

SUMMARY
We have investigated the ability of different regions of the left arm of Saccharomyces cerevisiae chromosome V to participate in the formation of gross chromosomal rearrangements (GCRs). We found that the 4.2 kb HXT13 DSF1 region sharing divergent homology with chromosomes IV, X, and XIV, similar to mammalian segmental duplications, was “at-risk” for participating in duplication-mediated GCRs generated by homologous recombination. Numerous genes and pathways, including SGS1, TOP3, RMI1, SRS2, RAD6, SLX1, SLX4, SLX5, MSH2, MSH6, RAD10 and the DNA replication stress checkpoint requiring MRC1 and TOF1 were highly specific for suppressing these GCRs compared to GCRs mediated by single copy sequences. These results indicate that the mechanisms for formation and suppression of rearrangements occurring in regions containing “at risk” sequences differ from those occurring in regions of single copy sequence. This explains how extensive genome instability is prevented in eukaryotic cells whose genomes contain numerous divergent repeated sequences.
INTRODUCTION
The importance of maintaining the stability of the genome is revealed by the numerous genetic diseases caused by inherited and de novo mutations ranging from base changes to genome rearrangements1, 2. In addition, many cancers are associated with ongoing genome instability and the continued accumulation of mutations and genome rearrangements3-7. Despite the problems introduced by genome instability, the human genome contains many features prone to be unstable, including microsatellite repeats, minisatellite repeats, triplet repeats, short separated repeats, mirror repeats, inverted repeats, and dispersed repetitive elements such as retroviral elements, SINEs, LINEs, segmental duplications and regions of copy number variation (CNVs)8, 9. Dispersed repetitive elements can underlie chromosomal rearrangements through non-allelic homologous recombination (HR) between elements at non-homologous chromosomal locations. The Alu elements, for example, cause HR-mediated deletions, duplications, and chromosomal translocations implicated in over 15 inherited diseases as well as rearrangements leading to cancer10. Similarly, more than 20 human diseases are caused by rearrangements mediated by non-allelic HR between segmental duplications11. Given the large numbers of repeated regions in the genome, it is surprising that the genome is as stable as it is.
Some types of “at-risk” sequences have been characterized in Saccharomyces cerevisiae9. Engineered duplications are targets of ectopic recombination, leading to both gene conversion and chromosomal rearrangements12. Similarly, Ty transposons, which are dispersed, repeated sequences, can recombine to produce genome rearrangements13, and inverted copies of Ty transposons can cause DSBs during replication resulting in genome rearrangements14. Consistent with this, “at-risk” sequences appear to be selected against15; however, the human genome still retains many such sequences. While “at-risk” sequences can induce genome instability, little is known about how such genome rearrangements are prevented and whether there are specific pathways that prevent such sequences from causing genome rearrangements.
We have described assays for measuring the rate of accumulating gross chromosomal rearrangements (GCRs)16. This assay system detects GCRs that occur in natural DNA sequences and does not depend on the introduction of artificial DNA sequences or the artificial induction of DSBs. Here, we applied this system to compare the rates and properties of GCRs in a chromosomal region devoid of “at-risk” sequences with that of a region of the genome containing a sequence homeologous to ectopic regions of the the genome reminiscent of segmental duplications.
RESULTS
Duplications alter the GCR spectrum and rate
We placed a CAN1/URA3 cassette in different locations on the non-essential left end of chromosome V to select for canavanine (Can) and 5-fluoroorotate (5FOA) resistant GCRs similar to our original GCR assay16 (Fig. 1A). GCRs, but not co-mutation or interstitial co-deletion of CAN1 and URA3, dominated the Canr 5FOAr products as evidenced by frequent loss of a telomeric hygromycin-resistance marker (Suppl. Table 1), similar to the original GCR assay17. Overall, the GCR rates increased approximately linearly with the size of the chromosome V breakpoint region except for the yel072w∷CAN1/URA3 assay, which had a higher rate than predicted based on the breakpoint region length (Table 1). YEL072W is telomeric to the HXT13 DSF1 region, which shares ~4.2 kb of imperfect homology with chromosome XIV and ~2 kb of imperfect homology with nearly identical regions of chromosomes IV and X (Fig. 1B), similar to mammalian segmental duplications18. Deletion of HXT13 DSF1 eliminated the duplication-associated GCR rate increase (Table 1). Homology-driven monocentric t(V;XIV) and t(V;IV or X) translocations accounted for 90% of the GCRs even though the HXT13 DSF1 region accounts for 13% of the breakpoint region (Fig. 2A). Sequencing of 20 t(V;XIV) junctions only revealed translocation breakpoints in the HXT13 DSF1 homology regions (Suppl. Fig. 1A)17. Array comparative genomic hybridization (aCGH) demonstrated that the target chromosomes were duplicated from the homology to the telomere (Fig. 1C), indicating that an intact copy of the target chromosomes were maintained; this was confirmed by PCR amplification of the native HXT13 DSF1 related junctions on the target chromosome (data not shown). Overall, the homology-driven GCRs were consistent with break-induced replication (BIR) or related mechanisms19, 20.
Figure 1. New assays for evaluating the genes that suppress the accumulation of GCRs.

A. The standard chromosome V GCR assay (top) contains URA3 integrated at HXT13 and selects for GCRs with Chr V breakpoints located between CAN1 and the essential PCM1 gene. The modified GCR assays (bottom) have a CAN1/URA3 cassette inserted into YEL062W, YEL064C, YEL068C, and YEL072W in a strain with ura3-52 and can1∷hisG mutations and a telomeric hygromycin resistance marker (hph). B. The average percent identity in 50 bp windows with the HXT13 DSF1 region with regions of chromosomes XIV, X, and IV is plotted against the Chr V position. C. aCGH data (log2 of the fluorescence ratio of individual GCR isolates to wild-type) indicates that the region from the Chr V homologies to the target chromosome telomere was duplicated. The two t(V;XIV) fusions lost unique Chr V signals telomeric to the HXT13 DSF1 region (Chr V 1-19500) and CAN1 from the CAN1/URA3 cassette (ChrV 31694-33466). Increased signals were observed with all probes for Chr XIV telomeric to YNR073C (Chr XIV 776300-787000). The two t(V;IV or X) fusions had Chr V signals similar to the t(V;XIV) fusions and essentially unchanged Chr XIV signals, excepting a subtle loss of signal in the DSF1 and YNR073C regions (Chr V 19589-21097; Chr XIV 774792-776300), consistent with loss of cross hybridization of DSF1 DNA to probes for DSF1-like genes. Increased fluorescence of the left arm of Chr IV and the right arm of Chr X demonstrated amplification and cross hybridization between these almost identical regions, despite the scarcity of probes. The aCGH data revealed no other significant copy number changes, excepting the region indicating loss of URA3 from the CAN1/URA3 cassette (data not shown).
Table 1.
GCR rates for different positions of the CAN1/URA3 cassette on chromosome V.
| Assay | Wild-type | rad27Δ | mre11Δ | sgs1Δ | Breakpoint Region Length (kb)** |
Wild-type Rate / Length (kb-1) |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Strain RDKY# |
Rate* | Strain RDKY# |
Rate* | Strain RDKY# |
Rate* | Strain RDKY# |
Rate* | |||
| Standard GCR*** | 3615 | 3.5×10-10 (1) | 3630 | 3.7×10-7 (1057) | 3633 | 2.2×10-7 (629) | 3813 | 2.5×10-8 (71) | 11.6 | 3.0×10-11 |
| yel062w::CAN1/URA3 | 6675 | 1.15 [0.0-5.6]×10-10 (0.3) | 6679 | 6.87×10-7 (2180) | 6680 | 3.23×10-7 (496) | 6681 | 1.19×10-8 (34) | 9.7 | 1.2 [0.0-5.7]×10-11 |
| yel064c::CAN1/URA3 | 6676 | 5.09 [2.5-7.7]×10-10 (1.6) | 6682 | 7.47×10-7 (2371) | 6683 | 2.63×10-7 (465) | 6684 | 1.77×10-8 (51) | 14.5 | 3.5 [1.7-5.3]×10-11 |
| yel068c::CAN1/URA3 | 6677 | 2.27 [1.3-4.8]×10-9 (7.2) | 6685 | 5.57×10-7 (1591) | 6686 | 5.75×10-7 (1643) | 6687 | 1.69×10-8 (48) | 19.2 | 12 [6.8-25]×10-11 |
| yel072::CAN1/URA3 | 6678 | 1.97 [1.6-4.3]×10-8 (56) | 6688 | 2.78×10-6 (7943) | 6689 | 1.52×10-6 (4345) | 6690 | 1.93×10-6 (5515) | 31.0 | 64 [52-140]×10-11 |
| yel068c::CAN1/URA3 hxt13-dsf1Δ | 6872 | 1.43 [0.0-4.2]×10-9 (4.1) | 6873 | 3.74×10-7 (1068) | - | n.d. | 6874 | 4.39×10-9 (12) | 19.2 | 7.5 [0.0-22]×10-11 |
| yel072::CAN1/URA3 hxt13-dsf1Δ | 6875 | 5.64 [4.1-12]×10-9 (16) | 6876 | 5.22×10-7 (1492) | - | n.d. | 6877 | 1.17×10-8 (33) | 27.7 | 20 [15-42]×10-11 |
Rate of accumulating CanR 5FOARprogeny. The number in parentheses is the fold increase relative to the standard wild-type rate (3.5×10-10; 16). Numbers in brackets are the 95% confidence interval limits.
The breakpoint region is defined as the length between the telomeric end of PCM1 and the telomeric end of CAN1.
Figure 2. Summary of the types of GCRs detected in the HXT13 DSF1 region mediated GCR assay.
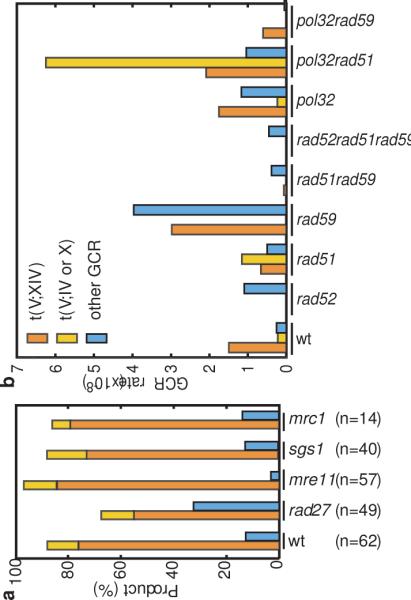
A. Percentage of the different types of GCRs in wild-type, rad27Δ, mre11Δ, sgs1Δ, and mrc1Δ yel072∷CAN1/URA3 strains. The homology-driven GCRs are shown as a stacked bar with t(V;XIV) in orange and t(V;IV or X) in yellow, and single-copy sequence mediated GCRs in blue. B. Mutations affecting both HR and BIR alter the rates of the formation of t(V;XIV), t(V;IV or X) and other GCRs detected in the yel072∷CAN1/URA3 assay. Rates for each class of GCR were calculated by multiplying the fraction of each kind of rearrangement by the overall rate.
Genotype affects the impact of the duplication
In the standard GCR assay, deletion of MRE11 or RAD27 caused ~600-1000 fold increased GCR rates16 and caused similar rate increases in strains where the CAN1/URA3 cassette was centromeric to HXT13 DSF1 (Table 1). When the cassette was telomeric to the duplication, the mutations only caused a modest increase in GCR rate relative to the yel068c∷CAN1/URA3 assay: five-fold for rad27Δ and three-fold for mre11Δ. The GCRs in the rad27Δ yel072w∷CAN1/URA3 background were a mix of 33 duplication-mediated and 16 single-copy sequence-mediated products (Fig. 2A). Like the wild-type strain that had 56 duplication-mediated and 6 single-copy sequence-mediated GCRs, the ratios of products were similar to the fold increase in rate caused by the duplication. In contrast, the mre11Δ yel072w∷CAN1/URA3 GCRs were dominated by duplication-mediated rearrangements (56:1). This suggests that an mre11Δ mutation alters the mechanism underlying GCRs such as by decreasing telomere maintenance21 resulting in increased degradation of chromosome ends, which would preferentially target telomeric duplicated sequences.
Deletion of SGS1, encoding a RecQ-family helicase, caused a moderate increase in the rate of GCRs in assays with the CAN1/URA3 cassette centromeric to the duplication similar to the standard GCR assay, but caused a dramatic increase in the yel072w∷CAN1/URA3 GCR rate that depended on the HXT13 DSF1 duplication (Table 1). The ratio of duplication-mediated to single copy sequence-mediated GCRs in the sgs1Δ mutant, 35:5 (Fig. 2A), was not as high as might be predicted from the 115-fold increase in GCR rates in the yel072w∷CAN1/URA3 assay vs. the yel068c∷CAN1/URA3 assay. Sequencing of 25 sgs1Δ t(V;XIV) breakpoint junctions revealed 21 t(V;XIV) and 4 complex translocations (Suppl. Fig. 1B). Three complex breakpoints resulted from t(V;XIV;V;XIV) translocations, and the fourth was consistent with a t(V;X;XIV) translocation. The complex junctions could be generated by template-switching during HR as implicated during BIR in wild-type strains22 and CAN1-LYP1-ALP1 translocations in sgs1Δ mutants23, or by formation of multiduplex joint molecules as observed in meiosis24.
Different HR pathways yield distinct GCR signatures
We next examined the role of the RAD52 epsistasis group genes (Table 2). As in the standard GCR assay25, the rad52Δ mutation increased the GCR rate in the yel068c∷CAN1/URA3 assay where GCRs are formed in single copy DNA sequences (Table 2). In contrast, the rad52Δ mutation modestly decreased the GCR rate in the yel072w∷CAN1/URA3 assay compared to wild-type (Table 2) and eliminated the duplication-mediated translocations (Fig. 2B). Deletion of RAD51 or RAD59, which define two distinct RAD52-dependent HR pathways26, had modest effects on the GCR rates in both assays, and non-reciprocal duplication-mediated translocations were observed in both single mutants (Table 2; Fig. 2B; Suppl. Fig. 2), indicating that these rearrangements are not strictly dependent on either pathway. t(V;IV or X) translocations were not observed in the rad59Δ strain, suggesting that efficient recombination with the translocation target that was shorter and had lower sequence identity was RAD59 dependent. Both the rad51Δ rad59Δ double mutant and the rad51Δ rad59Δ rad52Δ triple mutant had decreased rates of duplication-mediated GCRs (Table 2). Surprisingly, t(V;XIV) rearrangements were observed in the rad51Δ rad59Δ double mutant, unlike the rad52Δ single mutant and the rad51Δ rad59Δ rad52Δ triple mutant (Fig. 2B). Thus, it appears that at least one additional RAD52-dependent, RAD51- and RAD59- independent HR pathway can promote interchromosomal HR-mediated rearrangements at low rates; this is consistent with a more severe HR defect in a rad52Δ single mutant compared to a rad51Δ rad59Δ double mutant27.
Table 2.
Effect of homologous and homeologous recombination defective mutations on GCR rates
| Genotype | yel068c::CAN1/URA3 | yel072w::CAN1/URA3 | Ratio** | ||
|---|---|---|---|---|---|
| Strain | GCR Rate* | Strain | GCR Rate* | ||
| Wild-type | RDKY6677 | 2.27×10-9 (5.1) | RDKY6678 | 1.97×10-8 (56) | 8.7 |
|
| |||||
| rad52 Δ | RDKY6691 | 1.67×10-8 (48) | RDKY6708 | 1.09×10-8 (31) | 0.7 |
| rad51 Δ | RDKY6692 | <2.63×10-10 (<0.8) | RDKY6709 | 2.31×10-8 (66) | >88 |
| rad59 Δ | RDKY6693 | 5.85×10-9 (17) | RDKY6710 | 6.94×10-8 (198) | 11.9 |
| rad51Δ rad59Δ | RDKY6694 | 2.92×10-9 (8.3) | RDKY6711 | 4.48×10-9 (13) | 1.5 |
| rad51Δ rad59Δ rad52Δ | RDKY6695 | 3.84×10-9 (11) | RDKY6712 | 4.53×10-9 (13) | 1.2 |
|
| |||||
| msh2 Δ | RDKY6696 | 1.10×10-9 (3.1) | RDKY6713 | 1.75×10-7 (499) | 159 |
| msh6 Δ | RDKY6697 | 1.52×10-9 (4.4) | RDKY6714 | 2.10×10-7 (599) | 138 |
| msh3 Δ | RDKY6698 | 1.42×10-9 (4.1) | RDKY6715 | 3.67×10-8 (105) | 26 |
| mlhl Δ | RDKY6699 | 5.80×10-10 (1.7) | RDKY6716 | 3.85×10-8 (110) | 66 |
| sgsl Δ | RDKY6687 | 1.69×10-8 (48) | RDKY6690 | 1.93×10-6 (5515) | 114 |
| sgs1Δ rad52Δ | RDKY6700 | 7.75×10-8 (222) | RDKY6717 | 8.07×10-8 (231) | 1.0 |
| top3 Δ | RDKY6701 | <1.64×10-9 (<4.7) | RDKY6718 | 2.14×10-6 (6103) | >1300 |
| rmi1 Δ | RDKY6702 | 1.41×10-7 (404) | RDKY6719 | 1.27×10-5 (36700) | 98.3 |
|
| |||||
| pol32 Δ | RDKY6703 | 3.41×10-9 (9.4) | RDKY6720 | 3.15×10-8 (90.0) | 9.24 |
| pol32Δ rad51Δ | RDKY6704 | 3.00×10-8 (86) | RDKY6721 | 9.37×10-8 (268) | 3.1 |
| pol32Δ rad59Δ | RDKY6705 | 5.50×10-10 (1.6) | RDKY6722 | 6.52×10-9 (19) | 12 |
| pri2-1 (23 deg) | RDKY6706 | <3.93×10-10 (<1.1) | RDKY6723 | 7.10×10-10 (2.0) | >1.8 |
| pol12-100 (23 deg) | RDKY6707 | 6.35×10-9 (18.1) | RDKY6724 | 2.31×10-8 (66.1) | 3.7 |
Rate of accumulating CanR 5FOARprogeny. The number in parentheses is the fold increase relative to the standard wildtype rate (3.5×10-10;16).
The yel072w::CAN1/URA3 rate divided by the yel068c::CAN1/URA3 rate.
Mismatch repair (MMR) proteins28 and Sgs129 are predicted to suppress HR between the HXT13 DSF1 region and the imperfect homologies on chromosomes IV, X, and XIV. Elimination of mismatch detection by a msh2Δ mutation or impairment by msh6Δ or msh3Δ mutations specifically increased the GCR rates in the duplication-containing assay (Table 2). The larger effects of msh2Δ and msh6Δ relative to msh3Δ are consistent with the heteroduplexes formed during duplication-mediated HR, which would contain primarily base-base mispairs and fewer insertion/deletion mispairs. Similar to the effects of mlh1Δ in single-stranded annealing assays30, mlh1Δ caused a smaller but significant increase in the rate of duplication-mediated GCRs (Table 2). An sgs1Δ mutation caused an increase in duplication-mediated GCRs (Table 1 and Fig. 2), and a rad52Δ mutation eliminated this increase (Table 2), indicating that homeologous recombination mediates most of the GCRs in the yel072∷CAN1/URA3 assay in the sgs1Δ mutant. However, sgs1Δ caused a higher duplication-mediated GCR rate than msh2Δ (Table 2), despite the fact that Sgs1 is downstream of MMR during suppression of homeologous recombination30. Deletion of TOP3 and RMI1, which function in concert with SGS131, also caused higher rates of duplication-mediated GCRs than the msh2Δ mutation; the increased GCR rates caused by rmi1Δ relative to sgs1Δ and top3Δ suggests that RMI1 may have SGS1- and TOP3-independent roles (Table 2). These data, in combination with the synergistic increase in the GCR rate in the yel068c∷CAN1/URA3 assay in the sgs1Δ rad52Δ double mutant (Table 2) suggests that sgs1Δ as well as top3Δ and rmi1Δ cause defects in suppression of homeologous recombination and also affect other pathways that suppress duplication-mediated GCRs.
HR-mediated GCRs are POL32-independent
The t(V;XIV) and t(V;IV or X) translocation products and their dependence on HR genes are consistent with BIR or related mechanisms19, 20. POL32, encoding a DNA polymerase delta subunit, is essential for ectopic BIR induced by HO-mediated DSBs32, but not strictly required for allelic BIR20. Deletion of POL32 caused a small increase in the duplication-mediated GCR rate and did not change the rate of t(V;XIV) or t(V;IV or X) translocations (Table 2; Fig. 2B). The three pol32Δ t(V;XIV) translocations analyzed by aCGH were non-reciprocal (Suppl. Fig. 3). These results could be explained if previously observed POL32-dependent BIR was predominantly RAD51-dependent32, in contrast to both RAD51-dependent and RAD51-independent pathways observed here.
Both the pol32Δ rad51Δ and pol32Δ rad59Δ double mutants had low levels of duplication-mediated GCRs (Table 2, Fig. 2B). The pol32Δ rad51Δ double mutant had increased GCR rates in both assays, with the duplication causing a modest increase primarily due to accumulation of t(V;IV or X) translocations, consistent with the possibility that RAD51 is required to suppress GCRs in a pol32Δ mutant. In contrast, the pol32Δ rad59Δ double mutant had a lower GCR rate than the rad59Δ and pol32Δ single mutants, and, compared to the rad51Δ rad59Δ double mutant, had a similar GCR rate in the yel072w∷CAN1/URA3 assay and a somewhat lower rate in the yel068c∷CAN1/URA3 assay. In addition, the rate of t(V;XIV) translocations was reduced in the pol32Δ rad59Δ mutant relative to pol32Δ and rad59Δ single mutants, but not to the extent seen in rad51Δ rad59Δ double mutants (Fig. 2B). These results suggest that POL32 functions in the RAD51-dependent pathway but not the RAD59-dependent pathway that promotes duplication-mediated GCRs; however, in the RAD51-dependent pathway, the formation of duplication-mediated GCRs is not completely dependent on POL32. Thus, a subset of the RAD51-dependent duplication-mediated GCRs are likely produced by POL32-dependent BIR, whereas POL32-independent RAD51-dependent and RAD59-dependent duplication-mediated GCRs either result from other HR mechanisms, such as a half-crossover mechanism20, or are produced by a BIR pathway that has different genetic requirements than BIR driven by HO-induced DSBs. Two other replication-associated mutations, pri2-1, which suppresses HR-mediated BIR32, and pol12-100, which increases levels of Holliday junctions during replication33, generally decreased or weakly increased GCR rates, respectively.
Pathways that suppress HR-mediated GCRs
As analysis of sgs1Δ, top3Δ, rmi1Δ, msh2Δ and msh6Δ mutants (Table 2) indicated that the yel072w∷CAN1/URA3 assay can reveal pathways that specifically suppress duplication-mediated rearrangements, we screened for additional context-specific mutations (Table 3). Deletion of SGS1 causes synthetic growth defects with deletions of SLX1, SLX4, SLX5, SLX8, MUS81, SAE2, SRS2 or RRM3. Deletion of each of these genes, except SAE2, caused duplication specific increases in GCR rates whereas only deletion of SAE2 and MUS81 caused increases in GCRs mediated by single copy DNA sequences. Similarly, deletion of the repair genes RAD6, MPH1, RAD10 or EXO1 caused large increases in duplication-specific GCR rates, but little or no increase in single-copy sequence mediated GCRs. The duplication-specific effects of rad10Δ contrast with prior findings that the Rad1-Rad10 complex is required for single-copy DNA sequence mediated GCRs34. Deletion of ESC2 and ESC4/RTT107, which encodes a protein recruited to stalled replication forks38, caused a general increase in GCR rates and a preferential increase in the rate of HXT13 DSF1 duplication-mediated GCRs. Defects in chromatin modifying pathways caused by deletion of ASF1, RTT109, ARP8 or NHP10 also had duplication-specific effects; however, in contrast to deleting ASF1 or RTT109, deleting ARP8 and NHP10, which encode subunits of the Ino80 chromatin remodeling complex35, did not alter the rate of single-copy sequence mediated GCRs. In contrast, deletion of CTF18, which causes sister chromatid cohesion defects36, caused similar increases in both assays. These results demonstrate that the genetics of suppressing GCRs changes substantially depending on chromosomal features in the breakpoint region.
Table 3.
Effect of mutations on the accumulation of duplication-mediated rearrangements
| Genotype | yel068c::CAN1/URA3 | yel072w::CAN1/URA3 | Ratio** | ||
|---|---|---|---|---|---|
| Strain | GCR Rate* | Strain | GCR Rate* | ||
| Wild-type | RDKY6677 | 2.27×10-9 (5.1) | RDKY6678 | 1.97×10-8 (56) | 8.7 |
|
| |||||
| SGS1 interactors | |||||
| mus81 Δ | RDKY6731 | 1.26×10-8 (36) | RDKY6748 | 2.51×10-7 (717) | 20 |
| rrm3 Δ | RDKY6735 | 9.46×10-10 (2.7) | RDKY6752 | 3.87×10-8 (110) | 41 |
| sae2 Δ | RDKY6737 | 4.23Δ10-8 (120) | RDKY6754 | 1.65×10-7 (470) | 3.9 |
| slx1 Δ | RDKY6738 | <1.12×10-9 (<3.2) | RDKY6755 | 2.32×10-8 (66) | >20.6 |
| slx4 Δ | RDKY6739 | <7.94×10-10 (<2.3) | RDKY6756 | 9.26×10-8 (264) | >116 |
| slx5 Δ | RDKY6740 | 1.48×10-9 (4.2) | RDKY6757 | 4.82×10-7 (1378) | 326 |
| slx8 Δ | RDKY6846 | <1.81×10-9 (<5.2) | RDKY6847 | 9.65×10-7 (2757) | >532 |
| srs2 Δ | RDKY6741 | 7.18×10-10 (2.1) | RDKY6758 | 1.28×10-7 (365) | 178 |
|
| |||||
| Chromatin | |||||
| asf1 Δ | RDKY6725 | 1.34×10-8 (38) | RDKY6742 | 2.89×10-7 (825) | 22 |
| arp8 Δ | RDKY6726 | <6.05×10-10 (<1.73) | RDKY6743 | 4.84×10-8 (138) | >80 |
| nhp10 Δ | RDKY6732 | 1.39×10-9 (4.0) | RDKY6749 | 3.01×10-8 (86) | 22 |
| rtt109 Δ | RDKY6736 | 5.64×10-9 (16) | RDKY6753 | 1.84×10-7 (526) | 33 |
|
| |||||
| Cohesion | |||||
| ctf18 Δ | RDKY6727 | 2.40×10-8 (69) | RDKY6744 | 2.22×10-7 (633) | 9.2 |
|
| |||||
| Other Repair | |||||
| esc2 Δ | RDKY6878 | 4.36×10-8 (124) | RDKY6879 | 1.07×10-5 (30700) | 247 |
| esc4 Δ | RDKY6728 | 1.66×10-8 (48) | RDKY6745 | 3.07×10-7 (876) | 18.5 |
| exo1 Δ | RDKY6729 | 2.00×10-9 (5.7) | RDKY6746 | 8.44×10-8 (241) | 42 |
| mph1 Δ | RDKY6794 | 2.00×10-9 (5.7) | RDKY6795 | 1.05×10-7 (300) | 53 |
| rad6 Δ | RDKY6733 | 4.66×10-9 (13) | RDKY6750 | 6.03×10-7 (1724) | 130 |
| rad10 Δ | RDKY6734 | 8.49×10-10 (2.4) | RDKY6751 | 1.80×10-7 (404) | 212 |
|
| |||||
| Checkpoint | |||||
| mrc1 Δ | RDKY6730 | 3.35×10-9 (9.6) | RDKY6747 | 3.75×10-7 (1071) | 112 |
| mrc1-aq | RDKY6766 | 1.51×10-9 (4.3) | RDKY6775 | 1.23×10-7 (351) | 81 |
| tof1 Δ | RDKY6767 | 5.71×10-9 (16) | RDKY6776 | 4.25×10-7 (1214) | 74 |
| mrc1Δ tof1Δ | RDKY6779 | 6.41×10-8 (183) | RDKY6780 | 1.26×10-6 (3612) | 20 |
| mrc1-aq tof1 Δ | RDKY6848 | 3.69×10-9 (11) | RDKY6849 | 2.06×10-7 (589) | 56 |
| rad24 Δ | RDKY6759 | 2.00×10-8 (57.3) | RDKY6768 | 1.97×10-7 (555) | 9.7 |
| mec1Δ sml1Δ | RDKY6760 | 2.34×10-8 (67) | RDKY6769 | 1.50×10-7 (429) | 6.4 |
| tel1 Δ | RDKY6761 | 4.99×10-9 (14) | RDKY6770 | 2.87×10-8 (82) | 5.8 |
| rad53Δ sml1Δ | RDKY6762 | 5.60×10-8 (160) | RDKY6771 | 3.05×10-7 (871) | 5.4 |
| rad9 Δ | RDKY6765 | 2.17×10-8 (62) | RDKY6774 | 3.82×10-8 (109) | 1.8 |
| chk1 Δ | RDKY6764 | 1.76×10-8 (50) | RDKY6773 | 1.96×10-7 (560) | 11 |
| dun1 Δ | RDKY6763 | 1.63×10-8 (47) | RDKY6772 | 1.61×10-7 (461) | 9.9 |
Rate of accumulating CanR 5FOARprogeny. The number in parentheses is the fold increase relative to the standard wildtype rate (3.5×10-10;16).
The yel072w::CAN1/URA3 rate divided by the yel068c::CAN1/URA3 rate.
Checkpoint suppression of HR-mediated GCRs
Deletion of MRC1, which encodes a Rad53 coactivator with roles in DNA replication and replication stress checkpoint signaling37, caused a small increase in the rate of single copy sequence mediated GCRs and a large increase in HXT13 DSF1 duplication-mediated GCRs. The latter GCRs were primarily homology-driven translocations (Fig. 2A) and 2 GCRs predicted to be t(V;XIV) translocations by PCR were non-reciprocal translocations (Suppl. Fig. 3) similar to all other duplication-mediated GCRs analyzed by aCGH (Fig. 1C, Suppl. Fig. 2 & 3). The mrc1-aq allele, which specifically affects the checkpoint function of MRC137, had little effect on single copy sequence-mediated GCRs but caused a large increase in duplication-mediated GCRs (Table 3). Similarly, deleting TOF1, which encodes another replication fork and checkpoint protein39, caused a specific increase in HXT13 DSF1 duplication-mediated GCRs (Table 3). We found a synergistic interaction between mrc1Δ and tof1Δ but not between mrc1-aq and tof1Δ in both the yel068c∷CAN1/URA3 and yel072w∷CAN1/URA3 assays, indicating a partial redundancy of these genes (Table 3).
Mutations in the checkpoint genes RAD24, MEC1, RAD53, DUN1 and CHK1 increased the GCR rate in both the yel068c∷CAN1/URA3 and yel072w∷CAN1/URA3 assays (Table 3), although the affect on duplication-mediated GCR rates was possibly not as large as that of mrc1Δ or tof1Δ mutations, raising the possibility that mrc1Δ and tof1Δ mutations might increase DNA damage in addition to causing checkpoint defects. Mutations in TEL1, which encodes a protein kinase that is partially redundant with Mec1, resulted in small rate increases in both GCR assays, consistent with a small checkpoint role for Tel1 in the presence of Mec1; however, tel1Δ telomere maintenance defects could contribute to a low level of GCRs. Mutations in RAD9, which encodes an alternative Rad53 co-activator that responds to general DNA damage signaling, but not replication fork damage in strains with MRC139, were similar to the affects of damage checkpoint mutations on single copy sequence-mediated GCRs, but caused a much smaller increase than these mutations in the rate of duplication-mediated GCRs. Together, these data suggest that the DNA damage checkpoint primarily suppresses single copy sequence-mediated GCRs whereas both the DNA damage checkpoint to a lesser extent and the replication stress checkpoint to a much greater extent suppress duplication-mediated GCRs.
DISCUSSION
We have found that many genes play little or no role in suppressing GCRs in single-copy sequences but play a large role in suppressing GCRs mediated by non-allelic HR at the HXT13 DSF1 “at risk” sequence that resembles a segmental duplication in mammalian cells. One group of genes include the MMR genes and the genes encoding the Sgs1-Top3-Rmi1 complex that suppress HR between divergent sequences29. Another group included MRC1 and TOF1, and our analysis of checkpoint genes indicated that the replication stress checkpoint is critical in suppressing HXT13 DSF1 mediated GCRs but not single-copy sequence mediated GCRs. A third group of genes that almost all exclusively function in suppressing HXT13 DSF1 duplication-mediated GCRs include SRS2, RRM3, MUS81, SLX1, SLX2, SLX4, SLX5, and SLX8, which cause synthetic growth defects when deleted in combination with an sgs1Δ mutation, due to accumulation of toxic replication intermediates that in many cases can be suppressed by a HR defect40-43. Potentially related to these genes are: RAD10 and EXO1 that encode an endonuclease and an exonuclease, respectively, that can act in processing of HR and aberrant replication intermediates44, 45; MPH1 encoding a DNA helicase that may disrupt HR intermediates like Sgs146-48; and RAD6 that regulates processes that act on replication forks that encounter DNA damage49. Finally, ARP8, NHP10, ASF1 and RTT109, which function in chromatin remodeling and checkpoint regulation and can act during S-phase35, 50, strongly suppressed duplication-mediated GCRs. All of these genes may function in responses to replication stress, including checkpoint activation or shut-off, repair of aberrant replication intermediates and suppression of the formation of aberrant replication intermediates, and some clearly act to directly prevent aberrant HR. How might the products of these genes act so specifically to prevent duplication mediated GCRs? It is unlikely that they solely act to prevent aberrant DNA structures during replication such as DSBs as they would also suppress GCRs mediated by single copy DNA sequences. Rather, they may prevent aberrant HR such as homeologous recombination or aberrant BIR intermediates so that HR can selectively target homologous sequences on sister chromatids and homologs as well as restart damaged replication forks to prevent genome instability rather than result in HR-mediated GCRs.
Our results indicate that dispersed repetitive elements in DNA resembling segmental duplications are “at-risk” for causing genomic instability. The presence of multiple pathways that are highly specific for suppressing rearrangements between these elements explains how genomes remain stable despite the presence of sequences “at risk” for mediating genome rearrangements. These results complement previous studies that identified critical pathways and genes that suppress GCRs that target single copy sequences17. Overall our data suggest that defects in different DNA repair pathways result in distinct GCR signatures that may be diagnostic of the defects that underlie genome instability.
METHODS SUMMARY
Yeast strains were constructed by deleting CAN1 and integrating a telomeric hygromycin marker and a CAN1/URA3 cassette in the RDKY3023 background (MATa leu2Δ1 his3Δ200 trp1Δ63 lys2ΔBgl hom3-10 ade2Δ1 ade8 ura3-52). GCRs were selected using standard methods16. GCR products were analyzed by PCR and by aCGH (NimbleGen).
FULL METHODS
Plasmid construction
A can1∷hisG-URA3-hisG disruption cassette was constructed by first PCR amplifying fragments that are telomeric to CAN1 (Chr V 30187-30928) flanked by Apa I and Xho I sites and centromeric to CAN1 (Chr V 34339-34965) flanked by Xba I and Bam HI sites and inserting them into pRS31551 to generate pRDK1374. A hisG-URA3-hisG fragment was amplified from pNKY51,52 and was then inserted into Sma I-digested pRS315 by recombinational cloning in S. cerevisiae. Then the hisG-URA3-hisG fragment was then subcloned into pRDK1374 between Sal I and Bam HI sites to generate pRDK1375 containing the hisG-URA3-hisG fragment flanked by 626 bp of upstream and 741 bp of downstream homology to the CAN1 locus.
The CAN1/URA3 cassette was constructed by cloning fragments of CAN1 and URA3 into a plasmid with flanking Nhe I sites. The CAN1 gene and flanking sequence (Chr V 30952-34315) was amplified by PCR and cloned into pCR2.1-TOPO (Invitrogen) to generate pRDK1376. The URA3 gene and flanking sequence (Chr V 116011- 117061) was amplified by PCR with primers to introduce flanking Xba I sites, and cloned into pRDK1376; inserts with CAN1 and URA3 in divergent orientations were selected. The CAN1/URA3 cassette was then PCR amplified with primers adding flanking Nhe I sites, cloned into pCRT7CT (Invitrogen), and verified by sequencing to generate pRDK1377.
For each chromosome V integration site, integration constructs were generated by subcloning the pRDK1377 Nhe I fragment into plasmids containing the target genes of interest. The gene and flanking regions of YEL072W (Chr V 12961-14898) and YEL068C (Chr V 25222-26411) were amplified by PCR, cloned into pRS315, and modified by site-directed mutagenesis to introduce Nhe I sites into the center of the genes. Subcloning the CAN1/URA3 cassette into the engineered Nhe I sites in YEL072W and YEL068C generated the plasmids pRDK1378 and pRDK1379, respectively. Similarly, the gene and flanking regions of YEL064C (Chr V 30060-30928) and YEL062W (Chr V 36007-36992) were amplified by PCR, cloned into pET21a, and the Nhe I-digested CAN1/URA3 cassette was subcloned into compatible Spe I sites to generate plasmids pRDK1380 and pRDK1381, respectively.
Genetic Methods
YPD and synthetic drop-out media for propagation of strains have been described previously53. The can1∷hisG-URA3-hisG integration fragment was cut out from pRDK1375 using Kpn I and Sac I and transformed into RDKY3023 (MATa leu2Δ1 his3Δ200 trp1Δ63 lys2ΔBgl hom3-10 ade2Δ1 ade8 ura3-52). Uracil prototrophs were verified by PCR, and a can1∷hisG uracil auxotroph, RDKY5461, was selected on 5FOA containing medium. The CAN1/URA3 integration cassettes were amplified by PCR from plasmids described above and integrated into RDKY5461. These strains were then modified by integrating a hygromycin resistance cassette telomeric to YEL072W (Chr V 11081-11618) to generate RDKY6678 (yel072w∷CAN1/URA3), RDKY6677 (yel068c∷CAN1/URA3), RDKY6676 (yel064c∷CAN1/URA3), and RDKY6675 (yel062w∷CAN1/URA3). Additional mutations were added to these strains using standard PCR-based mutagenesis methods, pop-in, pop-out plasmid vectors or intercrossing with mutants derived from the same parental strain background (Supplemental Table 2). The media and protocol for measuring GCR rates were essentially as described previously53. 95% confidence intervals of the median were calculated by the a two-sided nonparametric test (http://www.math.unb.ca/~knight/utility/MedInt95.htm). The GCR rates determined using this method are highly reliable. Using data from a number of studies covering a broad range of mutants and GCR rates, we have calculated that the average upper and ower 95% confidence interval limits are 1.5 and 0.7, respectively, times the median GCR rate determined. In less than 8% of the measurements were the upper and lower 95% confidence intervals greather than 2 or less than 0.5, respectively, times the median GCR rate determined.
Analysis of GCR Isolates
GCR isolates were tested for loss of hygromycin resistance by growth on YPD media containing 300 μg/mL hygromycin B (Invitrogen). Genomic DNA was prepared from individual isolates and subjected to PCR analysis to categorize GCRs. The t(V;XIV) and t(V;IV or X) translocations were identified by amplification of the junction region with a Chr V-specific primer centromeric to the HXT13 DSF1 region and a Chr XIV or Chr IV/X specific primer telomeric to the HXT13 DSF1 homologies on those chromosomes under conditions where no product was generated with DNA from wild-type strains. A series of PCR reactions spanning the ~20 kb region between HXT13 and PCM1 on Chr V were used to map breakpoints for isolates that were not t(V;XIV) or t(V;IV or X) translocations by identifying the region where all telomeric reactions failed and all centromeric reactions succeeded. Breakpoint junctions from selected t(V;XIV) isolates were amplified as described above and sequenced by dye terminator DNA sequencing.
Array Comparative Genomic Hybridization
1 ug of genomic DNA was prepared from GCR isolates and wild-type RDKY6678 using the Purgene kit (Qiagen) and concentrated to over 100 ng/uL. GCR isolate DNA was amplified and labeled with Cy5 and the wild-type DNA was amplified and labeled with Cy3 and GCR isolate/wild-type pairs were applied to a NimbleGen 4-plex chip. Data were analyzed using the SignalMap software (NimbleGen).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the UCSD Microarray Core Facility for assistance in the aCGH experiments and Cathy Smith, Scarlet Shell, and John Petrini for helpful comments on the manuscript. This work was supported by NIH grant GM26017.
Footnotes
The authors declare no competing financial interests.
REFERENCES
- 1.OMIM (Institute of Genetic Medicine, Johns Hopkins University (Baltimore MD) and National Center for Biotechnology Information, National Library of Medicine, 1999)
- 2.Stankiewicz P, Lupski JR. The genomic basis of disease, mechanisms and assays for genomic disorders. Genome Dyn. 2006;1:1–16. doi: 10.1159/000092496. [DOI] [PubMed] [Google Scholar]
- 3.Mitelman F. Catalog of chromosome aberrations in cancer. Wiley Liss; New York, N. Y.: 1991. [Google Scholar]
- 4.Gorringe KL, et al. Evidence that both genetic instability and selection contribute to the accumulation of chromosome alterations in cancer. Carcinogenesis. 2005;26:923–30. doi: 10.1093/carcin/bgi032. [DOI] [PubMed] [Google Scholar]
- 5.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–7. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 6.Ribas M, et al. The structural nature of chromosomal instability in colon cancer cells. Faseb J. 2003;17:289–91. doi: 10.1096/fj.02-0425fje. [DOI] [PubMed] [Google Scholar]
- 7.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 8.Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67:183–93. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 9.Gordenin DA, Resnick MA. Yeast ARMs (DNA at-risk motifs) can reveal sources of genome instability. Mutat Res. 1998;400:45–58. doi: 10.1016/s0027-5107(98)00047-5. [DOI] [PubMed] [Google Scholar]
- 10.Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–9. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 11.Ji Y, Eichler EE, Schwartz S, Nicholls RD. Structure of chromosomal duplicons and their role in mediating human genomic disorders. Genome Res. 2000;10:597–610. doi: 10.1101/gr.10.5.597. [DOI] [PubMed] [Google Scholar]
- 12.Harris S, Rudnicki KS, Haber JE. Gene conversions and crossing over during homologous and homeologous ectopic recombination in Saccharomyces cerevisiae. Genetics. 1993;135:5–16. doi: 10.1093/genetics/135.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Umezu K, Hiraoka M, Mori M, Maki H. Structural analysis of aberrant chromosomes that occur spontaneously in diploid Saccharomyces cerevisiae: retrotransposon Ty1 plays a crucial role in chromosomal rearrangements. Genetics. 2002;160:97–110. doi: 10.1093/genetics/160.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell. 2005;120:587–98. doi: 10.1016/j.cell.2004.12.039. [DOI] [PubMed] [Google Scholar]
- 15.Lobachev KS, et al. Inverted Alu repeats unstable in yeast are excluded from the human genome. Embo J. 2000;19:3822–30. doi: 10.1093/emboj/19.14.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–5. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 17.Putnam CD, Pennaneach V, Kolodner RD. Saccharomyces cerevisiae as a model system to define the chromosomal instability phenotype. Mol Cell Biol. 2005;25:7226–38. doi: 10.1128/MCB.25.16.7226-7238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eichler EE. Recent duplication, domain accretion and the dynamic mutation of the human genome. Trends Genet. 2001;17:661–9. doi: 10.1016/s0168-9525(01)02492-1. [DOI] [PubMed] [Google Scholar]
- 19.Bosco G, Haber JE. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics. 1998;150:1037–47. doi: 10.1093/genetics/150.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deem A, et al. Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics. 2008;179:1845–60. doi: 10.1534/genetics.108.087940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boulton SJ, Jackson SP. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. Embo J. 1998;17:1819–28. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447:102–5. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt KH, Wu J, Kolodner RD. Control of translocations between highly diverged genes by Sgs1, the Saccharomyces cerevisiae homolog of the Bloom's syndrome protein. Mol Cell Biol. 2006;26:5406–20. doi: 10.1128/MCB.00161-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oh SD, et al. BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell. 2007;130:259–72. doi: 10.1016/j.cell.2007.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myung K, Chen C, Kolodner RD. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature. 2001;411:1073–6. doi: 10.1038/35082608. [DOI] [PubMed] [Google Scholar]
- 26.Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–71. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 27.Bai Y, Symington LS. A Rad52 homolog is required for RAD51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev. 1996;10:2025–37. doi: 10.1101/gad.10.16.2025. [DOI] [PubMed] [Google Scholar]
- 28.Datta A, Adjiri A, New L, Crouse GF, Jinks Robertson S. Mitotic crossovers between diverged sequences are regulated by mismatch repair proteins in Saccaromyces cerevisiae. Mol Cell Biol. 1996;16:1085–93. doi: 10.1128/mcb.16.3.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myung K, Datta A, Chen C, Kolodner RD. SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet. 2001;27:113–6. doi: 10.1038/83673. [DOI] [PubMed] [Google Scholar]
- 30.Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci U S A. 2004;101:9315–20. doi: 10.1073/pnas.0305749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullen JR, Nallaseth FS, Lan YQ, Slagle CE, Brill SJ. Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-Top3 complex. Mol Cell Biol. 2005;25:4476–87. doi: 10.1128/MCB.25.11.4476-4487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–3. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 33.Zou H, Rothstein R. Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell. 1997;90:87–96. doi: 10.1016/s0092-8674(00)80316-5. [DOI] [PubMed] [Google Scholar]
- 34.Hwang JY, Smith S, Myung K. The Rad1-Rad10 complex promotes the production of gross chromosomal rearrangements from spontaneous DNA damage in Saccharomyces cerevisiae. Genetics. 2005;169:1927–37. doi: 10.1534/genetics.104.039768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conaway RC, Conaway JW. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci. 2009;34:71–7. doi: 10.1016/j.tibs.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 36.Mayer ML, Gygi SP, Aebersold R, Hieter P. Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol Cell. 2001;7:959–70. doi: 10.1016/s1097-2765(01)00254-4. [DOI] [PubMed] [Google Scholar]
- 37.Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003;17:1755–67. doi: 10.1101/gad.1098303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts TM, Zaidi IW, Vaisica JA, Peter M, Brown GW. Regulation of rtt107 recruitment to stalled DNA replication forks by the cullin rtt101 and the rtt109 acetyltransferase. Mol Biol Cell. 2008;19:171–80. doi: 10.1091/mbc.E07-09-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katou Y, et al. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003;424:1078–83. doi: 10.1038/nature01900. [DOI] [PubMed] [Google Scholar]
- 40.Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics. 2001;157:103–18. doi: 10.1093/genetics/157.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt KH, Kolodner RD. Requirement of Rrm3 helicase for repair of spontaneous DNA lesions in cells lacking Srs2 or Sgs1 helicase. Mol Cell Biol. 2004;24:3213–26. doi: 10.1128/MCB.24.8.3213-3226.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fabre F, Chan A, Heyer WD, Gangloff S. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci U S A. 2002;99:16887–92. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torres JZ, Schnakenberg SL, Zakian VA. Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: viability of rrm3 cells requires the intra-S-phase checkpoint and fork restart activities. Mol Cell Biol. 2004;24:3198–212. doi: 10.1128/MCB.24.8.3198-3212.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugawara N, Paques F, Colaiacovo M, Haber JE. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc Natl Acad Sci U S A. 1997;94:9214–9. doi: 10.1073/pnas.94.17.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiorentini P, Huang KN, Tishkoff DX, Kolodner RD, Symington LS. Exonuclease I of Saccharomyces cerevisiae functions in mitotic recombination in vivo and in vitro. Mol Cell Biol. 1997;17:2764–73. doi: 10.1128/mcb.17.5.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krejci L, et al. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–9. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- 47.Prakash R, et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009;23:67–79. doi: 10.1101/gad.1737809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veaute X, et al. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–12. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- 49.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–41. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 50.Chen CC, et al. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–43. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics. 1987;116:541–5. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–5. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.