

Abstract

A one-pot, asymmetric multicatalytic formal [3+2] reaction between 1,3-dicarbonyls and α,β-unsaturated aldehydes is described. The multicatalytic process involves a secondary amine catalyzed Michael addition followed by a N-heterocyclic carbene catalyzed intramolecular crossed benzoin reaction to afford densely functionalized cyclopentanones with high enantioselectivities. The reaction proceeds with a variety of alkyl and aryl enals as well as a range of 1,3-dicarbonyls (diketones and β-ketoesters). The functionalized products are obtained from cheap, readily available starting materials in a rapid and efficient manner in a one-pot, one-step operation.
Increased focus has recently been placed on the development of organic transformations that allow for the rapid construction of densely functionalized molecules from simple readily available starting materials. One approach in which these demands can be achieved is through the expanding field of cascade catalysis.1 Although a number of examples of asymmetric cascade catalysis have been developed, most rely on a single catalyst1a,d–g,2 to perform two sequential operations. More recently, multiple catalyst systems for cascade reactions have been realized.1b,c,3 Although these reactions showcase the potential power in this field, relatively few asymmetric examples exist.4 The major challenge in developing multiple catalyst systems for cascade catalysis is that each catalyst must be compatible with all reagents, intermediates and other catalysts present from the onset of the reaction. This circumstance is occasionally avoided by addition of reagents and/or catalysts at the midpoint of a given reaction.
Stimulated by the inherent basicity of many common organic catalysts, most notably secondary amines,5 Cinchona alkaloids,6 and nucleophilic carbenes,7 we speculated that these catalysts could co-exist in a single flask to exert their influence over complementary bond-forming events without mutual interference. Indeed, we have already revealed this by utilizing carbene and acyl transfer co-catalysis in the synthesis of amides.8 To generalize this concept, we initiated a research program to develop multicatalytic asymmetric processes.
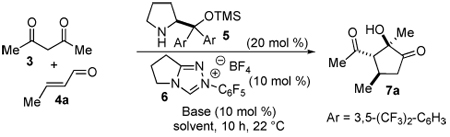
At the outset of our studies, we considered the use of active methylene compounds and α,β-unsaturated aldehydes as the first opportunity to test our hypothesis. A number of examples involving the enantioselective secondary-amine-catalyzed conjugate addition of malonates and β-ketoesters to α,β-unsaturated aldehydes9 and ketones10 have been described. Reaction between a 1,3-diketone and an enal generates aldehyde 1 bearing a tethered ketone (eq 1). We envisioned that this intermediate would undergo an intramolecular crossed benzoin11 reaction in the presence of a carbene catalyst to afford highly functionalized cyclopentanone 2 via a formal [3+2] process in a multicatalytic cascade sequence.
 |
(1) |
While both secondary amines and carbenes are inherently basic, initiation of the sequence by carbene reaction could be detrimental given the possibility of the carbene-catalyzed benzoin,12 Stetter13 and redox14,15 pathways. The test reaction involved treatment of acetylacetone with crotonaldehyde, 3,5-bistrifluoromethyl diphenyl prolinol TMS ether catalyst 5,16 and triazolium salt 617 (Table 1). Combining the two catalysts in the absence of exogenous base yields trace amounts of the desired cyclopentanone (entry 1). However, intermediate 1 is observed by crude 1H NMR, suggesting that the Michael addition occurs but the carbene is not being generated under the reaction conditions. To alleviate this problem, catalytic triethylamine was added to the reaction. In the presence of base, the desired cyclopentanone 7a is formed in moderate yield and excellent enantioselectivity (entry 2). A brief screen of the reaction conditions revealed sodium acetate to be the optimal base, while chloroform gives the highest level of diastereoselectivity (entries 3–6). Increasing the concentration in the presence of two equivalents of dicarbonyl 3 provides optimal yields without loss of enantioselectivity or diastereoselectivity (entry 7).
Table 1.
Catalyst and Solvent Screen.
 | |||||
|---|---|---|---|---|---|
| Entrya | Solvent | Base | Yield (%) | drb | ee (%)c,d |
| 1 | CH2Cl2 | none | trace | nd | nd |
| 2 | CH2Cl2 | Et3N | 31 | 80:20:<1:<1 | 84 |
| 3 | CH2Cl2 | NaOAc | 55 | 80:20:<1:<1 | 86 |
| 4 | EtOH | NaOAc | <20 | 80:20:<1:<1 | 70 |
| 5 | PhMe | NaOAc | 53 | 80:20:<1:<1 | 84 |
| 6 | CHCl3 | NaOAc | 60 | 85:15:<1:<1 | 86 |
| 7e | CHCl3 | NaOAc | 93 | 85:15:<1:<1 | 86 |
All reactions conducted using 20 mol % 5, 10 mol % 6, and 10 mol % base at 22 °C (0.06M) unless otherwise stated.
Diastereomeric ratio determined by GC analysis of unpurified reaction mixture.
Enantiomeric excess determined by GC analysis on chiral stationary phase.
Major diastereomer.
20 mol % 5, 10 mol % 6, 10 mol % NaOAc, and 2 equiv 3 (0.2M) at 22 °C.
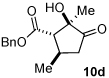
In all cases, only two of the four possible diastereomers are observed by GC/MS analysis. The relative configuration of the major diastereomer was assigned on the basis of nOe studies, confirmed by analogy to X-ray crystal structures of major and minor diastereomers (13 and 10d respectively). The absolute configuration is based on previous work by Jørgensen and coworkers.9d
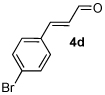
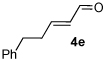
A series of α,β-unsaturated aldehydes with varying substitution was subjected to the optimized reaction conditions (Table 2). Alkyl as well as aromatic substitution is tolerated in the reaction (entries 1–5). Protected alcohols and amines participate in the desired reaction to give products readily amenable to further functionalization (entries 6–8). The sterically larger iso-propyl enal 4i provides reduced yields and diastereoselectivity while maintaining good levels of enantioselectivity (entry 8).
Table 2.
α,β-Unsaturated Aldehyde Scope.
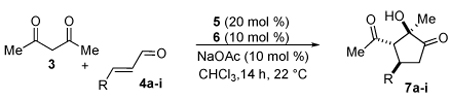
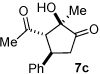
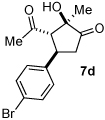
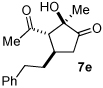
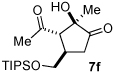
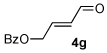
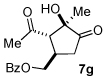
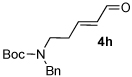
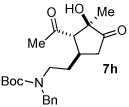
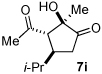
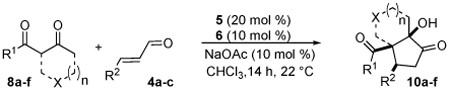
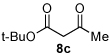
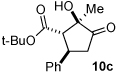
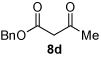
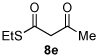
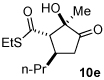
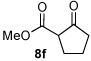
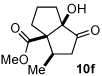
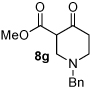
Next we examined unsymmetrical 1,3-dicarbonyls under the optimized reaction conditions (Table 3). Methyl, ethyl, tert-butyl, and benzyl acetoacetate provide the desired products with alkyl and aromatic enals giving high levels of enantioselectivity (entries 1–4). β-Ketothioester 8e also gives the desired product with good selectivity (entry 3). However, these unsymmetrical dicarbonyls afford all four possible diastereomers with moderate selectivity. Cyclic β-ketoesters also react under the optimized conditions to give both [3.3.0] and [4.3.0] bicyclic systems (entries 6–7). Benzyl protected nitrogen is tolerated in the cyclic β-ketoester backbone to afford the highly functionalized bicyclic structure 10g (entry 5).
Table 3.
β-Ketoester Scope
 | |||||
|---|---|---|---|---|---|
| Entrya | R2 | β-Ketoester | Product | Yield (%) (drb) |
eec,d |
|
1 |
Me 4a |
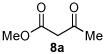 |
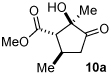 |
90 (64:33:3:<1) |
91 |
|
2 |
n-Pr 4b |
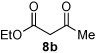 |
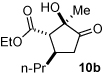 |
80 (60:30:8:2) |
93 |
|
3 |
Ph 4c |
 |
 |
86 (60:35:5:<1) |
97 |
| 4 | Me 4a |
 |
 |
90 (58:39:2:<1) |
82 |
|
5 |
n-Pr 4b |
 |
 |
56 (69:22:6:3) |
81 |
|
6 |
Me 4a |
 |
 |
79 (80:20:<1:<1) |
94 |
|
7 |
Me 4a |
 |
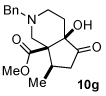 |
76 (85:15:<1:<1) |
90 |
See Table 2.
To shed light on the observed reactivity, we conducted a control experiment leaving the azolium catalyst out of the reaction (eq 2). Prolinol catalyst 5 mediates the asymmetric Michael reaction furnishing aldehyde 11 in 70% yield. Surprisingly, subjection of this intermediate to the azolium catalyst and base generates the desired product in only 58% ee again with diminished yield.18 Experiments aimed at elucidating the source of this variance revealed that aldehyde 11 undergoes a retro-Michael in the presence of prolinol 5, thus eroding ee. This situation is exacerbated in the presence of silica gel.19 The carbene catalyst serves to shuttle the intermediate aldehyde on to product thus limiting the prolinol-mediated retro-Michael.
To lend further credence to this hypothesis, we monitored the reaction between acetylacetone and crotonaldehyde by withdrawing aliquots at regular intervals (Figure 1). The GC data reveals only moderate buildup of intermediate aldehyde 11 and continuous formation of product, indicating both catalytic cycles are operating concurrently. The lower yield observed for cyclopentanone 7a obtained from the stepwise process (46% vs 93% in the one-step process) suggests a symbiotic relationship between the two catalysts wherein both catalysts function more efficiently in the presence of each other than they do independently. This type of reactivity highlights the power of the multicatalytic cascade process wherein the one pot reaction actually outperforms the sequential process, allowing for reactivity that is otherwise not easily accessible.
Figure 1.

One-pot cascade reaction monitored by GC analysis utilizing 20 mol % 5, 10 mol % 6, 10 mol % NaOAc, with 1 equiv acetylacetone 3 and crotonaldehyde 4a in CHCl3 (0.2M) at 22 °C
 |
(2) |
Finally, in an effort to probe the possibility of controlling the regio-selectivity of unsymmetrical 1,3 diketones, 1-benzoyl-acetone 12 was subjected to the reaction conditions (eq 3). To our delight the desired product was obtained in 74% yield, 87% ee in a ratio of 4:1 (major : the sum of all other potential regioisomers and diastereomers). It seems that sterics play a more significant role than eletronics in the carbene catalyzed intramolecular benzoin reaction, since the sterically smaller methyl ketone reacts preferentially over the larger yet more electrophilic phenyl ketone.
 |
(3) |
In conclusion, we have developed an operationally simple single step multicatalytic cascade process20,21 for the preparation of α-hydroxycyclopentanones containing three contiguous stereocenters. The highly functionalized products are obtained from cheap, readily available reagents in high enantioselectivities and moderate diastereoselectivities. We further provide conclusive evidence that both catalysts are mutually compatible and are operating concurrently. Experiments aimed at extending the scope of these types of transformations are currently underway.
Supplementary Material
Acknowledgment
We thank NIGMS for support (GM72586). T.R. thanks the Monfort Family Foundation for a Monfort Professorship. We thank Derek Dalton and Kevin Oberg (CSU) for solving the crystal structures of 10d and 13. We thank a reviewer for helpful suggestions.
Footnotes
Supporting Information Available: Experimental procedures, characterization 1H/13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews see: Ajamian A, Gleason JL. Angew. Chem. Int. Ed. 2004;43:3754. doi: 10.1002/anie.200301727. Lee JM, Na Y, Han H, Chang S. Chem. Soc. Rev. 2004;33:302. doi: 10.1039/b309033g. Wasilke J-C, Obrey SJ, Baker RT, Bazan GC. Chem. Rev. 2005;105:1001. doi: 10.1021/cr020018n. Chapman CJ, Frost CG. Synthesis. 2007:1. Enders D, Grondal C, Hüttl MRM. Angew. Chem. Int. Ed. 2007;46:1570. doi: 10.1002/anie.200603129. Walji AM, MacMillan DWC. Synlett. 2007:1477. Xinhong Y, Wang W. Org. Biomol. Chem. 2008;6:2037. doi: 10.1039/b800245m.
- 2.For select recent examples see: Enders D, Hüttl MRM, Grondal C, Raabe G. Nature. 2006;441:861. doi: 10.1038/nature04820. Marigo M, Bertelsen S, Landa A, Jørgensen KA. J. Am. Chem. Soc. 2006;128:5475. doi: 10.1021/ja058490o. Enders D, Narine AA, Benninghaus T, Raabe G. Synlett. 2007:1667. Wang J, Li H, Xie H-X, Zu L-S, Shen X, Wang W. Angew. Chem. Int. Ed. 2007;46:9050. doi: 10.1002/anie.200703163. Zu L-S, Li H, Xie H-X, Wang J, Jiang W, Tang Y, Wang W. Angew. Chem. Int. Ed. 2007;46:3732. doi: 10.1002/anie.200700485. Hayashi Y, Toyoshima M, Gotoh H, Ishikawa H. Org. Lett. 2009;11:45. doi: 10.1021/ol802330h. Ishikawa H, Suzuki T, Hayashi Y. Angew. Chem. Int. Ed. 2009;48:1304. doi: 10.1002/anie.200804883. Zu L, Zhang S, Xie H, Wang W. Org. Lett. 2009;11:1627. doi: 10.1021/ol9003433. Jiang H, Elsner P, Jensen KL, Falcicchio A, Marcos V, Jørgensen KA. Angew. Chem. Int. Ed. 2009;48:6844. doi: 10.1002/anie.200901446.
- 3.Cernak TA, Lambert TH. J. Am. Chem. Soc. 2009;131:3124. doi: 10.1021/ja809897f. [DOI] [PubMed] [Google Scholar]
- 4.For select recent examples see: Huang Y, Walji AM, Larsen CH, MacMillan DWC. J. Am. Chem Soc. 2005;127:15051. doi: 10.1021/ja055545d. Simmons B, Walji AM, MacMillan DWC. Angew. Chem. Int. Ed. 2009;48:4349. doi: 10.1002/anie.200900220.
- 5.(a) Erkkilä A, Majander I, Pihko PM. Chem. Rev. 2007;107:5416. doi: 10.1021/cr068388p. [DOI] [PubMed] [Google Scholar]; (b) Mukherjee S, Yang JW, Hoffmann S, List B. Chem. Rev. 2007;107:5471. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]
- 6.Tian S-K, Chen Y, Hang J, Tang L, McDaid P, Deng L. Acc. Chem. Res. 2004;37:621. doi: 10.1021/ar030048s. [DOI] [PubMed] [Google Scholar]
- 7.(a) Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (b) Moore JL, Rovis T. Top. Curr. Chem. 2009:291. doi: 10.1007/978-3-642-02815-1_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vora HU, Rovis T. J. Am. Chem. Soc. 2007;129:13796. doi: 10.1021/ja0764052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Brandau S, Landa A, Franzén J, Marigo M, Jørgensen KA. Angew. Chem. Int. Ed. 2006;45:4305. doi: 10.1002/anie.200601025. [DOI] [PubMed] [Google Scholar]; (b) Carlone A, Cabrera S, Marigo M, Jørgensen KA. Angew. Chem. Int. Ed. 2007;46:1101. doi: 10.1002/anie.200604479. [DOI] [PubMed] [Google Scholar]; (c) Carlone A, Marigo M, North C, Landa A, Jørgensen KA. Chem. Commun. 2006:4928. doi: 10.1039/b611366d. [DOI] [PubMed] [Google Scholar]; (d) Rueping M, Sugiono E, Merino E. Chem. Eur. J. 2008;14:6329. doi: 10.1002/chem.200800836. [DOI] [PubMed] [Google Scholar]; (e) Rueping M, Kuenkel A, Tato F, Bats JW. Angew. Chem. Int. Ed. 2009;48:3699. doi: 10.1002/anie.200900754. [DOI] [PubMed] [Google Scholar]
- 10.(a) Halland N, Aburel PS, Jørgensen KA. Angew. Chem. Int. Ed. 2003;42:661. doi: 10.1002/anie.200390182. [DOI] [PubMed] [Google Scholar]; (b) Halland N, Aburel PS, Jørgensen KA. Angew. Chem. Int. Ed. 2004;43:1272. doi: 10.1002/anie.200353364. [DOI] [PubMed] [Google Scholar]; (c) Halland N, Hansen T, Jørgensen KA. Angew. Chem. Int. Ed. 2003;42:4955. doi: 10.1002/anie.200352136. [DOI] [PubMed] [Google Scholar]; (d) Knudsen KR, Mitchell CET, Ley SV. Chem. Commun. 2006:66. doi: 10.1039/b514636d. [DOI] [PubMed] [Google Scholar]
- 11.For recent references on the crossed aldehyde-ketone benzoin reaction, see: Hachisu Y, Bode JW, Suzuki K. J. Am. Chem. Soc. 2003;125:8432. doi: 10.1021/ja035308f. Enders D, Niemeier O. Synlett. 2004:2111. Enders D, Niemeier O, Balensiefer T. Angew. Chem. Int. Ed. 2006;45:1463. doi: 10.1002/anie.200503885. Takikawa H, Hachisu Y, Bode JW, Suzuki K. Angew. Chem. Int. Ed. 2006;45:3492. doi: 10.1002/anie.200600268. Takikawa H, Suzuki K. Org. Lett. 2007;9:2713. doi: 10.1021/ol070929p.
- 12.Ide WS, Buck JS. Org. React. 1948;4:269. [Google Scholar]
- 13.(a) Stetter H, Kuhlmann H. Org. React. 1991;40:407. [Google Scholar]; (b) Read de Alaniz J, Rovis T. Synlett. 2009:1189. doi: 10.1055/s-0029-1216654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Reynolds NT, Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2004;126:9518. doi: 10.1021/ja046991o. [DOI] [PubMed] [Google Scholar]; (b) Reynolds NT, Rovis T. J. Am. Chem. Soc. 2005;127:16406. doi: 10.1021/ja055918a. [DOI] [PubMed] [Google Scholar]; (c) Vora HU, Moncecchi JR, Epstein O, Rovis T. J. Org. Chem. 2008;73:9727. doi: 10.1021/jo8020055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Chow KY-K, Bode JW. J. Am. Chem. Soc. 2004;126:8126. doi: 10.1021/ja047407e. [DOI] [PubMed] [Google Scholar]; (b) Sohn SS, Rosen EL, Bode JW. J. Am Chem. Soc. 2004;126:14370. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]; (c) Sohn SS, Bode JW. Org. Lett. 2005;7:3873. doi: 10.1021/ol051269w. [DOI] [PubMed] [Google Scholar]
- 16. Marigo M, Wabnitz TC, Fielenbach D, Jørgensen KA. Angew. Chem. Int. Ed. 2005;44:794. doi: 10.1002/anie.200462101. Also see: Hayashi Y, Gotoh H, Hayashi T, Shoji M. Angew. Chem. Int. Ed. 2005;44:4212. doi: 10.1002/anie.200500599.
- 17.Kerr MS, Read de Alaniz J, Rovis T. J. Org. Chem. 2005;70:5725. doi: 10.1021/jo050645n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Addition of 10 mol % sodium acetate to the first step results in similar yield and enantioselectivity. We also observed that carrying out the reaction in a stepwise manner led to more complex reaction mixtures.
- 19.See Supporting Information.
- 20.General Procedure: a 1 dram vial was equipped with a magnetic stir bar under argon and charged with catalyst 6 (0.024 mmol). CHCl3 (1 ml), 1,3 dicarbonyl (0.448 mmol), and enal (0.224 mmol) were added sequentially followed by catalyst 5 (0.044 mmol) and NaOAc (0.024 mmol) in one portion. The reaction was allowed to stir at room temperature for 14 h at which point the reaction mixture was filtered through a short pad of silica gel and eluted with Et2O (~10 ml), and then concentrated in vacuo. The resulting crude product was purified by flash silica gel chromatography.
- 21.Reaction of 4a and 8a was conducted on 2.4 mmol scale using 10 mol % 5, 5 mol % 6, and 5 mol% NaOAc to afford 90% yield, 91% ee and identical diastereomer ratio of 10a.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.