

Effect of sialic acid residues on the steady-state activation of EGFP-Kv12.2 expressed in CHO cells. A, shown is an immunoblot of lysate from EGFP-Kv12.2-transfected cells after neuraminidase treatment. Cells transfected with wild-type EGFP-Kv12.2 were treated as in Fig. 5A, except that three different concentrations of neuraminidase were used in this experiment: 0, 50, or 100 units/ml. The black arrow indicates the fully glycosylated form of EGFP-Kv12.2, and the white arrow indicates the desialylated EGFP-Kv12.2 channel. B, shown is an activation curve of untreated (filled circles) and neuraminidase-treated (open circles) EGFP-Kv12.2. To remove sialic acid residues, 50 units/ml neuraminidase were added to live CHO cells transfected with EGFP-Kv12.2. The activation curves were obtained following the same tail-current protocol used in Fig. 5C. Error bars correspond to the S.E. For neuraminidase-treated cells, V1/2 was 12.3 ± 2.8 mV, and the slope factor was 25.8 ± 1.8 mV (n = 4); for control cells, the values are the same as those in Fig. 5C.
Unglycosylated Kv12.2 Is Not Expressed on the Cell Surface
We next focused on the trafficking of unglycosylated channels to know whether unglycosylated channels, which have a different voltage dependence from glycosylated channels (Fig. 5C), are utilized in vivo. To address this issue we analyzed CHO cells expressing EGFP-Kv12.2 that were treated with tunicamycin, which inhibits N-linked glycosylation. Immunoblots revealed a single band of 167 kDa, corresponding to the size of EGFP-Kv12.2 after PNGase F treatment (Fig. 7A). This finding corroborated our conclusion that the 188- and 172-kDa bands correspond to glycosylated protein, and the 167-kDa bands correspond to unglycosylated protein and confirmed that tunicamycin effectively inhibits N-linked glycosylation in this system.
FIGURE 7.

Effects of tunicamycin treatment on the glycosylation and function of Kv12.2 in CHO cells. 8–10 h after transfection with EGFP-Kv12.2, CHO cells were treated with 5 μg/ml tunicamycin or DMSO as control. A, shown is an immunoblot of lysates from tunicamycin- and DMSO-treated CHO cells. The arrow indicates the mature form of EGFP-Kv12.2, the open arrowhead indicates the immature form, and the filled arrowhead indicates unglycosylated EGFP-Kv12.2. B, representative currents recorded from tunicamycin- and DMSO-treated CHO cells are shown. Tail current was elicited by a voltage step at −100 mV after a conditioning pulse of 120 mV for 500 ms. C, shown are average amplitudes of peak tail currents measured from tunicamycin- and DMSO-treated CHO cells. Error bars correspond to the S.E., and the numbers in parentheses indicate the number of cells measured.
To study the expression of unglycosylated Kv12.2 channels on the cell surface, we performed patch clamp studies on transfected CHO cells after tunicamycin treatment. If unglycosylated channels are expressed on the cell surface, tail currents would be observable because Kv12.2 channels lacking sugar chains are functional (Fig. 5B). Fig. 7B shows representative tail currents measured from EGFP-Kv12.2-transfected cells after treatment with tunicamycin or DMSO. Treatment of the cells with tunicamycin resulted in the loss of Kv12.2 current. Quantitative analysis of the data showed that tunicamycin treatment caused a dramatic reduction in the average current amplitude of EGFP-Kv12.2 (Fig. 7C). This result implies that unglycosylated Kv12.2 is not trafficked to the cell surface.
Expression of Kv12.2 on the Cell Surface Requires at Least One of the Three Glycosylation Sites to Carry a Sugar Chain
Kv12.2 has three consensus sequences for N-linked glycosylation at asparagines 421, 428, and 447 (Fig. 1B). Tunicamycin treatment inhibits the glycosylation of all three sites. To investigate how the number and position of N-glycosylation at these three sites affect maturation and cell surface expression of Kv12.2, we used site-directed mutagenesis to generate glutamine substitution mutants, specifically the three single mutants N421Q, N428Q, and N447Q (designated as EGFP-Kv12.2 QNN, NQN, and NNQ), the three double mutants N421Q,N428Q, N421Q,N447Q, and N428Q,N447 (designated as EGFP-Kv12.2 QQN, QNQ, and NQQ), and the triple mutant N421Q,N428Q,N447Q (designated as EGFP-Kv12.2 QQQ).
Immunoblots of the single mutants EGFP-Kv12.2 QNN, NQN, and NNQ showed three bands corresponding to the mature, immature, and unglycosylated form of the wild-type channel (Fig. 8A). In contrast, immunoblots of the double mutants EGFP-Kv12.2 QQN, QNQ, and NQQ showed only the bands representing the immature and unglycosylated form, but the mature form could no longer be detected (Fig. 8A). This result demonstrates that all three sites are potentially glycosylated and suggests that the maturation of the double mutants was less efficient than that of the wild type and single mutants. The triple mutant EGFP-Kv12.2 QQQ showed only one sharp band with the same mobility as the wild-type protein treated with PNGase F (Fig. 8A). Furthermore, PNGase F treatment had no effect on the electrophoretic mobility of the triple mutant (data not shown). These results suggest that N-linked glycosylation at sites Asn-421, Asn-428, and Asn-447 account for all the N-linked oligosaccharides on the Kv12.2 channel.
FIGURE 8.

Biochemical and electrophysiological analyses of Kv12.2 substitution mutants. CHO cells were transfected with mutants in which the asparagine residues in the three glycosylation sites were substituted with glutamine residues (see “Results” for details). A, shown is an immunoblot of wild-type (WT) EGFP-Kv12.2 and the substitution mutants. The arrow indicates the mature form of EGFP-Kv12.2, the open arrowhead indicates the immature form, and the filled arrowhead indicates unglycosylated EGFP-Kv12.2. B, average current amplitudes are shown of the peak tail currents measured from the transfected CHO cells as in Fig. 5B. Error bars correspond to the S.E., and the numbers in parentheses indicate the number of cells measured.
The function of the substitution mutants was analyzed by measuring their tail currents (Fig. 8B). CHO cells transfected with single mutants showed average current amplitudes similar to cells transfected with wild-type EGFP-Kv12.2. The average current amplitude was slightly reduced in cells transfected with double mutants and significantly reduced in cells transfected with the triple mutant (Fig. 8B). This result indicates that expression of Kv12.2 on the cell surface requires at least one of the three glycosylation sites to carry a sugar chain irrespective of their positions.
Intracellular Distribution and Surface Expression of Unglycosylated Kv12.2
The lack of current seen in CHO cells expressing wild-type EGFP-Kv12.2 after tunicamycin treatment (Fig. 7, B and C) and cells expressing the EGFP-Kv12.2 QQQ triple mutant (Fig. 8B) must be due to inefficient trafficking of the channels to the cell surface, because removal of sugar chains did not diminish the current amplitude (Fig. 5B). To further corroborate our conclusion, we used fluorescence microscopy to analyze the subcellular localization of the channels. In CHO cells expressing EGFP-Kv12.2 wild type and QQQ triple mutant, most of the signal localized to intracellular or perinuclear compartments, co-localizing mainly with endoplasmic reticulum marker (supplemental Fig. S3). To better detect the signal on the cell surface, we then used confocal microscopy, which revealed the EGFP signal at the cell membrane in cells expressing wild-type EGFP-Kv12.2 (Fig. 9, A, left panel and B, arrows). In contrast, no EGFP signal was seen at the cell surface in cells expressing the triple mutant (Fig. 9A, right panel). This subcellular distribution was further analyzed by immunoelectron microscopy with anti-EGFP antibodies, which confirmed the plasma membrane localization of wild-type EGFP-Kv12.2 (Fig. 9, C, left panel and D, arrows) but not of the triple mutant (Fig. 9C, right panel). We, therefore, conclude that N-glycosylation is necessary for intracellular trafficking of the Kv12.2 channel to the cell surface.
FIGURE 9.

Intracellular distribution of wild-type EGFP-Kv12.2 and QQQ mutant channels in CHO cells. A, shown are confocal microscope images of CHO cells transfected with wild-type (WT) EGFP-Kv12.2 (left panel) or the QQQ mutant (right panel). White arrows point to the EGFP signals on the cell surface of a CHO cell expressing wild-type EGFP-Kv12.2. Scale bars represent 10 μm. B, shown is a higher magnification image of the cell membrane of the cell expressing wild-type EGFP-Kv12.2 shown in panel A. The scale bar represents 1 μm. C, electron microscope images are shown of CHO cells transfected with wild-type EGFP-Kv12.2 (left panel) or the QQQ mutant (right panel). The cells were labeled with gold-conjugated antibodies against EGFP. Black arrows point to immunogold localized on the cell surface. Scale bars represent 1 μm. D, shown is a higher magnification image of the cell membrane of the cells expressing wild-type EGFP-Kv12.2 shown in panel C. Scale bar represents 0.5 μm.
DISCUSSION
In this study we demonstrate that N-linked glycosylation plays two important roles in Kv12.2. 1) N-linked glycans regulate the voltage-dependent activation of Kv12.2 through a mechanism that is independent of the presence of negatively charged sialic acid residues in the sugar chains. 2) Although the position is not crucial, at least one of the three glycosylation sites has to carry a sugar chain for Kv12.2 to be trafficked to the cell surface. In addition, our results provide evidence that Kv12.2 exists mostly in a glycosylated form in the mouse brain. These results suggest that N-glycosylated Kv12.2 is likely to function in vivo, whereas unglycosylated Kv12.2, which has different channel properties, is unlikely to have any functional significance.
We showed that removal of N-glycans results in a depolarizing shift in the voltage-dependent activation of Kv12.2 by ∼30 mV (Fig. 5C). A similar effect of sugar chains, especially of negatively charged sialic acid residues, has been reported for voltage-dependent potassium channels (Kv) and sodium channels (Nav) (19, 23, 28, 30). Enzymatic removal of the entire sugar chain or only the sialic acid residues from purified, transfected, or endogenous Kv and Nav channels shifted gating in the depolarized direction (23, 31, 32). Moreover, when some Kv and Nav α-subunit isoforms were expressed in the Lec2 mutant CHO cell line, which exhibits reduced sialylation, the channels opened at more depolarized potentials (19, 28, 29, 31). Reduced sialylation of these channels is thought to change their external surface charge, causing a shift in their activation curve by 10∼20 mV. Unexpectedly, in the case of Kv12.2, removal of sialic acids did not affect the steady-state activation curve (Fig. 6B). It is possible that Kv12.2 is modified by sialic acids that are resistant to neuraminidase added to live cells. Alternatively, the observed shift may be caused by a novel mechanism that is independent of the sialic acid residues. Possibly, the sugar chains directly interact with the channel itself and regulate its function. It has been reported that the S5-P loop in members of the EAG family contains an amphipathic α-helix, which interacts with the voltage sensor and contributes to the different inactivation characteristics of the channels (33–35). This suggests that the sugar chains in the long S5-P loop could also interact with a part of the channel itself such as the voltage sensor. The recent structure of a Kv1.2–2.1 chimera channel together with its β-subunit provided important insights into the gating of voltage-dependent channels, but it did not provide structural information on the sugar chains (36). Ideally, to understand channel gating under the most native conditions, structural information should be obtained for voltage-dependent channels carrying their native sugar chains. However, the heterogeneous and flexible nature of sugar chains often prevent the growth of three-dimensional crystals and/or are too disordered in crystals to be resolved in the resulting density map.
Our results from the inhibition of N-glycosylation by tunicamycin (Fig. 7) and the analysis of glutamine mutants (Figs. 8 and 9) showed that unglycosylated Kv12.2 channels are not trafficked to the cell surface in CHO cells. The Eag (Kv10.1) channel and the HERG (Kv11.1) channel, other members of the EAG family (Kv10.x ∼ Kv12.x, Fig. 1B), also require glycosylation for their proper cell surface trafficking (18, 20, 25). In the Kv10.1 channel the two glycosylation sites in the S5-P loop appear not to be equivalent, as it has been shown that proper complex glycosylation of at one of the two sites is crucial for proper trafficking of the channel and its functional properties (18). In contrast, the three sites of Kv12.2 appear to be equivalent at least for channel trafficking because all double mutants showed current amplitudes only slightly lower than that of the wild-type EGFP-Kv12.2 (Fig. 9B). The recognition mechanism of sugar chains or the structure of the S5-P loop may, thus, vary, even between members of the EAG family.
Here we presented the first biochemical data for N-glycosylation of Kv12.2. In the adult mouse Kv12.2 protein is expressed in the brain but not in the liver (Fig. 3C). This result is consistent with the brain-specific expression of its mRNA (6–8). The level of mature Kv12.2 protein increases with development (Fig. 3D). This result is also consistent with the up-regulation of Kv12.2 mRNA in the rat brain (6). A similar increase in protein level with development has been reported for other potassium channels, including Kv1.4, Kv1.5, Kv2.1, and Kv2.2 (37). This increase in the expression of Kv12.2 may be partly due to the postnatal increase in synaptogenesis and suggests that Kv12.2 is involved in the excitability of neurons. In our experiments the unglycosylated form of Kv12.2 was not observed in the mouse brain. This finding together with the fact that unglycosylated EGFP-Kv12.2 was not trafficked to the cell surface in CHO cells suggests that unglycosylated Kv12.2 is not utilized in vivo. Unlike in mouse brain and in cultured neurons, the unglycosylated and immature forms are predominant in the non-neuronal CHO cells even for the wild-type channels. This result suggests that Kv12.2 does not mature efficiently when transiently expressed in non-neuronal cells like CHO cells. Because the final maturation steps of sugar chains occur in the Golgi apparatus, the preponderance of immature and unglycosylated channels suggested that the channel is retained in the endoplasmic reticulum (13). Immunofluorescence microscopy of CHO cells expressing EGFP-Kv12.2 supported this notion, because the majority of the signal co-localized with endoplasmic reticulum markers (supplemental Fig. S3). These results indicate that Kv12.2 may require some factors for efficient maturation and/or efficient trafficking and these factors might only be present in neuronal cells but are lacking in non-neuronal cells, such as CHO cells. Certain channels are known to require auxiliary molecules, such as β-subunits, for proper folding and/or trafficking (38, 39). For example, Kvβ2 promotes co-translational N-linked glycosylation of the nascent Kv1.2 polypeptide and increases the efficiency of its cell surface expression (40). Kv12.2 may, thus, also need a β-subunit for efficient glycosylation and cell surface expression. Another possibility is that, as a recent study showed using Xenopus oocytes (41), intrinsic KCNE1 and KCNE3 may down-regulate the cell surface expression of Kv12.2 in CHO cells.
Finally, we discuss why Kv12.2 features an unusually long S5-P loop and has as many as three glycosylation sites. These structural features are unique to mammalian Kv12.2. The fruit fly ortholog Elk, the zebrafish ortholog Elk1, and the mammalian paralogs Kv12.1 and Kv12.3 lack both of them (Fig. 1B), whereas they are conserved in all known mammalian Kv12.2 channels. This suggests that all three N-glycans would play important roles in the regulation of the mammalian Kv12.2 channel. One N-glycosylation site is sufficient for trafficking (Fig. 8B), and the additional two N-glycans are, therefore, likely to have some other roles. As we have shown here, one role is to regulate the steady-state activation of Kv12.2. Further analysis is required to elucidate how the number and/or position of the sugar chains affect the functions of Kv12.2. N-Glycosylation of these three sites may also be involved in the folding and/or stability of the channel. Many membrane proteins are known to require N-glycosylation for efficient folding and/or stability (42). These N-glycans are recognized by molecular chaperons such as calnexin and calreticulin (43). It has been reported that the number of N-glycans correlates with the affinity of these chaperons and the folding rate (44, 45). Here, we observed a small reduction in the current amplitude (Fig. 8B) and inefficient maturation of double mutants (Fig. 8A) that have only one glycosylation site (Fig. 1B) compared with the wild-type protein and the single mutants. This observation may suggest that the three glycosylation sites are required for proper folding by mediating interactions with molecular chaperons. The three glycosylation sites of Kv12.2 would, thus, serve several distinct functions. The glycosylation sites are located in an extended region of the S5-P loop. The S5-P loop may have to be long to properly display the three glycosylation sites, in particular because efficient N-glycosylation typically only occurs on sites that are 10 amino acids removed from a transmembrane region (46, 47). It is also possible that the long S5-P loop itself has a specific, yet to be identified function.
Supplementary Material
Acknowledgments
We thank Drs. Hitoshi Niwa and Masatoshi Takeichi (RIKEN, Center for Developmental Biology) for kindly providing pCA plasmid and Drs. Christoph Gerle and Tomoko Doi for valuable discussions. We are grateful to Dr. Thomas Walz for critical reading of the manuscript.
This work was supported by Grants-in-aid for Specially Promoted Research and the Japan New Energy and Industrial Technology Development Organization.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
- Kv channels
- voltage-dependent potassium channels
- CHO
- Chinese hamster ovary
- Eag
- ether-a-go-go
- EGFP
- enhanced green fluorescent protein
- Elk
- ether-a-go-go-like
- Erg
- ether-a-go-go-related gene
- HERG
- human ether-a-go-go-related gene
- PB
- phosphate buffer
- PBS
- phosphate-buffered saline
- Endo H
- endoglycosidase H
- PNGase F
- peptide N-glycosidase F.
REFERENCES
- 1.Hille B. (2001) Ion Channels of Excitable Membranes, 3rd Ed., Sinauer Associates Sunderland,MA [Google Scholar]
- 2.Coetzee W. A., Amarillo Y., Chiu J., Chow A., Lau D., McCormack T., Moreno H., Nadal M. S., Ozaita A., Pountney D., Saganich M., Vega-Saenz de Miera E., Rudy B. (1999) Ann. N.Y. Acad. Sci. 868, 233–285 [DOI] [PubMed] [Google Scholar]
- 3.Yu F. H., Catterall W. A. (2004) Sci. STKE 2004, re15. [DOI] [PubMed] [Google Scholar]
- 4.Ganetzky B., Robertson G. A., Wilson G. F., Trudeau M. C., Titus S. A. ( 1999) Ann. N.Y. Acad. Sci. 868, 356– 369 [DOI] [PubMed] [Google Scholar]
- 5.Bauer C. K., Schwarz J. R. (2001) J. Membr. Biol. 182, 1–15 [DOI] [PubMed] [Google Scholar]
- 6.Engeland B., Neu A., Ludwig J., Roeper J., Pongs O. (1998) J. Physiol. 513, 647–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyake A., Mochizuki S., Yokoi H., Kohda M., Furuichi K. (1999) J. Biol. Chem. 274, 25018–25025 [DOI] [PubMed] [Google Scholar]
- 8.Trudeau M. C., Titus S. A., Branchaw J. L., Ganetzky B., Robertson G. A. (1999) J. Neurosci. 19, 2906–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou A., Lin Z., Humble M., Creech C. D., Wagoner P. K., Krafte D., Jegla T. J., Wickenden A. D. (2003) Am. J. Physiol. Cell Physiol. 285, C1356–C1366 [DOI] [PubMed] [Google Scholar]
- 10.Gutman G. A., Chandy K. G., Grissmer S., Lazdunski M., McKinnon D., Pardo L. A., Robertson G. A., Rudy B., Sanguinetti M. C., Stühmer W., Wang X. (2005) Pharmacol. Rev. 57, 473–508 [DOI] [PubMed] [Google Scholar]
- 11.Becchetti A., De Fusco M., Crociani O., Cherubini A., Restano-Cassulini R., Lecchi M., Masi A., Arcangeli A., Casari G., Wanke E. (2002) Eur. J. Neurosci. 16, 415–428 [DOI] [PubMed] [Google Scholar]
- 12.Grosso S., Pucci L., Farnetani M., Di Bartolo R. M., Galimberti D., Mostardini R., Anichini C., Balestri M., Morgese G., Balestri P. (2004) J. Child Neurol. 19, 604–608 [DOI] [PubMed] [Google Scholar]
- 13.Helenius A., Aebi M. (2001) Science 291, 2364–2369 [DOI] [PubMed] [Google Scholar]
- 14.Burda P., Aebi M. (1999) Biochim. Biophys. Acta 1426, 239–257 [DOI] [PubMed] [Google Scholar]
- 15.Watanabe I., Zhu J., Sutachan J. J., Gottschalk A., Recio-Pinto E., Thornhill W. B. (2007) Brain Res. 1144, 1–18 [DOI] [PubMed] [Google Scholar]
- 16.Watanabe I., Zhu J., Recio-Pinto E., Thornhill W. B. (2004) J. Biol. Chem. 279, 8879–8885 [DOI] [PubMed] [Google Scholar]
- 17.Brooks N. L., Corey M. J., Schwalbe R. A. (2006) FEBS J. 273, 3287–3300 [DOI] [PubMed] [Google Scholar]
- 18.Napp J., Monje F., Stühmer W., Pardo L. A. (2005) J. Biol. Chem. 280, 29506–29512 [DOI] [PubMed] [Google Scholar]
- 19.Watanabe I., Wang H. G., Sutachan J. J., Zhu J., Recio-Pinto E., Thornhill W. B. (2003) J. Physiol. 550, 51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrecca K., Atanasiu R., Akhavan A., Shrier A. (1999) J. Physiol. 515, 41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misonou H., Trimmer J. S. (2004) CRC Crit. Rev. Biochem. Mol. Biol. 39, 125–145 [DOI] [PubMed] [Google Scholar]
- 22.Vacher H., Mohapatra D. P., Trimmer J. S. (2008) Physiol. Rev. 88, 1407–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thornhill W. B., Wu M. B., Jiang X., Wu X., Morgan P. T., Margiotta J. F. (1996) J. Biol. Chem. 271, 19093–19098 [DOI] [PubMed] [Google Scholar]
- 24.Sutachan J. J., Watanabe I., Zhu J., Gottschalk A., Recio-Pinto E., Thornhill W. B. (2005) Brain Res. 1058, 30–43 [DOI] [PubMed] [Google Scholar]
- 25.Gong Q., Anderson C. L., January C. T., Zhou Z. (2002) Am. J. Physiol. Heart Circ. Physiol. 283, H77–H84 [DOI] [PubMed] [Google Scholar]
- 26.Mizoguchi A., Nakanishi H., Kimura K., Matsubara K., Ozaki-Kuroda K., Katata T., Honda T., Kiyohara Y., Heo K., Higashi M., Tsutsumi T., Sonoda S., Ide C., Takai Y. (2002) J. Cell Biol. 156, 555–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tretter V., Altmann F., März L. (1991) Eur. J. Biochem. 199, 647–652 [DOI] [PubMed] [Google Scholar]
- 28.Bennett E., Urcan M. S., Tinkle S. S., Koszowski A. G., Levinson S. R. (1997) J. Gen. Physiol. 109, 327–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett E. S. (2002) J. Physiol. 538, 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson D., Bennett E. S. (2008) Pflugers Arch. 456, 393–405 [DOI] [PubMed] [Google Scholar]
- 31.Ufret-Vincenty C. A., Baro D. J., Santana L. F. (2001) Am. J. Physiol. Cell Physiol. 281, C464–C474 [DOI] [PubMed] [Google Scholar]
- 32.Recio-Pinto E., Thornhill W. B., Duch D. S., Levinson S. R., Urban B. W. (1990) Neuron 5, 675–684 [DOI] [PubMed] [Google Scholar]
- 33.Liu J., Zhang M., Jiang M., Tseng G. N. (2002) J. Gen. Physiol. 120, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dun W., Jiang M., Tseng G. N. (1999) Pflugers Arch. 439, 141–149 [DOI] [PubMed] [Google Scholar]
- 35.Jiang M., Zhang M., Maslennikov I. V., Liu J., Wu D. M., Korolkova Y. V., Arseniev A. S., Grishin E. V., Tseng G. N. (2005) J. Physiol. 569, 75–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Long S. B., Tao X., Campbell E. B., MacKinnon R. (2007) Nature 450, 376–382 [DOI] [PubMed] [Google Scholar]
- 37.Maletic-Savatic M., Lenn N. J., Trimmer J. S. (1995) J. Neurosci. 15, 3840–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pongs O., Leicher T., Berger M., Roeper J., Bähring R., Wray D., Giese K. P., Silva A. J., Storm J. F. (1999) Ann. N.Y. Acad. Sci. 868, 344–355 [DOI] [PubMed] [Google Scholar]
- 39.Torres Y. P., Morera F. J., Carvacho I., Latorre R. (2007) J. Biol. Chem. 282, 24485–24489 [DOI] [PubMed] [Google Scholar]
- 40.Shi G., Nakahira K., Hammond S., Rhodes K. J., Schechter L. E., Trimmer J. S. (1996) Neuron 16, 843–852 [DOI] [PubMed] [Google Scholar]
- 41.Clancy S. M., Chen B., Bertaso F., Mamet J., Jegla T. (2009) PLoS ONE 4, e6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varki A. ( 1993) Glycobiology 3, 97– 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Helenius A., Aebi M. (2004) Ann. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 44.Vanoni O., Paganetti P., Molinari M. (2008) Mol. Biol., Cell 19, 4086–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hebert D. N., Zhang J. X., Chen W., Foellmer B., Helenius A. (1997) J. Cell Biol. 139, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu J., Recio-Pinto E., Hartwig T., Sellers W., Yan J., Thornhill W. B. (2009) Brain Res. 1251, 16–29 [DOI] [PubMed] [Google Scholar]
- 47.Landolt-Marticorena C., Reithmeier R. A. (1994) Biochem. J. 302, 253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.