

Abstract
Although terminal differentiation of intestinal epithelium is essential for the efficient digestion and absorption of nutrients, little is known about the molecular mechanisms underlying this process. Recent studies have shown that Elf3 (E74-like factor 3), a member of the ETS transcription factor family, has an essential role in the terminal differentiation of absorptive enterocytes and mucus-secreting goblet cells. Here, we demonstrated that Crif1 (CR6-interacting factor 1) functions as transcriptional coactivator of Elf3 in intestinal epithelium differentiation. The intestinal epithelium-specific Crif1-deficient mice died soon after birth and displayed severe alterations of tissue architecture in the small intestine, including poor microvillus formation and abnormal differentiation of absorptive enterocytes. Strikingly, these phenotypes are largely similar to that of Elf3-deficient mice, suggesting that Elf3 signaling in the intestinal epithelium depends on the Crif1 expression. We dissected this relationship further and found that Crif1 indeed interacted with Elf3 through its ETS DNA binding domain and enhanced the transcriptional activity of Elf3 by regulating the DNA binding activity. Knockdown of Crif1 by RNA interference conversely attenuated the transcriptional activity of Elf3. Consistently, the expression level of Tgf-βRII (transforming growth factor β type II receptor), a critical target gene of Elf3, was dramatically reduced in the Crif1-deficient mice. Our results reveal that Crif1 is a novel and essential transcriptional coactivator of Elf3 for the terminal differentiation of absorptive enterocytes.
Introduction
The surface of the small intestine is covered with a simple columnar epithelium with invaginations, known as the crypts of Liberkühn, as well as finger-like projections called villi. The upper two-thirds of the crypts are occupied by transiently proliferating cells. Upon exiting the cell cycle, these cells undergo differentiation into four different cell types. These cell types include enterocytes, which are absorptive cells, and three secretary cell types: mucus-secreting goblet cells, hormone-secreting enteroendocrine cells, and microbial peptide-secreting Paneth cells. Among them, enterocytes are the most abundant cell type. They form a tight epithelial monolayer along the crypt-villus axis of the intestine. They also serve as a permeability barrier between luminal contents and the blood supply as well as being a conduit for nutrient absorption and enzyme secretion (1–3).
Wnt/β-catenin signaling is necessary for stem cell renewal, proliferation, and differentiation (4). Notch signaling regulates the secretory/absorptive lineage commitment (4). Progenitors that express Hes1 will differentiate into absorptive enterocytes, whereas progenitors that express Math1 are committed to the secretary lineage as goblet, Paneth, or enteroendocrine cells (4, 5). Further commitment toward specific cell types is dependent on other transcription factors. For example, the activation of Ngn3, Beta2, Pax4, Pax6, Pdx1, Neurod1, and Gfi1 is associated with the enteroendocrine lineage (6, 7). Wnt signaling and Sox9 are required for the differentiation of Paneth cells, and the differentiation of goblet cells is influenced by Klf4 (8–10). Although the molecular mechanisms of the differentiation to secretory lineages have been well known, specification and differentiation mechanisms to enterocytes remain to be elucidated besides the transcriptional factor Elf3 (E74-like factor 3; also known as Ese-1, Esx, Ert, and Jen) (11).
Elf3 is a member of the ETS transcription factor family, which is characterized by a highly conserved DNA binding domain, known as the ETS domain (12). Inactivation of Elf3 leads to fetal death (11). One-third of null fetuses die around embryonic day 11.5, whereas the rest of mutant embryos survive to birth and exhibit severe alterations in the cellular architecture of the small intestine, including poor villus formation and defective terminal differentiation of mucus-secreting goblet cells and absorptive enterocytes. Moreover, enterocytes from Elf3-deficient embryos show reduced expression of Tgf-βRII (transforming growth factor β type II receptor), a potent inhibitor of cell proliferation and an inducer of cell differentiation (11). Recent studies reported that Elf3 activates the TGF-βRII promoter by binding to two overlapping ETS sites (13, 14). Intriguingly, the expression of the TGF-βRII transgene rescues the intestinal defects of Elf3-deficient mice, demonstrating that TGF-βRII is a critical downstream mediator of Elf3.
Crif1 (CR6-interacting factor 1), also termed Gadd45gip1 (Gadd45 γ-interacting protein 1), is a nuclear protein that interacts with members of the Gadd45 protein family, Nur77, androgen receptor and has been suggested as a potential regulator of cell cycle progression and cell growth (15–17). Recently, we identified that Crif1 is essentially required for early embryogenesis and cellular transformation activity of STAT3 as a transcriptional coactivator of STAT3 (18). However, the tissue-specific function of Crif1 remains to be identified. In this study, we found that conditional inactivation of Crif1 in intestinal epithelial cells using the Cre-LoxP system resulted in perinatal lethality and severe alterations in the cellular architecture of the small intestine, including poor microvilli formation and defective terminal differentiation of absorptive enterocytes, which are similar to the phenotypes of Elf3 knock-out mice (11). We found that Crif1 readily interacted with Elf3 through the ETS DNA binding domain and co-localized with Elf3 in the nucleus. In addition, knockdown experiments with Crif1 siRNA2 revealed that endogenously expressed Crif1 is required for the DNA binding activity of Elf3. Furthermore, the expression of Tgf-βRII was markedly decreased in the Crif1-null mice. Thus, our findings reveal Crif1 to be an essential transcriptional coactivator of Elf3 that is required for the terminal differentiation of absorptive enterocytes.
EXPERIMENTAL PROCEDURES
Mice
Villin-Cre (Vil-Cre) mice were purchased from Jackson Laboratories and maintained in our animal colony under institutional guidelines. Crif1f/f mice (18) were crossed with the Vil-Cre mice to generate Vil-Cre;Crif1f/f mice. All of the mouse experiments were performed in the animal facility under POSTECH institutional guidelines.
Tissue Preparation and Immunohistochemistry
The intestinal tract was flushed gently with cold phosphate-buffered saline, followed by a flush with 4% paraformaldehyde in phosphate-buffered saline. For histological analysis, tissues were fixed in 4% paraformaldehyde overnight at 4 °C and embedded in paraffin wax for sectioning. The sections (3–4 μm) were stained with hematoxylin and eosin. For immunohistochemistry, paraffin-embedded sections were rehydrated, and the antigenic epitopes were exposed using a 10 mm citrate buffer and microwaving. Sections were incubated in a blocking solution (3% bovine serum albumin, 5% goat serum or horse serum, and 0.5% Tween 20 in phosphate-buffered saline) at room temperature for 4 h, followed by an additional incubation with antibodies to villin (1:100; Chemicon), intestinal fatty acid-binding protein (1:100; gift from Dr. J. I. Gordon), Ki-67 (1:300; Novo Castra), and E-cadherin (1:200; BD Biosciences). Specific binding was detected with an Alexa-488-labeled (green) and/or Alexa-594-labeled (red) antibody (Molecular Probes).
Luciferase Assay
For the luciferase assay, HCT116 and DLD-1 cells were transiently transfected using the Lipofectamine PLUS reagent (Invitrogen) with 0.5 μg of plasmid DNA (mTgf-βRII (−108/+56)-Luc), with control pcDNA3, HA-Crif1, GFP-Elf3a, mock siRNA (Bioneer), or hCrif1 siRNA in each well of 12-well plates. The synthetic hCrif1 siRNA duplex was prepared to the following target region: 5′-AAGAACGCGAAUGGUACCCGA-3′. The Renilla reporter construct pRL-TK (Promega) was used to normalize the transfection efficiency. The cells were incubated for 36 h in DMEM containing 10% FBS and were then harvested. Luciferase activity was determined using the dual luciferase reporter assay system (Promega).
Chromatin Immunoprecipitation (ChIP) Analysis and Re-ChIP Analysis
ChIP analyses of the HCT116 cells were performed using the EZ-Chip kit according to the manufacturer's protocol (Upstate Biotechnology). Cells were co-transfected using Lipofectamine PLUS reagent (Invitrogen) with the promoter/reporter vector mTgf-βRII (−108/+56) and a FLAG-tagged Elf3a expression vector and HA-Crif1. Immunoprecipitation was done with the anti-FLAG antibodies. Specific PCR primers were designed to contain a putative Ets family transcription factor-binding motif. The FOR primer (5′-CGCTCTCCATCAAAACAAAA-3′), which is specific for a sequence located in the plasmid backbone, and the REV primer (5′-ACCTCACTCATCAACTTTACCC-3′), which is specific for a sequence within the −108/+56 region of mTgf-βRII, were used to amplify the mTgf-βRII promoter region located in the transfected promoter/reporter construct. Real-time PCR was performed with a MyiQ single color real-time PCR detection system (Bio-Rad) with iQ SYBR Green Supermix (Bio-Rad).
Re-ChIP analyses were carried out as described previously (19). Briefly, protein-DNA complexes after the first ChIP with anti-FLAG antibodies were washed with washing buffer and Tris-EDTA buffer. Complexes were eluted by incubation for 30 min at 37 °C in elution buffer (1× TE containing 2% SDS, 15 mm dithiothreitol, and protease inhibitors). After centrifugation, the supernatant was diluted 30 times with dilution buffer (50 mm Tris, 5 mm EDTA, 200 mm NaCl, 0.5% Nonidet P-40) supplement with 50 μg of bovine serum albumin and protease inhibitors. The samples were then subjected to the ChIP procedure with anti-HA antibodies.
In Situ Hybridization
Details of the RNA in situ hybridizations on whole mount embryos have been described (20). Antisense digoxigenin-labeled riboprobes were generated from pGEM-T vectors (Promega) containing amplified mCrif1 cDNA fragments (18).
Western Blot Analysis and Co-immunoprecipitation Assays
Cultured cells were resuspended in an immunoprecipitation buffer (50 mm HEPES/NaOH (pH 7.5), 3 mm EDTA, 3 mm CaCl2, 80 mm NaCl, 1% Triton X-100, 5 mm dithiothreitol). Generally, the protein in the supernatants (25–40 μg) was separated by size, blotted with primary and secondary antibodies, and visualized with ECL Plus (Amersham Biosciences). The primary antibodies used were as follows: anti-Crif1 (18), anti-GFP (Invitrogen), anti-HA, anti-FLAG, anti-Tgf-βRII, and anti-actin (Santa Cruz Biotechnology). Immunoprecipitation was performed as described previously (20).
RESULTS
The Expression of Crif1 in the Intestinal Epithelium and Its Disruption in Vil-Cre;Crif1f/f Mutant Intestine
During early development, Crif1 was expressed in the inner cell mass region of blastocysts, and embryonic and extraembryonic tissues of embryonic day 7.5 (E7.5) embryos (18), whereas it was ubiquitously expressed in adult human tissues (15). To characterize the spatial expression pattern of Crif1 in late developmental stage, we performed a whole mount in situ hybridization using E18.5 embryos. Crif1 mRNA is expressed ubiquitously but highly in the brain and intestinal epithelium (Fig. 1A). Examination at high magnification revealed that Crif1 is strongly expressed in the intestinal epithelium (Fig. 1B).
FIGURE 1.

Conditional inactivation of the Crif1 locus in the mouse intestine. A, whole mount in situ hybridization of a wild-type E18.5 embryo. Note that Crif1 is highly expressed in the brain and intestinal epithelium. B, higher magnification image of the boxed region in A. C, the relative abundance of Crif1 transcripts in E18.5 embryos was evaluated using quantitative real-time RT-PCR. The Crif1 mRNA expression upon Cre expression driven by the rat villin promoter was seen in two independent embryos. β-Actin was used for normalization. Error bars, S.D. D, duodenum; J, jejunum; I, ileum. D, genotype analysis of the progeny from Vil-Cre;Crif1+/f and Crif1f/f mating. P1 and P21, postnatal day 1 and 21, respectively.
To examine the biological function of Crif1 in the intestinal development, we disrupted the Crif1 gene in the intestinal epithelium by crossing Crif1flox mice with Vil-Cre transgenic mice (18, 21). The villin promoter directs gene expression in all epithelial cells of the intestine starting on E12.5 (21). The disruption of the Crif1 gene was verified using quantitative real time RT-PCR (Fig. 1C). Vil-Cre;Crif1+/f mice showed no apparent differences when compared with their wild-type littermates. When they were mated with Crif1f/f mice, only 2 of 157 offspring were Vil-Cre;Crif1f/f mice at postnatal day 21, and only 6 of 52 were Vil-Cre;Crif1f/f mice at postnatal day 1 (Fig. 1D). Moreover, only 16 of 83 Vil-Cre;Crif1f/f embryos survived to E18.5. This represented 77% of the expected frequency (Fig. 1D). These data show that Crif1 deficiency leads to perinatal lethality and suggest that Crif1 may have a critical role in intestinal development.
Dysmorphogenesis of the Small Intestine in Vil-Cre;Crif1f/f Mice
Because the Crif1 is expressed in the intestinal epithelium and the Vil-Cre;Crif1f/f mice exhibited a perinatal lethal phenotype, we performed a histological analysis using the small intestine from Vil-Cre;Crif1f/f embryos at E18.5. The entire gut lumen of wild-type embryos was filled with numerous well developed villi (Fig. 2A). In contrast, much shorter, fewer, and abnormally shaped villi were present in the small intestine of Vil-Cre;Crif1f/f embryos (Fig. 2B). Intriguingly, the small intestine of Vil-Cre;Crif1f/f embryos showed a peculiar protrusion or tufting of the apical brush border of the absorptive enterocytes.
FIGURE 2.

The small intestine of Vil-Cre;Crif1f/f embryos display abnormal morphogenesis. Paraffin sections of the small intestine from E18.5 Vil-Cre;Crif1+/f (A and C) or Vil-Cre;Crif1f/f (B and D) embryos were stained with hematoxylin and eosin. The arrows indicate the tufted apical brush border (D). Original magnification was ×100 (A and B) and ×400 (C and D).
Examination at high magnification revealed that the absorptive enterocytes lining the villi were markedly disturbed in the Vil-Cre;Crif1f/f intestines (Fig. 2D). In the wild-type small intestine, absorptive enterocytes were highly polarized and acquired a tall, columnar cell shape. In addition, the basally located nucleus and a uniform cytoplasm were seen in the absorptive enterocytes (Fig. 2C). In contrast, the Crif1-deficient enterocytes had irregular cell shape and size, heterogeneously stained cytoplasm, and disorganized cellular nuclei (Fig. 2D). The peculiar protrusion or tufting of the apical cell membrane in a dome-like manner was also seen in the small intestine of Vil-Cre;Crif1f/f embryos (Fig. 2D, arrows). These results suggest that Crif1 is required for proper development of absorptive enterocytes.
Abnormal Differentiation of Small Intestinal Epithelium in Vil-Cre;Crif1f/f Mice
For detailed analysis of Crif1-deficient enterocytes, we performed an immunohistochemical analysis with anti-villin antibody. Villin is a cytoskeletal protein of the microvilli found on the brush border of epithelial cells (22). Enterocytes in wild-type embryos have uniform villin localization on the brush border (Fig. 3A). In contrast, villin expression was dramatically reduced in Vil-Cre;Crif1f/f enterocytes (Fig. 3B). Despite a drastic reduction of villin expression, the Vil-Cre;Crif1f/f enterocytes showed relatively intact FABP expression, which is expressed only in intestinal enterocytes (Fig. 3, C and D). These results suggest that the Vil-Cre;Crif1f/f enterocytes have a defect in final differentiation step, whereas they retained their epithelial characteristics.
FIGURE 3.

Abnormal differentiation of absorptive enterocytes in Vil-Cre;Crif1f/f embryos. A–D, immunohistochemical analysis of the small intestine from E18.5 Vil-Cre;Crif1+/f (A and C) or Vil-Cre;Crif1f/f (B and D) embryos. Paraffin sections were immunostained with anti-villin (A and B, in red) and anti-intestinal fatty acid-binding protein antibodies (C and D, in red). Original magnification was ×200 (A–D). E and F, transmission electron photomicrographs of microvilli on the apical cell membranes. Note that Vil-Cre;Crif1f/f intestines have abnormal sizes and numbers of microvilli (F). Bars, 0.2 μm.
To further examine the status of microvilli at the apical membrane of enterocytes, we performed an ultrastructural examination using transmission electron microscopy. In wild-type enterocytes, the microvilli were uniform and densely packed. In contrast, Vil-Cre;Crif1f/f enterocytes contained fewer, shorter microvilli at the apical membrane (Fig. 3, E and F). Taken together, our results show that Vil-Cre;Crif1f/f embryos exhibit various defects, such as severe alterations in cellular architecture, poor villus formation, and defective terminal differentiation of enterocytes, whereas they retained intestinal enterocyte characteristics, which is similar to characteristic defects in Elf3-deficnent mice (11).
Elf3 Interacts and Co-localizes with Crif1
Because Vil-Cre;Crif1f/f embryos showed phenocopy of Elf3-deficient mice, we first sought to determine whether Crif1 interacts with Elf3. There are two forms of full-length Elf3, Elf3a and Elf3b. Elf3b contains a 20-amino acid insertion upstream of the pointed domain (23). However, functional differences between the variants remain unknown. GFP-tagged Elf3a or GFP-tagged Elf3b was co-transfected with HA-tagged Crif1 into HEK 293 cells. Lysates were immunoprecipitated with an anti-HA antibody and then immunoblotted with an anti-GFP antibody to detect Elf3. Both isoforms of Elf3 interacted readily with Crif1 (Fig. 4, A and B). To identify which domain of Elf3 is responsible for Crif1 binding, we used three GFP-Elf3a deletion mutants (Fig. 4A). Immunoprecipitation experiments revealed that Crif1 interacts with the C-terminal ETS DNA binding domain of Elf3a (Fig. 4C).
FIGURE 4.

Interaction and co-localization of Crif1 with Elf3. A, schematic representation of the Elf3 wild-type and deletion mutants, fused to the GFP. The pointed domain, transactivation domain (TAD), serine-aspartic acid-rich domain (SAR), AT-hook domain, and ETS domain are indicated. B and C, interaction between Crif1 and Elf3. GFP-tagged Elf3a and GFP-tagged Elf3b plasmids were each co-transfected with HA-tagged Crif1 into HEK 293T cells and then immunoprecipitated with an anti-HA antibody (B). GFP-tagged Elf3a deletion mutants were co-transfected with HA-tagged Crif1 into HEK293T cells and then immunoprecipitated with anti-HA antibody (C). Total cell extracts and immunoprecipitates (IP) were blotted (IB) with anti-HA and anti-GFP antibodies. D, Crif1 and Elf3 co-localize in the nucleus as a speckle. GFP-tagged Elf3a and GFP-tagged Elf3b plasmids were each co-transfected with HA-tagged Crif1 into COS-7 cells and then stained with anti-HA antibody to detect Crif1 expression. Original magnification was ×400 (D).
Because both Elf3 and Crif1 localize exclusively in nuclei and are predominantly concentrated in dotlike structures (24, 25), we next examined whether they co-localize in the nucleus. The GFP-tagged Elf3a or GFP-tagged Elf3b gene was co-transfected with HA-tagged Crif1 into COS-7 cells. As expected, the merged pictures showed the co-localization of both proteins in the nucleus with a prominent speckled pattern (Fig. 4D). These results show that the Elf3 interacts and co-localizes with Crif1.
Crif1 Enhances the Transcriptional Activity of Elf3 by Modulating Its DNA Binding Activity
The ETS DNA binding domain is known to direct not only protein-DNA interactions but also protein-protein interactions. These interactions regulate DNA binding, target gene selection, and transcriptional activity of ETS proteins (26). Since Crif1 interacts with the ETS DNA binding domain of Elf3, we speculated that it might affect the transcriptional activity of Elf3. To examine this possibility, we performed reporter gene assays using the promoter/reporter gene construct mTgf-βRII −108/+56, which contains the mouse Tgf-βRII promoter region (13). Crif1 alone slightly enhanced the Elf3-mediated transcriptional activity in HCT116 and DLD-1 human colon cancer cell lines. However, when Crif1 was co-expressed with full-length Elf3a, Crif1 substantially increased the Elf3-mediated transcriptional activity in both cell lines (Fig. 5, A and B).
FIGURE 5.

Crif1 enhances the transcriptional activity of Elf3. A and B, enhanced transcriptional activity of Elf3a by Crif1. HCT116 cells (A) and DLD-1 cells (B) were co-transfected with the Crif1 expression vector and the Elf3-responsive mTgf-βRII (−108/+56)-luciferase construct in the absence of a FLAG-tagged Elf3a expression vector. Luciferase activity was measured 36 h after transfection. C, the DNA binding activity of Elf3a by Crif1. HCT116 cells were co-transfected with the promoter/reporter construct Tgf-βRII (−108/+56), a FLAG-tagged Elf3a, and HA-tagged Crif1 plasmids. Soluble chromatin prepared from each transfectant was immunoprecipitated (IP) with an anti-FLAG antibody. The final DNA extracts were amplified using pairs of primers that cover the Elf3a binding sites in the mTgf-βRII (−108/+56) construct. D, co-occupancy of Crif1 with Elf3 on the mTgf-βRII promoter. Elf3-bound chromatin was subjected to a second round of immunoprecipitation, using anti-HA antibodies or nonspecific IgG and analyzed by real-time PCR. A representative of three independent experiments is shown. Error bars, S.D. Significant differences are as follows: *, p < 0.002; **, p < 0.002.
Because Crif1 resides in the nucleus and enhances the transcriptional activity of Elf3, it may affect the DNA binding activity of Elf3. To examine this possibility, HCT116 cells were transiently transfected with the mTgf-βRII −108/+56 promoter/reporter gene construct and FLAG-tagged Elf3a expression vector. Consistent with a previous report, Elf3 alone could bind to the mTgf-βRII promoter (13). However, Crif1 enhanced the occupancy of Elf3 on the mTgf-βRII promoter in a dose-dependent manner (Fig. 5C). To address whether Crif1 is also recruited to the mTgf-βRII promoter, we performed a re-ChIP assay. Protein-DNA complexes from the first immunoprecipitation with anti-FLAG antibodies were subjected to an additional immunoprecipitation with anti-HA antibodies or nonspecific IgGs (Fig. 5D). This result clearly revealed that both Crif1 and Elf3 were present at mTgf-βRII promoter. Therefore, Crif1, as a component of Elf3 complex, enhances the transcriptional activity of Elf3 by modulating its DNA binding activity.
Although Crif1 significantly enhanced the transcriptional activity of Elf3, Elf3 alone readily activated promoter activity of Tgf-βRII. This suggests that endogenous Crif1 may have a role in the transcriptional activity of Elf3. To examine this possibility, we used siRNA to knockdown hCrif1 in HCT116 cells and then performed the Elf3 reporter assay. The expression levels of Crif1 mRNA were dramatically decreased by hCrif1 siRNA (Fig. 6A, bottom). As expected, the transcriptional activity of Elf3a was significantly decreased with hCrif1 siRNA treatment compared with the control siRNA treatment (∼0.6-fold) (Fig. 6A, top). This suggests that the transcriptional activity of Elf3 is dependent on endogenous Crif1 expression.
FIGURE 6.
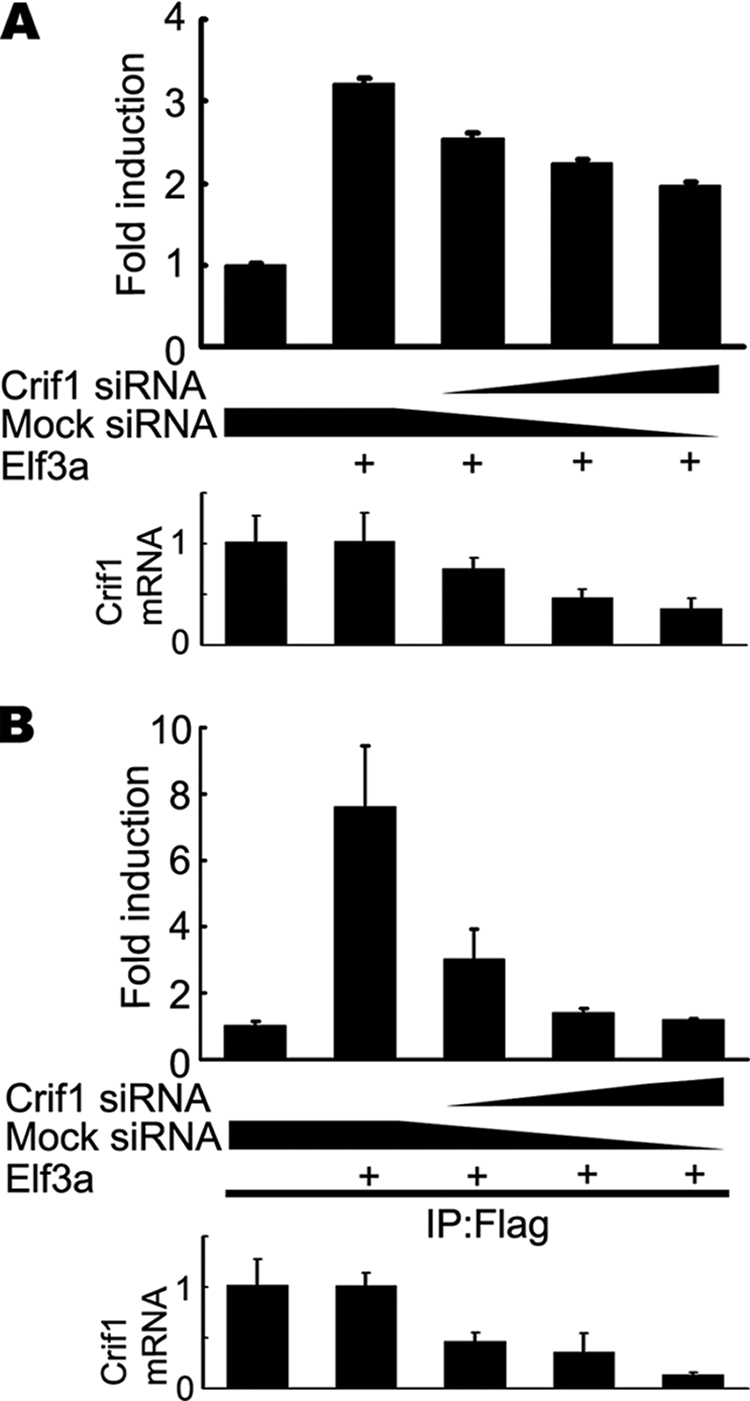
Crif1 is required for the DNA binding activity of Elf3. A, decreased transcriptional activity of Elf3a by Crif1 siRNA. HCT116 cells were co-transfected with the Crif1 siRNA duplex, the Elf3-responsive mTgf-βRII (−108/+56)-luciferase construct, and the FLAG-tagged Elf3a expression vector. Luciferase activity was measured 36 h after transfection (top). Endogenously expressed Crif1 mRNA levels were measured by real-time RT-PCR after transfection of the Crif1 siRNA duplex (bottom). B, decreased DNA binding activity of Elf3a by Crif1 siRNA. HCT116 cells were co-transfected with the Crif1 siRNA duplex and the promoter/reporter construct Tgf-βRII (−108/+56). They were then co-transfected with the FLAG-tagged Elf3a expression vector. Soluble chromatin prepared from each transfectant was immunoprecipitated (IP) with the anti-FLAG antibody. The final DNA extracts were amplified using pairs of primers which cover the Elf3a binding sites in the mTgf-βRII (−108/+56) construct (top). Endogenously expressed Crif1 mRNA levels were measured by real-time RT-PCR after transfection with the Crif1 siRNA duplex (bottom). A representative of three independent experiments is shown. Error bars, S.D. Significant differences are as follows: *, p < 0.01; **, p < 0.002.
To further examine the role of Crif1 on the DNA binding activity of Elf3, we co-transfected HCT116 cells with FLAG-tagged Elf3a and mTgf-βRII promoter/reporter constructs along with siRNA for hCrif1. Thirty-six hours after transfection, cross-linked chromatin derived from HCT116 cells was immunoprecipitated with anti-FLAG antibody. Intriguingly, the recruitment of Elf3 onto the mTgf-βRII promoter was completely abolished in the hCrif1 siRNA-treated HCT116 cells (Fig. 6B). These results show that Crif1 plays a critical role in the DNA binding activity of Elf3 as a transcriptional co-activator.
Decreased Expression of Tgf-βRII in the Vil-Cre;Crif1f/f Epithelia
Given that Elf3 transactivates the Tgf-βRII promoter, and Elf3-deficient epithelia showed reduced expression of Tgf-βRII, we examined whether Vil-Cre;Crif1f/f epithelia also show a reduced expression of Tgf-βRII. As expected, a Western blot analysis with lysates from the small intestine of E18.5 Vil-Cre;Crif1f/f embryos revealed that the expression of Tgf-βRII was markedly reduced compared with that of wild-type embryos (Fig. 7A), suggesting that Crif1 is also required for the proper expression of the Elf3 target gene Tgf-βRII in vivo. We next examined whether the expression of Elf3 is affected in the Vil-Cre;Crif1f/f mice. The expression level of Elf3 mRNA in the small intestine of Vil-Cre;Crif1f/f mice was assessed by quantitative real-time RT-PCR. The expression levels of Elf3 mRNA were not affected by the deletion of the Crif1 gene (Fig. 7B), indicating that the differentiation defect of enterocytes in the Vil-Cre;Crif1f/f mice is not due to the reduction of Elf3 expression. Taken together, our results show that Crif1 is required for the transcription of Tgf-βRII.
FIGURE 7.

Reduced expression of Tgf-βRII in Vil-Cre;Crif1f/f embryos. A, Western blot analysis of the total protein lysates from the jejunum of Vil-Cre;Crif1+/f or Vil-Cre;Crif1f/f embryos with anti-Tgf-βRII antibody and anti-Crif1 antibody. Note that the expression levels of both precursor (∼60 kDa; arrow) and mature (∼70 kDa; arrowhead) forms of Tgf-βRII were reduced in the Crif1-deficient small intestine. β-Actin was used for normalization. A representative of two independent experiments is shown. B, the relative abundance of the Elf3 transcript in E18.5 Vil-Cre;Crif1f/f embryos was evaluated using quantitative real-time RT-PCR. Note that the expression level of Elf3 mRNA was not affected by the deletion of the Crif1 gene. β-Actin was used for normalization. Error bars, S.D. D, duodenum; J, jejunum; I, ileum. A representative of three independent experiments is shown.
Normal Goblet Cell Differentiation and Decreased Crypt Cell Proliferation in Vil-Cre;Crif1f/f Mice
Since Elf3-deficient embryos showed a defect in the maturation of sialomucin-containing goblet cells (11), we also analyzed the differentiation of the sialomucin- and sulfomucin-containing goblet cell lineages. Alcian blue staining, used to identify the distinct acid mucin subgroups, revealed that the number of goblet cells producing either sialomucins or sulfomucins was not changed in the Vil-Cre;Crif1f/f embryos (Fig. 8, A and B). These results suggest that Crif1 is not implicated in the differentiation of the goblet cell lineages with Elf3.
FIGURE 8.

Normal differentiation of acid mucin-producing goblet cells and normal crypt cell proliferation in Vil-Cre;Crif1f/f embryos. A and B, serial small intestinal sections were stained with Alcian blue, pH 2.5 or pH 1.0, to identify sialomucin- or sulfomucin-containing goblet cells, respectively. The numbers of sialomucin-containing (A) and sulfomucin-containing goblet cells (B) were then counted and expressed as relative proportions of the total number of villus epithelial cells counted. C and D, paraffin sections from Vil-Cre;Crif1+/f or Vil-Cre;Crif1f/f embryos were analyzed using Ki-67 (red) and E-cadherin (green) double immunostaining. Original magnification was ×200. E, Ki-67-positive cells were counted and expressed as a relative proportion of the total number of crypt cells counted. Error bars, S.D. values. Significant difference is as follows: *, p < 0.054.
We further examined whether crypt cells in the Vil-Cre;Crif1f/f intestines have intact proliferation activity as in the Elf3-deficient mice (11). The proliferation marker Ki-67 was co-immunostained with E-cadherin, an epithelial cell marker. Quantification of the number of cells staining positive for Ki-67 revealed a slight decrease (∼10.7%) in Crif1-deficient embryos (Fig. 8E). Decreased proliferation of crypt cells in Crif1-deficient embryos might result in much shorter and fewer villi in Crif1-deficient embryos.
DISCUSSION
Our results from the Vil-Cre;Crif1f/f embryos show that Crif1 has an essential role in the maturation of intestinal epithelium. Strikingly, Crif1-deficient embryos show largely phenocopy of Elf3-deficient embryos, suggesting that Crif1 participates in the regulation of Elf3 signaling. We have dissected this relationship further and identified that Crif1 is a novel and essential transcriptional coactivator of Elf3 by modulating its DNA binding ability.
The highly conserved ETS domain contains a winged helix-turn-helix structural fold that binds DNA as a monomer. The ETS domain confers sequence-specific DNA binding to sites containing an ETS binding site (27). In addition, the ETS domain is also a target for protein-protein interactions that are mediated either intramolecularly or in trans by co-regulatory proteins (28). Dozens of binding partners for the ETS domain have been discovered. These interactions have important regulatory consequences, such as the regulation of DNA binding, subcellular localization, target gene selection, and transcriptional activity of ETS proteins (28). In this study, we found that Crif1 interacts with the ETS DNA binding domain of Elf3. Runt-domain proteins (e.g. AML-1/CBFα2), Pax proteins (e.g. Pax-5), basic helix-loop-helix proteins (e.g. USF-1), and GABPβ enhance DNA binding ability and transcriptional activity of ETS proteins by binding the ETS domain (28). Similarly, Crif1 enhances the transcriptional activity and DNA binding ability of Elf3. The siRNA-induced knockdown of Crif1 reduced the transcriptional activity and DNA binding ability of Elf3 in the human colon cancer cell lines. Since Crif1 itself does not contain a DNA binding motif, it might stabilize the binding of Elf3 by forming a large transcription complex or making the epitope more accessible for DNA binding. Indeed, we found that Crif1 is recruited onto the mTgf-βRII promoter with Elf3 complex. These findings suggest that Crif1 acts as a transcriptional co-activator of Elf3 by modulating its DNA binding activity via the ETS domain.
Elf3-deficient mice showed intestinal abnormalities, such as poor differentiation of absorptive enterocytes and goblet cells (11). The Crif1-null small intestines also displayed severe alterations of the tissue architecture, including a tufted appearance of the absorptive enterocytes and poor microvilli formation. However, the number of goblet cells that produce sialomucins was not affected by the deletion of the Crif1 gene, whereas the Elf3-null small intestines showed a reduced number of sialomucin-producing goblet cells (11, 29). The discrepancy in the generation of sialomucin-producing goblet cells might be due to a different requirement of co-regulators in the transcriptional activity of Elf3: Crif1 for the differentiation of enterocytes and unknown co-regulator for the differentiation of sialomucin-producing goblet cells.
We previously reported that Crif1 is a novel transcriptional co-activator of STAT3 and that Crif1 specifically interacts with STAT3 but not with STAT1 or STAT5a (18). STAT3 plays crucial roles in early embryogenesis as well as in a broad spectrum of biological activities, including cytokine-induced responses, differentiation, cell growth, and antiapoptosis (30, 31). However, despite the clear importance of STAT3 during very early development, the ablation of STAT3 in adult tissues leads to surprisingly mild phenotypes. Mice with interferon γ-inducible disruption of STAT3 genes in both macrophages and intestinal epithelial cells develop enterocolitis. However, pathological changes in the small intestine were not detected (32). Thus, intestinal abnormalities in Vil-Cre;Crif1f/f mice are due to the defective transcriptional activity of Elf3 but not STAT3.
Elf3 has previously been shown to bind to the positive regulatory element 2 of the TGF-βRII gene promoter and activate its transcription in vitro (14). A reduction in TGF-βRII gene expression was achieved in a colon cancer cell line by the exogenous addition of antisense Elf3 RNA. In addition, the overexpression of TGF-βRII genetically corrected the abnormal development of the small intestinal epithelium in Elf3-deficient mice (29). TGF-βRII has been found to be a tumor suppressor gene in a variety of human tumor types, including colon, gastric, and pancreatic cancer (33). In this study, we found that Vil-Cre;Crif1f/f mice showed reduced expression of Tgf-βRII in the intestinal epithelium. This result suggests that Crif1 may have tumor suppressor properties by regulating Elf3-mediated TGF-βRII gene expression. However, we previously demonstrated that Crif1 has an indispensable role in cellular transformation by the oncogenic activation of STAT3 in mouse embryonic fibroblast cell lines (18). Interestingly, controversial properties of Elf3 in carcinogenesis have also been reported. Elf3 expression alone confers a transformed and in vitro metastatic phenotype to the normal breast epithelium cell line MCF-12A (34). Thus, Elf3 may act as a proto-oncogene in certain carcinomas while behaving like a tumor suppressor in other carcinomas. Similar to Elf3, Crif1 may function as a proto-oncogene or a tumor suppressor in a cellular context-dependent manner.
In summary, our results demonstrate a novel and essential function of Crif1 in the development of intestinal epithelium. The remarkable similarities between the Crif1-null and Elf3-null embryonic phenotypes lead us to identify Crif1 as a novel transcriptional coactivator of Elf3 in intestinal development.
Acknowledgments
We thank the members of Professor Kong's laboratory for discussions; Drs. M. Shong, H. S. Lim, J. K. Han, and J. M. Kim for critical comments; and M. P. Kong and H. S. Hwang for technical support.
This work was supported by Center for Biological Modulators of the 21C Frontier R&D Program Grant CBM32-A2300-01-00-01, Korea Science and Engineering Foundation Grant 2009-0062800, 21C Frontier Functional Human Genome Project Grant FG06-42-1 from the Ministry of Science and Technology of Korea, and Basic Science Research Program Grant 2009-0079371 through the National Research Foundation of Korea.
- siRNA
- small interfering RNA
- ChIP
- chromatin immunoprecipitation
- HA
- hemagglutination antigen
- GFP
- green fluorescent protein
- En
- embryonic day n
- GFP
- green fluorescent protein
- RT
- reverse transcription.
REFERENCES
- 1.Reya T., Clevers H. (2005) Nature 434, 843–850 [DOI] [PubMed] [Google Scholar]
- 2.Mills J. C., Gordon J. I. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 12334–12336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bach S. P., Renehan A. G., Potten C. S. (2000) Carcinogenesis 21, 469–476 [DOI] [PubMed] [Google Scholar]
- 4.Sancho E., Batlle E., Clevers H. (2004) Annu. Rev. Cell Dev. Biol. 20, 695–723 [DOI] [PubMed] [Google Scholar]
- 5.Yang Q., Bermingham N. A., Finegold M. J., Zoghbi H. Y. (2001) Science 294, 2155–2158 [DOI] [PubMed] [Google Scholar]
- 6.Schonhoff S. E., Giel-Moloney M., Leiter A. B. (2004) Endocrinology 145, 2639–2644 [DOI] [PubMed] [Google Scholar]
- 7.Shroyer N. F., Wallis D., Venken K. J., Bellen H. J., Zoghbi H. Y. (2005) Genes Dev. 19, 2412–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mori-Akiyama Y., van den Born M., van Es J. H., Hamilton S. R., Adams H. P., Zhang J., Clevers H., de Crombrugghe B. (2007) Gastroenterology 133, 539–546 [DOI] [PubMed] [Google Scholar]
- 9.Gregorieff A., Clevers H. (2005) Genes Dev. 19, 877–890 [DOI] [PubMed] [Google Scholar]
- 10.Katz J. P., Perreault N., Goldstein B. G., Lee C. S., Labosky P. A., Yang V. W., Kaestner K. H. (2002) Development 129, 2619–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng A. Y., Waring P., Ristevski S., Wang C., Wilson T., Pritchard M., Hertzog P., Kola I. (2002) Gastroenterology 122, 1455–1466 [DOI] [PubMed] [Google Scholar]
- 12.Hsu T., Trojanowska M., Watson D. K. (2004) J. Cell. Biochem. 91, 896–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim J. H., Wilder P. J., Hou J., Nowling T., Rizzino A. (2002) J. Biol. Chem. 277, 17520–17530 [DOI] [PubMed] [Google Scholar]
- 14.Choi S. G., Yi Y., Kim Y. S., Kato M., Chang J., Chung H. W., Hahm K. B., Yang H. K., Rhee H. H., Bang Y. J., Kim S. J. (1998) J. Biol. Chem. 273, 110–117 [DOI] [PubMed] [Google Scholar]
- 15.Chung H. K., Yi Y. W., Jung N. C., Kim D., Suh J. M., Kim H., Park K. C., Song J. H., Kim D. W., Hwang E. S., Yoon S. H., Bae Y. S., Kim J. M., Bae I., Shong M. (2003) J. Biol. Chem. 278, 28079–28088 [DOI] [PubMed] [Google Scholar]
- 16.Park K. C., Song K. H., Chung H. K., Kim H., Kim D. W., Song J. H., Hwang E. S., Jung H. S., Park S. H., Bae I., Lee I. K., Choi H. S., Shong M. (2005) Mol. Endocrinol. 19, 12–24 [DOI] [PubMed] [Google Scholar]
- 17.Suh J. H., Shong M., Choi H. S., Lee K. (2008) Mol. Endocrinol. 22, 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwon M. C., Koo B. K., Moon J. S., Kim Y. Y., Park K. C., Kim N. S., Kwon M. Y., Kong M. P., Yoon K. J., Im S. K., Ghim J., Han Y. M., Jang S. K., Shong M., Kong Y. Y. (2008) EMBO J. 27, 642–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bararia D., Trivedi A. K., Zada A. A., Greif P. A., Mulaw M. A., Christopeit M., Hiddemann W., Bohlander S. K., Behre G. (2008) Leukemia 22, 800–807 [DOI] [PubMed] [Google Scholar]
- 20.Koo B. K., Lim H. S., Song R., Yoon M. J., Yoon K. J., Moon J. S., Kim Y. W., Kwon M. C., Yoo K. W., Kong M. P., Lee J., Chitnis A. B., Kim C. H., Kong Y. Y. (2005) Development 132, 3459–3470 [DOI] [PubMed] [Google Scholar]
- 21.Madison B. B., Dunbar L., Qiao X. T., Braunstein K., Braunstein E., Gumucio D. L. (2002) J. Biol. Chem. 277, 33275–33283 [DOI] [PubMed] [Google Scholar]
- 22.Khurana S., George S. P. (2008) FEBS Lett. 582, 2128–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tymms M. J., Ng A. Y., Thomas R. S., Schutte B. C., Zhou J., Eyre H. J., Sutherland G. R., Seth A., Rosenberg M., Papas T., Debouck C., Kola I. (1997) Oncogene 15, 2449–2462 [DOI] [PubMed] [Google Scholar]
- 24.Görnemann J., Hofmann T. G., Will H., Müller M. (2002) Virology 303, 69–78 [DOI] [PubMed] [Google Scholar]
- 25.Do H. J., Song H., Yang H. M., Kim D. K., Kim N. H., Kim J. H., Cha K. Y., Chung H. M., Kim J. H. (2006) FEBS Lett. 580, 1865–1871 [DOI] [PubMed] [Google Scholar]
- 26.Li R., Pei H., Watson D. K. (2000) Oncogene 19, 6514–6523 [DOI] [PubMed] [Google Scholar]
- 27.Verger A., Duterque-Coquillaud M. (2002) BioEssays 24, 362–370 [DOI] [PubMed] [Google Scholar]
- 28.Sharrocks A. D. (2001) Nat. Rev. Mol. Cell Biol. 2, 827–837 [DOI] [PubMed] [Google Scholar]
- 29.Flentjar N., Chu P. Y., Ng A. Y., Johnstone C. N., Heath J. K., Ernst M., Hertzog P. J., Pritchard M. A. (2007) Gastroenterology 132, 1410–1419 [DOI] [PubMed] [Google Scholar]
- 30.Levy D. E., Darnell J. E., Jr. (2002) Nat. Rev. Mol. Cell Biol. 3, 651–662 [DOI] [PubMed] [Google Scholar]
- 31.Levy D. E., Lee C. K. (2002) J. Clin. Invest. 109, 1143–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alonzi T., Newton I. P., Bryce P. J., Di Carlo E., Lattanzio G., Tripodi M., Musiani P., Poli V. (2004) Cytokine 26, 45–56 [DOI] [PubMed] [Google Scholar]
- 33.Grady W. M., Markowitz S. D. (2002) Annu. Rev. Genomics Hum. Genet. 3, 101–128 [DOI] [PubMed] [Google Scholar]
- 34.Schedin P. J., Eckel-Mahan K. L., McDaniel S. M., Prescott J. D., Brodsky K. S., Tentler J. J., Gutierrez-Hartmann A. (2004) Oncogene 23, 1766–1779 [DOI] [PubMed] [Google Scholar]