

Abstract
The performance of the protein structure prediction server pro-sp3-TASSER in CASP8 is described. Compared to CASP7, the major improvement in prediction is in the quality of input models to TASSER. These improvements are due to the PRO-SP3 threading method, the improved quality of contact predictions provided by TASSER_2.0, multiple short TASSER simulations for building the full length model, and the accuracy of model selection using the TASSER-QA quality assessment method. Finally, we analyze the overall performance and highlight some successful predictions of the pro-sp3-TASSER server.
Keywords: protein structure prediction, TASSER, chunk-TASSER
Introduction
During the past few years, our laboratory has developed the protein structure prediction method TASSER (Threading/ASSembly/Refinement)1 and its improved version chunk-TASSER for targets of Medium/Hard difficulty2. The full TASSER procedure involves identification of template fragments by threading method followed by assembly and refinement of the fragments. The primary usage of TASSER is in large-scale automatic protein structure prediction1,3-5, although TASSER could be used for manual protein structure predictions by manually identifying and modifying template fragments. In past CASPs, TASSER was used for both server and human predictions3,6.
The performance of TASSER depends strongly on the energy functions derived from the input models. For automatic servers, the input models can be built from templates identified by threading methods as in METATASSER6 which uses SPARKS7, SP3 8 and PROSPECTOR_39 threading methods. Threading is also used for generation of side-chain contact potentials in addition to template identification. For human predictions in CASP8, we also include models selected either manually or automatically from other server predictions that were not available for automated server. For very difficult targets, where the identified templates are unreliable, we developed chunk-TASSER. In chunk-TASSER, ab initio folded fragments or chunks of three consecutive regular secondary structure segments are also included in the input models. To improve our TASSER based approaches, we have made effort in two directions: First, we have improved the accuracy of predicted contacts while maintaining reasonable coverage in TASSER 2.0 10 by a composite sequence approach; second, by improving the input model accuracy as in pro-sp3-TASSER by selecting template models from an ensemble of diversified models built by short TASSER simulations. The main difference between pro-sp3-TASSER and our previously developed METATASSER6 is in the identification and selection of template models input into TASSER for refinement. METATASSER6 uses three state-of-the-art threading methods, SPARKS7, SP3 8 and PROSPECTOR_39, for template identification and a 3D-jury11 approach for template rank and selection, while pro-sp3-TASSER employs a new threading approach, PRO-SP3 for template identification and TASSER-QA12 for template model selection.
Method
Since a detailed description of pro-sp3-TASSER method has been given elsewhere13, here we just provide a brief overview. As shown in Figure 1, pro-sp3-TASSER has three main steps: (A) threading and alignment by the newly developed PRO-SP3 threading method; (B) multiple short TASSER simulations that generate an ensemble of diversified full length models; (C) selection of top models with TASSER-QA from the ensemble, followed by full TASSER refinement and final model selection. PRO-SP3 consists of the SP3 threading score and four other scores derived from PROSPECTOR_39 and SP3 score components8. These five different scores independently rank and align the sequence to templates. PRO-SP3 threading was shown in Ref.13 to perform better than the combination of SPARKS, SP3 and PROSPECTOR_3. For targets of Medium/Hard difficulty (i.e. those with poor quality templates/alignments), alternative sequence to template alignments for top ranked templates are generated by a parametric alignment method14. Target difficulty is classified by the Z-score of the top template in the SP3 threading score: targets with Z-score ≥ 6.0 are classified as Easy (those likely to have a good template/alignment), with a Z-score ≤ 4.5 as Hard (having poor quality templates with inaccurate alignments), and those with 4.5 < Z-score < 6.0 as Medium targets, respectively. Template models (including alternative alignments for Medium/Hard targets) generated independently by the five threading scores are then grouped into different sets and inputted into TASSER for short simulations (∼10 hrs each simulation) to generate full length models. For Easy targets, up to 105 models and for Medium/Hard targets, up to 155 models are generated. These models are then ranked by the protein quality assessment prediction method TASSER-QA 12, and the top 20 models are selected for further full TASSER refinement. Special attention is paid to possible multiple domain and extremely easy targets. To deal with possible multiple domain proteins, we first check the coverage of the top template identified by SP3. If more than 50 continuous residues are unaligned in the top scoring template, in addition to modeling the full-length target sequence, the unaligned and aligned regions are modeled separately. The separately modeled, putative domains are then superimposed onto the full-length models in the full TASSER refinement (step (C)). Another special case is that when the top ranked template by SP3 has a Z-score more than 2.0 units larger than the second ranked template or if it has sequence identity to the target of more than 50%, then we only use the single, top scoring template (with five alignments, one each provided by the five individual threading scores) in the subsequent modeling procedure. We do this so as not to diminish the accuracy of the prediction due to contamination by the dominating poorer quality templates
Figure 1.
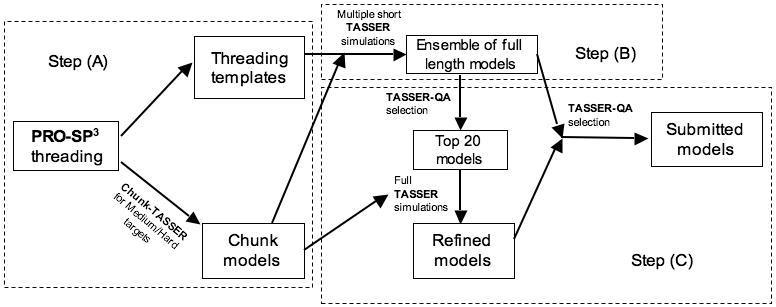
Flowchart of the pro-sp3-TASSER server.
In the above short TASSER simulations as well as the subsequent full TASSER refinements, we always use the chunk-TASSER protocol2 for Medium/Hard targets where ab initio folded chunk structures are included in TASSER inputs. Furthermore, for Medium/Hard targets, as these are of higher accuracy, the new predicted contacts and contact potential of TASSER 2.010 is also included in the full TASSER or chunk-TASSER refinements. The final five submitted models are selected from both the ensemble of models by short TASSER runs and by full TASSER refinements. Since TASSER models are represented with C α atoms only, main-chain atoms are added by the PULCHRA program15 and side-chains are re-built by an in-house method based on DFIRE energy16 optimization. For server predictions, the whole procedure is limited to 72 hours. A summary of the methods/protocols used in pro-sp3-TASSER server has been given in Table 1.
Table 1.
Summary of TASSER-based methods/protocols used in pro-sp3-TASSER server
| Method/protocol | Brief description |
|---|---|
| TASSER | Original TASSER method and force field based on Cα and side-chain-center of mass representation and PROSPECTOR_3 threading method1. |
| TASSER_2.0 | TASSER method enhanced with a new side-chain contact potential predicted by the composite method, specifically for Medium/Hard targets10. The composite method combines predictions from both native template sequences and designed template sequences. |
| Chunk-TASSER | TASSER method supplemented with ab initio folded chunk models in the derivation of contact and distance restraints, specifically for Medium/Hard targets2. Chunks are defined as 3 consecutive regular secondary structure segments and folded by a fragment insertion method. |
| METATASSER | TASSER method based on SPARKS, SP3, PROSPECTOR_3 threading methods and 3D-jury for template model selection6. |
| Pro-sp3-TASSER | Method used in this report. A new threading method, PRO-SP3, and TASSER-QA for template model selection are used13. It also uses TASSER, chunk-TASSER, and the new side-chain contact predictions in TASSER_2.0 for model building and refinement. |
| TASSER-QA | Protein structure quality assessment prediction method that combines a fragment comparison score and TASSER Cα atom contact energy12. It has comparable or better performance compared to the state-of-the-art methods in the literature for selecting good quality models. |
| Short TASSER | A simulation protocol of TASSER, or chunk-TASSER that limits the simulation time (here to 10 hours) regardless of the size of the target, i.e., TASSER or chunk-TASSER stops when the time limit is reached. |
| Full TASSER | A simulation protocol that uses the original settings of TASSER or chunk-TASSER which means longer time simulations for larger targets for a given number of Monte Carlo sampling steps. |
Results
We compared our current method with the previously developed METATASSER method6 now updated with chunk-TASSER2 on a benchmark set of 723 proteins less than 250 residues in length that has 348 Easy, 155 Medium and 220 Hard targets, respectively. Pro-sp3-TASSER shows on average a statistically significant, 2-3% TM-score improvement over METATASSER17. In CASP8, METATASSER also implemented a special treatment for putative multiple domain proteins and extremely Easy targets as well as for the selection of final models for submission from multiple TASSER simulations by TASSER-QA. As shown in Table 2, the overall performance of pro-sp3-TASSER in CASP8 (a relatively smaller target set compared to the 723 protein benchmark set) shows that it performs slightly better than METATASSER according to both the official assessment and our own analysis. Therefore, the difference between pro-sp3-TASSER and METATASSER comes mainly from input models that are selected for TASSER refinement. Pro-sp3-TASSER shows slightly better performance for Medium/Hard targets; however, due to small statistics, the difference is insignificant.
Table 2.
Comparison of pro-sp3-TASSER and METATASSER on the 124 whole chain CASP8 targets#.
| First model | Best of top five | |||||||
|---|---|---|---|---|---|---|---|---|
| Target set (# of targets) | METATASSER | Pro-sp3-TASSER | METATASSER | pro-sp3-TASSER | ||||
| Easy (99) | 98 | 76.52 | 97 | 76.38 | 98 | 78.64 | 98 | 78.77 |
| Medium (11) | 9 | 4.71 | 8 | 4.98 | 9 | 5.19 | 9 | 5.23 |
| Hard (14) | 2 | 4.00 | 3 | 4.23 | 4 | 4.66 | 8 | 5.14 |
| All (124) | 109 | 85.23 | 108 | 85.60 | 111 | 88.49 | 115 | 89.14 |
Numbers in the first columns under each method are numbers of targets having first/best of top five models with TM-score to native > 0.4. TM-score of 0.4 is a statistically significant threshold for structural similarity. The numbers in second columns are TM-scores.
Figure 2A presents the pro-sp3-TASSER server prediction of T0472, which is one of the best predictions among all servers and human groups for this target. While T0472 is a two-domain protein, the PRO-SP3 identified template covers only one domain. In step (B), some good whole chain models in the ensemble of structures were generated by TASSER, and TASSER-QA was able to pick out these good models for further TASSER refinement. Another example of an outstanding prediction by pro-sp3-TASSER server is shown in Figure 2B for T0478-D1. This is a hard target for PRO-SP3 threading and the best template model built from 1n1b_A has a TM-score17 to native of 0.40. The fifth model of the pro-sp3-TASSER server prediction has a TM-score to native of 0.55, owing to the good performance of chunk-TASSER on α-helical proteins2. A third example shown in Figure 2C is target T0514, a 145 residue single domain α/β protein that is a Hard target. Pro-sp3-TASSER has the best server prediction with a TM-score of the first model 0.615 (RMSD=5.48 Å). The best template model for this target has TM-score to native of 0.5 (RMSD=6.93 Å) from template 2zf8_A. From the structure ensemble generated by short TASSER simulations, TASSER-QA has selected a number of models with TM-score > 0.5, but the best one, which is ranked first, has a TM-score=0.62. After the full TASSER refinement, the final model is further improved compared to TASSER-QA selected ones.
Figure 2.
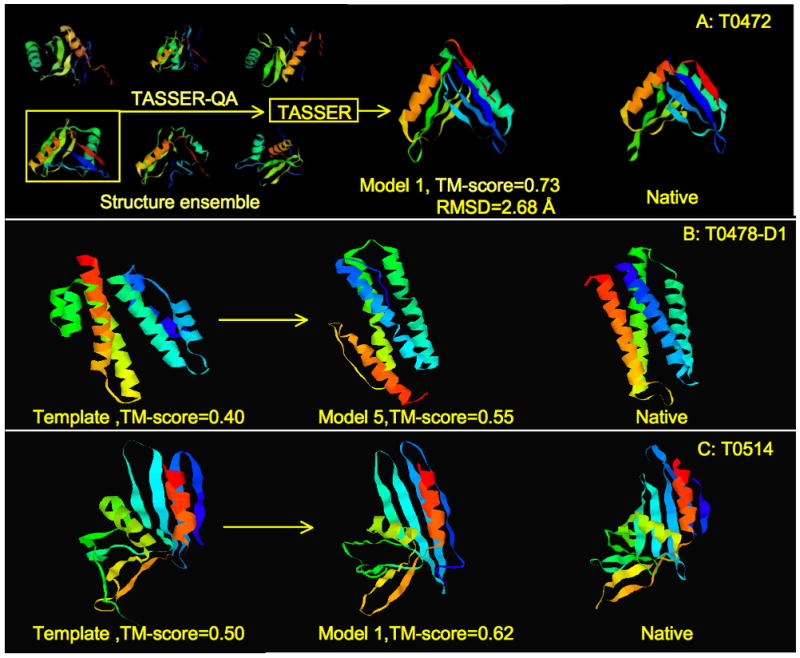
Pro-sp3-TASSER server prediction examples: (A) prediction for target T0472; (B) prediction for target T0478-D1; (C) prediction for target T0514.
Discussion
The overall performance of pro-sp3-TASSER server, particularly for the best of top five models, is among the top ranked servers according to the official assessment and our own evaluation. There are also several outstanding individual predictions by the pro-sp3-TASSER server. One reason for its good performance is that the new threading method PRO-SP3 used here is even better than the combination of the three state-of-the-art methods: SPARKS, SP3 and PROSPECTOR_313. The other reason is the use of multiple simulations of TASSER that generate much more diversified model quality than a single simulation does. The diversified ensemble of models in most cases has a few good models. Subsequent use of TASSER-QA selection enriches the good models among the top 20 selected models13 as in the case of target T0472; although in other cases, it may not be as apparent, as in T0472. According to our benchmark, chunk-TASSER and the new side-chain contact potential in TASSER_2.0 also show incremental improvement for Medium/Hard targets over original TASSER2,10.
Although pro-sp3-TASSER performed well among all servers, there are still a number of problems that need to be addressed including ranking, especially for Medium/Hard targets, where there are often better models in the ensemble of structures that are not selected by TASSER-QA. Lack of a perfect ranking method also results in the big difference between the first submitted models and the best submitted models (3% for Easy, 5% for Medium, and 22% for Hard targets, respectively). There also remain significant issues with domain parsing as indicated by the worse relative performance compared to other servers when only domains are used by official assessment. Furthermore, the threading component of pro-sp3-TASSER still fails to identify good templates for a number of Medium/Hard targets despite the fact that these templates are in the PDB. Another critical issue that needs improvement is the construction of an acceptable detailed atomic model from the TASSER reduced protein representation. Both the backbone and side chain geometries need significant improvement. Also the hydrogen bond scheme in both TASSER and in the detailed atomic model needs significant refinement. These issues are currently being addressed in our ongoing research.
Acknowledgments
This research was supported in part by grant Nos. GM-37408 and GM-48835 of the Division of General Medical Sciences of the National Institutes of Health.
References
- 1.Zhang Y, Skolnick J. Automated structure prediction of weakly homologous proteins on genomic scale. Proc Natl Acad Sci (USA) 2004;101:7594–7599. doi: 10.1073/pnas.0305695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou H, Skolnick J. Ab initio protein structure prediction using chunk-TASSER. Biophys J. 2007;93:1510–1518. doi: 10.1529/biophysj.107.109959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Arakaki A, Skolnick J. TASSER: An automated method for the prediction of protein tertiary structures in CASP6. Proteins. 2005;(suppl 7):91–98. doi: 10.1002/prot.20724. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, DeVries ME, Skolnick J. Structure modeling of all identified G protein-coupled receptors in the human genome. PLoS Computational Biology. 2006;2:0088–0099. doi: 10.1371/journal.pcbi.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pandit SB, Zhang Y, Skolnick J. TASSER-Lite: An automated tool for protein comparative modeling. Biophysical Journal. 2006;91:4180–4190. doi: 10.1529/biophysj.106.084293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou H, Pandit SB, Lee S, Borreguerro J, Chen H, Wroblewska L, Skolnick J. Analysis of TASSER based CASP7 protein structure prediction results. Proteins. 2007;69(S8):90–97. doi: 10.1002/prot.21649. [DOI] [PubMed] [Google Scholar]
- 7.Zhou H, Zhou Y. Single-body residue-level knowledge-based energy score combined with sequence-profile and secondary structure information for fold recognition. Proteins. 2004;55:1005–1013. doi: 10.1002/prot.20007. [DOI] [PubMed] [Google Scholar]
- 8.Zhou H, Zhou Y. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins. 2005;58:321–328. doi: 10.1002/prot.20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skolnick J, Kihara D, Zhang Y. Development and large scale benchmark testing of the PROSPECTOR 3.0 threading algorithm. Proteins. 2004;56:502–518. doi: 10.1002/prot.20106. [DOI] [PubMed] [Google Scholar]
- 10.Lee S, Skolnick J. Benchmarking of TASSER_2.0: An improved protein structure prediction algorithm with more accurate predicted contact restraints. Biophys J. 2008;95:1956–1964. doi: 10.1529/biophysj.108.129759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 12.Zhou H, Skolnick J. Protein model quality assessment prediction by combining fragment comparisons and a consensus Cα contact potential. Proteins. 2007;71:1211–1218. doi: 10.1002/prot.21813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou H, Skolnick J. Protein structure prediction by pro-sp3-TASSER. Biophys J. 2009;96:2119–2127. doi: 10.1016/j.bpj.2008.12.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chivian D, Baker D. Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucl Aci Res. 2006;34:e112. doi: 10.1093/nar/gkl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rotkiewicz P, Skolnick J. Fast procedure for reconstruction of full-atom protein models from reduced representations. Journal of Computational Chemistry. 2008;29:1460–1465. doi: 10.1002/jcc.20906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Science. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Skolnick J. A scoring function for the automated assessment of protein structure template quality. Proteins. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]