

Abstract
Nearly degenerate asymmetric stretches with perpendicular transition dipole moments of the deuterated guanidinium cation (DGdm+) in D2O and D-glycerol/D2O mixtures at 1600 cm−1 were investigated by linear FTIR spectroscopy and polarization dependent femtosecond pump-probe spectroscopy. The vibrational coupling of the asymmetric stretches of guanidinium occurs within 0.5 ps and leads to fast decay of the anisotropy to a level of 0.1. A systematic study of the influence of the coherence transfer on pump-probe signals is given. Following this decay the anisotropy decays with a time constant of 4.1 ps in D2O by rotational diffusion about an axis perpendicular to the DGdm+ mean plane. The presence of aggregation was demonstrated for concentrations higher than 0.2 M.
Keywords: Ultrafast IR spectroscopy, guanidinium chloride, pump-probe, anisotropy
INTRODUCTION
The ability of various salts to denature proteins 1–9 or modify the protein associated water structures 10–12 related to protein stability has been widely demonstrated. In particular, guanidinium (C(NH2)3+, HGdm+) salts have particular interest mainly because the chloride, HGdmCl, is a very effective protein denaturant that can cause partial unfolding at concentrations well below 1 M 13–15. Experiments based on neutron diffraction 16, 17, calorimetry 18–20, solubility 21, densimetry 22, and FTIR spectroscopy 23 have been employed to clarify this denaturing power and two important effects have been identified: the direct interaction between amphiphilic HGdm+ and the nonpolar groups of the proteins 18, 21, 24; and the modification of the water hydrogen bonding network upon the addition of the ions 23. A more complete description of the structure and properties of HGdm+ will have important applications in understanding the unfolding and aggregation of peptides and proteins that are involved in essential biological processes related to diseases 25, 26.
Recent studies of HGdm+ include a report on the kinetics that shows HGdm+ influencing the end-to-end diffusion of a small peptide to which it transiently binds 24 and a spectroscopic study reporting the significant effects of HGdm+ on the water OH stretching vibrations 23. Evidently the various thermodynamic, spectroscopic and kinetic properties of the HGdm+ salts in water and in their association with proteins present significant challenges for modern physical methods that are able to access both structural and dynamical features in a single experiment. One such method is polarization dependent pump-probe spectroscopy 27 which provides the transition dipole reorientational motions and, as this paper will show, the environmentally induced intermode coupling.
Computations 28 of gas phase HGdm+ predict a D3 symmetry ion with the C and N atoms on the plane perpendicular to a three fold symmetry axis. The H-N-H bond angle is 120° and the plane of each of the NH2 groups make an angle ~ 12° with the CN3 plane. The normal mode analysis 29 for the free ion demonstrates that there is a degenerate mode around 1600 cm−1 corresponding to the CN3 stretch and NH2 scissors motions that has a large dipole derivative. The presence of two nearly equal frequency modes in the mid-IR range for HGdm+ in water provides a novel opportunity to expose hitherto unknown properties of its local structure and dynamics.
The goal of the present paper is to investigate the interactions of monomeric HGdm+ with water aqueous solution by probing the vibrational transitions at ca. 1600 cm−1 on ultrafast time scales by linear FTIR spectroscopy and polarization dependent femtosecond IR pump-probe method. Particular attention is given to the roles of coherence and population transfer between vibrational modes involved in pump-probe spectroscopy.
EXPERIMENTAL METHODS
Sample
The guanidinium salts are fully deuterated (C(ND2)3+, DGdm+) and dissolved in D2O (1.19 cP 30). A deuterated sample is required because the OH bending mode overlaps spectrally with the ca. 1600 cm−1 degenerate mode of the DGdm+ guanidinium salt. The guanidinium chloride was purchased from Sigma-Aldrich (http://www.sigmaaldrich.com) and completely deuterated by successive dissolution in excess D2O followed by lyophilization. Samples were held between two 2 mm CaF2 windows. For concentrations 1 M and below, a 6 or 12 micron spacer was used while for the higher concentrations, no spacer was used in the experiments. Mixtures of D-glycerol and D2O with weights of D-glycerol 59%, 81% and 100% having viscosity 10 cP, 70 cP and 1400 cP 31 were also used in the experiments.
Pump-probe experiment
The frequency selective pump-probe experiment was based on the generation of femtosecond laser pulses in the 6 μm region from a Ti:sapphire regenerative amplifier (Spitfire, Spectra-Physics) and home-made IR optical parametric amplifier (OPA) 32. The near transform-limited 75 fs pulse was centered at 1600 cm−1 and had ~2.4 μJ energy. The output was divided into pump (k⃑1) and probe (k⃑2) pulses. The pump pulse was vertically polarized and contained ~500 nJ energy. An attenuator composed of two polarizers was used to decrease the intensity of the probe to about a tenth of k⃑1 and to rotate to its polarization by 45° with respect to that of the pump pulse. The pump and probe beams were focused onto the sample by a parabolic mirror and the probe beam was re-collimated by a CaF2 lens. The probe was focused onto the entrance slit of a spectrometer and spectrally dispersed onto a liquid-nitrogen-cooled 32 element MCT array detector. A polarizer was placed after the sample to select parallel, perpendicular and magic angle polarization components of the probe pulse. The pump-probe interaction with the sample generates a field that heterodynes with the probe field on the detector. The interval between pump and probe pulses is denoted as T, and free evolution of the generated field occurs on the interval t. The first 0.25 ps (0.4 ps in case of neat D-glycerol) were excluded from data processing to completely avoid contamination by the CaF2 non-resonant signal. Additionally, the signals of the neat solvents were subtracted from the pump-probe data sets. The processing yields the transient absorptions Δα||(ω,T), Δα⊥(ω,T), and ΔαMA(ω,T) for parallel (XXXX tensor), perpendicular (XXYY tensor) and magic angle (MA) polarization components of the probe pulse, respectively. These signals are used to compute the anisotropy r (ω,T), which is independent of population relaxation and reflects transition dipole reorientation:
| (1) |
RESULTS
Figure 1 shows the FTIR spectra of DGdmCl in the 1500 ~ 1700 cm−1 region at different concentrations and different solvents. This vibrational transition is atop of a weak and broad D2O combination mode which has been subtracted in Figure 1a and Figure 1b. The center frequency (1600 cm−1) of the DGdm+ transition does not change significantly with concentration, however, its full width half maximum (FWHM) is concentration dependent. In the more dilute solutions, less than 0.25 M, the FWHM is ~ 28 cm−1. The variation of width with concentration is shown in Figure 1c. Compared with dilute solution, at 8 M, the FWHM increases by ~ 39 %. Figure 1b shows the FTIR spectrum of 0.1 M DGdmCl in a 59% D-glycerol/water mixture where the band at 1600 cm−1 is now clearly split into two components. The curves represented by squares in Figures 1a and 1b are simulated linear spectra based on parameters that are discussed later.
Figure 1.

Concentration dependent FTIR spectra of guanidinium chloride. (a) the normalized FTIR spectra of guanidinium chloride at 0.1 M (solid curve) and 8 M (dashed curve) after the water (D2O) combination mode is subtracted. (b) FTIR spectrum of 0.1 M guanidinium chloride in 59% D-glycerol/D2O (solid curve). (c) FWHM of absorption band for different concentration in D2O (inset is a blow-up of the low concentration regime). Simulated FTIR spectra of DGdm+ absorption (parameters from Table 2) are shown by squares on (a) and (b).
The pump-probe signal shows bleaching and absorption components separated by 35 cm−1 with almost equal amplitudes as shown in Figure 2. The ratio of the amplitudes of bleaching and excited state absorption is preserved during the scanned time range of T as discussed below. This ratio is an important observable since simulations show it is sensitive to certain parameters. The kinetics of the pump-probe data shows the rise and decay of the transient absorption signal (Figure 3a). The rising part of the signal is described by perturbed free induction decay 33 and the rise time is close to the inverse of the width of the absorption band in the linear FTIR spectrum. The lifetime (T1) of the ca. 1600 cm−1 vibrational state was obtained from pump-probe experiments for 0.1 M and 6 M concentrations in D2O and a 0.1 M sample in a D-glycerol/D2O mixture (Figure 3a). The maxima of bleaching and new absorption regions were probed and no significant frequency dependence of the decay time was found. The values of T1 obtained from these magic angle pump-probe experiments (see Figure 3a) for samples in D2O are 1.82±0.01 ps (at 0.1 M) and 1.87±0.02 ps (at 6 M). For the 59%D-glycerol/D2O mixture T1= 2.44±0.02 ps, for 81% D-glycerol/D2O mixture T1= 2.64±0.02 ps and for neat D-glycerol T1= 2.89±0.03 ps.
Figure 2.
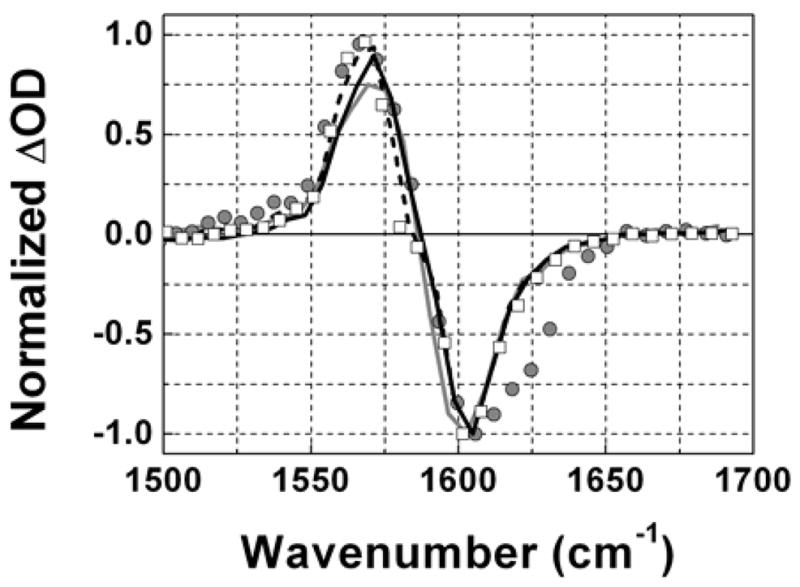
Normalized pump-probe signal of DGdm+ for parallel polarizations of pump and probe pulses at T=0.3 ps. Gray curve – 0.1 M in D2O, Gray circles – 6 M in D2O, Black curve – 0.1 M in 59%D-glycerol/D2O, Dashed curve – 0.1 M in 81%D-glycerol/D2O, Squares – 0.1 M in neat D-glycerol.
Figure 3.

Polarization dependent pump probe data for DGdm+. (a) Magic angle pump-probe signals at different concentrations and in different solvents probed at 1603 cm−1; (red) 0.1 M in D2O, (black) 6 M in D2O, (green) 0.1 M in 59%D-glycerol/D2O, (blue) 0.1 M in 81%D-glycerol/D2O, (purple) 0.1 M in D-glycerol. (b) Anisotropy decay at 1603 cm−1 and (c) anisotropy decay at 1557 cm−1; color coding is the same as in part (a). Red smooth curves are the two exponential decay fits.
The transition dipole reorientation dynamics of DGdm+ were obtained from the pump-probe anisotropy for which data are presented in Figures 3b–c for two different probe frequencies: one at the maximum of the bleaching and the other on the low frequency edge of the excited state absorption. The latter experiment permits measurement of the anisotropy in a region where it is less influenced by the overlap of the bleaching with the new absorption component. The pump-probe anisotropy exhibits decay on two well separated time scales and all experiments have been fit to the double exponential (see Table 1):
| (2) |
where AF and AS are the amplitudes of the fast and slow processes. The slowest decay with amplitude AS ~0.1 describes the overall rotational diffusion for the DGdm+ ion as discussed below. The significant subpicosecond decay (Figure 3b and 3c) in the ~ 500 fs range for all samples except for neat D-glycerol where the fast decay is slowed to 1.2 ps, is clearly not controlled by overall rotation because it is nearly as fast in more viscous solutions and in the higher concentration sample that contains aggregates. For all samples except neat D-glycerol the anisotropy from fits appears to have an initial value of at least ~0.34 when probed at the bleaching, and close to the limit of 0.4 when probed at the exited state absorption. But in neat D-glycerol a value of r(~0) ≈0.3 is obtained for both spectral components.
Table 1.
Parameters of double exponential fit of anisotropy decay for different concentrations of DGdm+ and different solvents.
| Sample (M) | Solvent | AF | τF (ps) | AS | τS (ps) | |
|---|---|---|---|---|---|---|
| 0.1 | D2O | Bleach | 0.20±0.02 | 0.53±0.11 | 0.13±0.03 | 4.1±1.3 |
| Abs | 0.34±0.03 | 0.49±0.07 | 0.12±0.01 | >6 | ||
| 6 | D2O | Bleach | 0.25±0.02 | 0.45±0.09 | 0.08±0.02 | >6 |
| Abs | 0.29±0.01 | 0.60±0.05 | 0.1±0.01 | >6 | ||
| 0.1 | 59%D-Glycrol/D2O | Bleach | 0.26±0.01 | 0.63±0.03 | 0.09±0.01 | >6 |
| Abs | 0.35±0.03 | 0.50±0.05 | 0.09±0.01 | >6 | ||
| 0.1 | 81%D-Glycrol/D2O | Bleach | 0.21±0.01 | 0.77±0.06 | 0.11±0.01 | >6 |
| Abs | 0.36±0.03 | 0.48±0.05 | 0.09±0.01 | >6 | ||
| 0.1 | D-glycerol | Bleach | 0.17±0.01 | 1.27±0.13 | 0.10±0.01 | >6 |
| Abs | 0.21±0.01 | 1.22±0.17 | 0.09±0.01 | >6 |
The main purpose of this paper is to present, and provide a quantitative explanations for the time dependence of the anisotropy.
DISCUSSION
Linear Spectroscopy
The experiments of Figure 1 show that the vibrational linewidth and center frequency are independent of DGdm+ concentrations below ca. 0.2 M which suggests that at the lowest concentrations the solution is mainly monomeric DGdm+ ions. Ab initio calculations were performed on [C(ND2)3]+ − n D2O clusters (n=0 – 8) with Gaussian03 34 and B3LYP density-functional theory (DFT) method with the basis set 6–31G to optimize the geometry. It was confirmed that the mode near 1600 cm−1 is a degenerate mode as illustrated in Figure 4. The Raman spectrum assignments of this band also indicate that it is a degenerate mode 35. The Gaussian computation indicates that the degeneracy can be split by up to 80 cm−1 due to the interaction of DGdm+ with asymmetrically disposed water molecules interacting mainly with the ND2 groups. The ratio of the lower to the higher frequency transition dipole moment of the transitions of these clusters varied from 1 to 0.82. The transitions to the different components of the degenerate state in vacuum have dipole moments perpendicular to one another. Therefore we can expect that in solution there will exist a significant frequency distribution of hydrogen bonded states and that at any instant, each ion may have a different water cluster surrounding it. These solvation shells would be expected to reduce the symmetry such that two “degenerate” modes are no longer interchangeable by a three-fold rotation and a phase shift. However any particular splitting pattern should only persist as long as the asymmetric solvent configurations remain intact. The existence of two overlapping transitions in the 1600 cm−1 region in solutions, as evidenced most clearly by the glycerol spectra, implies that the mean structure of DGdm+ in solution does not have three-fold symmetry. The splitting of the degenerate states in the linear spectrum is not so obvious in D2O but the band fits well to two nearly identical bands split by 10.8 cm−1. The FTIR spectrum of [C(ND2)3]Cl in D-glycerol/D2O on Figure 1b clearly shows the splitting of the 1600 cm−1 band into two transitions having slightly different intensities: these components must correspond to the two components of DGdm+ that are degenerate in the gas phase molecule. Although the detailed structural-energetic aspects, in particular the relative disposition of the three ND2 groups of these split states are not known yet, there are clearly a number of factors that can contribute. For the D-glycerol/D2O mixtures, in addition to hydrogen bonds between DGdm+ and the glycerol-OD and D-OD hydroxyls and oxygens, the hydrophobic faces of glycerol and DGdm+ may associate. Such complexation may not be symmetric relative to the ND2 groups. Such an asymmetric interaction with the environment could be a factor in causing a larger splitting of the degeneracy and the small modifications of the intensities of the two transitions that are observed.
Figure 4. Energy diagram for the separated normal modes model.

Also shown are displacements of the atoms of the CN3 group of DGdm+ for the two degenerate components of the 1600 cm−1 mode. The atomic displacements illustrate the orthogonal polarization of the two transitions.
Simulation model
In the analysis the two nearly degenerate modes will be treated as independent normal modes such as those depicted in Figure 4. These components are described in the notation |n1 n2〉 where n1 and n2 are the numbers of quanta of modes 1 and 2. The ground state is |00〉. There are two one quantum states (|01〉 and |01〉) which are the nearly degenerate component harmonic states ψ1 (Q1)ψ0 (Q2) and ψ0 (Q1)ψ1 (Q2). The transition dipole moments to them from |00〉 are assumed to be perpendicular to one another. There are three two quanta states (|02〉, |20〉 and |11〉), corresponding to ψ0 (Q1)ψ2 (Q2), ψ2 (Q1)ψ0 (Q2) and ψ1 (Q1)ψ1 (Q2), which are the overtones and a combination. The Liouville path diagrams considered in the present analysis are shown in Figure 5. The six basis states |00〉, |10〉, |01〉, |20〉, |02〉 and |11〉 are written on the diagrams as 0,1, 2, 1+1, 2+2 and 1+2 to simplify the visualization of the time dependent steps in the evolution. The interpretation of the diagrams will be deferred until the specific cases are discussed. The Liouville path diagrams are discussed in general in reference 36.
Figure 5.

Feynman diagrams contributing to the pump-probe signal. Row 1 describes stimulated emission (a, b) and ground state bleaching (c – f). Row 2 describes excited state absorption. Rows 3 describes excited state absorption to a combinational mode. Rows 4 and 5 describe the paths with coherence during the waiting time T. The factors of two before the pathway symbols, 〈abcd〉, indicate there are two diagrams with the time ordering of the first two pulses exchanged: in addition, when a ≠ b, a and b must also be interchanged. A set of comparable diagrams with the numerical indices (1 and 2) interchanged are also implied
We have explored other possibilities for the eigenstates in detail. For example, another approach would be to start from a two dimensional harmonic oscillator with modes that have some vibrational angular momentum. In such modes the motions of each atom follow elliptical paths and they are strongly coupled to overall rotation through the Coriolis interaction 37: they are three-fold symmetry adapted linear combinations of the separated normal modes with complex coefficients. There are two one quantum states: and , and three two quanta states: an A state and E states and 38. A simple analysis based on the diagrams of Figure 5 with ψ1,1 and ψ1,−1 replacing |10〉 and |01〉, ψ2,0 replacing |11〉, and ψ2,±2 replacing |20〉 and |02〉 finds that the magnitudes of the signals and anisotropies of the new absorption and the bleach in the pump-probe spectrum are equal for a variety of state energy distributions. The anisotropy is 0.3 if the A and E two quanta states both contribute to the additional absorption signal. The anisotropy is 0.7 if only the A state contributes to the additional absorption, and the anisotropy is 0.1 if only the E states contribute to additional absorption. In each of these examples the bleach and new absorption signals are equal. The predicted theoretical anisotropies at zero delay from this two dimensional oscillator model do not correspond to the experimental values which are found to be significantly higher than 0.3 at the peaks of the absorption and the bleach. It is concluded that solvent interactions strongly perturb these eigenstates of the vibrational angular momentum. Once the solvent induced decay of the angular momentum correlation function is completed, the ensemble should be treatable as if there are separated normal modes having zero mean values of the vibrational angular momentum, such as we have used in the treatment below. This correlation decay must be faster than the 250 fs time resolution of the anisotropy experiment and is probably caused by the strong restriction of overall motion by hydrogen bonding to the ND2 groups and the removal of three-fold symmetry by these solvent interactions.
Pump-probe data and linear spectra for dilute solutions of DGdm+ were therefore simulated from the two independent modes model (Figure 4). Each of the frequencies of the |01〉 → |02〉 or |10〉 → |20〉 transitions are considered to be downshifted by a diagonal anharmonic shift Δ. The frequency of the combinational mode, |11〉, is anharmonically shifted by ΔC. The simulation of the frequency resolved pump-probe spectra was based on known response functions 39 assuming delta pulses. The T1 factors for the two degenerate components were assumed to be equal, and those of the overtones were assumed to follow a harmonic approximation 39. The central frequencies, ω1 and ω2, of the two modes, their transition dipole moments, μ1 and μ2, and parameters to describe autocorrelation functions C(t) = 〈δωi(t)δωi(0)〉 were made consistent with linear FTIR spectra by simulations (see Figure 1a and 1b). The linear spectrum is adequately, though perhaps not uniquely fit in the peak region and in the spectral wings by assuming a frequency correlation function that follows Bloch dynamics having the specific form:
| (3) |
The pump-probe experiment does not add much to knowledge of the vibrational frequency correlations. The fits used parameters γ = 0.9 ps−1 and σ = 1.1 ps−1 for DGdm+ in D2O. These parameters essentially represent a Voigt profile and they fit the linear spectrum with good precision. They are reasonable approximations to the more accurate frequency correlations obtained from 2D IR spectra that are in the course of publication 40. The parameters of the autocorrelation function for glycerol mixtures were: γ = 0.98, 0.95 and 0.86 ps−1 and σ = 0.83, 0.79 and 0.77 ps−1 for 59%, 81% glycerol mixtures and neat glycerol. The changes in σ with viscosity are small but they indicate that the inhomogeneous distribution of structures does not increase with viscosity as expected. This suggests that the preferred local D-glycerol/D2O configuration is dominant even in the 50% mixture.
Ultrafast anisotropy decay
There are three processes that need to be considered in a description of the fast anisotropy decay. The most obvious is rotational diffusion but it will be shown that rotational diffusion only dominates the later part of the anisotropy decay but not the early part and therefore it will be treated in the next section.
The second possible source of fast decay of the anisotropy arises because there are three orientations of any given ion that is distorted from three-fold symmetry: they represent the distorted solute structure in identical solvation shell configurations each with its unique vibrational transition dipoles rotated from each other by ±2π / 3 in the approximate plane of an ion. As a result of solvent reconfiguration and no other dynamics, the anisotropy would gradually decay to 0.1 as is observed in the experiment. The long time anisotropy limit for this process is the average of the pumped state anisotropy of 0.4 (for, say, one of the configurations) with 0.4P2(−1/2) contributions from the other two configurations rotated by ±2π/3. A complete redistribution of water molecules in the solvation shell of ion is needed to cause the equilibration of these configurations which we refer to as 2π/3 flips. The many picosecond time scale of the exchange of solvent configurations, which are not unrelated to the rotations of the mode dipoles, are discussed further below in relation to our MD simulations. As shown in Figures 3b and 3c the subpicosecond anisotropy loss occurs in the glycerol mixtures as well as in water. This result makes it unlikely that the fast anisotropy decay is caused by complete exchange of the solvent configurations. It is possible that this process might contribute to the anisotropy decay in water, particularly at much higher temperatures, but the vibrational dynamics discussed below is considered to dominate the anisotropy decay at short times.
The anisotropy is sensitive to the solvent induced transfers between the two nearly degenerate components and the coherence transfers that occur in the detection period. Intermode coupling has previously been invoked to explain anisotropy decays 41–43. Recent experimental examples include CN vibrations of hexacyanides 41 and CO vibrations of hexacarbonyls 42, 43. In these cases the degenerate states can be described by the coupling patterns of symmetrically placed CN or CO modes and the relaxation kinetics of the states has been directly related to the coupling and energy transfer between equivalent ligands. The present example is conceptually different in that DGdm+ has no identifiable ligands between which excitations are transferring: the relaxation of DGdm+ is intrinsically related to solvent induced mixing of the delocalized degenerate vibrations that involve atomic displacements that are present on all the atoms of the molecule.
In view of the fact that the 1600 cm−1 transition involves a nearly degenerate pair of states it is also necessary to incorporate coherence transfer into a description of the pump-probe spectra and kinetics. Transfer between vibrational coherences between carbonyl transitions in a metalcarbonyl have been examined in detail by 2D IR methods 44 and evaluated by a Redfield approach: some of the physical assumptions used in the present analysis are similar. In a pump-probe experiment, assuming delta-pulse excitation, the pump pulse field interacts twice with the system to create either a population state (Figure 5, lines 1 to 3) or a coherent superposition of states (Figure 5, lines 4 and 5) at T=0. The system then evolves during the waiting time T and the detection time t. The signal acquisition involves a Fourier integration over t by the monochromator and slow detector but the coherence dynamics during that period must still be incorporated to obtain the correct signal: indeed the effects on the pump-probe signals of coherence transfer during t could be rather significant.
Description of the pump-probe signal
A highly simplified, schematic approach is now used to keep track of the various terms that contribute to the pump-probe signal. The system-bath interaction is usefully displayed as an expansion in the normal modes Q1 and Q2 of the DGdm+ degenerate state:
| (4) |
Where q is intended to represent the coordinates of a bath of harmonic oscillators. The reduced density matrix elements in the basis of the ion eigenmodes undergoes dynamics in both the T and t periods of the pump-probe experiment in accordance with the equation of motion:
| (5) |
Where R is the Redfield matrix 36, 45. The secular approximation used in reaching Equation 5 requires that |ωij − ωkl| ≪ Δt−1, where Δt is approximately the evolution time of the density matrix of the system. The near degeneracy results in many pairs of states satisfying this inequality and many density matrix elements need to be considered in the simulation of the pump-probe spectra dynamics and anisotropy. The frequency differences are small enough that we will assume the Redfield matrix elements to be real and representative of kinetic models that approximately describe the dynamics during the evolution in T and t. The coherence transfers between all the different v=1 -> v=2 transitions of the pump-probe experiment involve nearly equal frequencies since the values of |ωij − ωkl| arise from the small diagonal or off-diagonal anharmonic shifts or from the small splitting of the degeneracy. All these differences are small compared with kBT so the forward and backward rate coefficients are always nearly equal. The T1 relaxations from any of the six basis states described above into the background states from the combination and overtone states of the other modes of the molecule are introduced empirically. The measured T1 times are near 2 ps which are are substantially longer than the fastest anisotropy decays.
In the present approximation of separated but identical harmonic modes, the rate of population flow between degenerate components, R11,22 and R22,11 and the coherence exchange R12,21 rate coefficients are equal, and both arise from the quadratic interaction in Equation 4. These processes act during the T period. If V is linear in q and the bath is characterized by a spectral density g(ω), the rate coefficients are all given by the Golden Rule:
| (6) |
In order to illustrate the underlying processes, Equation 7 shows the time evolution of density matrix elements during the T time period assuming a motionally narrowed limit:
| (7) |
With similar equations for ρ22(T) and ρ21(T), where ρ(0) represents the density matrix immediately after the pump pulse has interacted with the system, 1/T1 is the population decay rate of a V=1 state assumed to be equal for both components, and k12 makes explicit, for the purpose of illustration, that the coherence decays by pure dephasing which competes with whatever effects the coherence transfers would have on the signal.. The actual coherence loss computed from our simulations discussed below is nonexponential.
Because the Redfield terms are not reliably calculated by inclusion of only the harmonic terms, the transfer during t amongst coherences (1,1+1), (1,1+2), (1,2+2), (2,1+2), (2,2+2), and (2,1+1) was simulated by a kinetic model with one rate coefficient, kct, as a variable parameter, except for transitions between coherences that vanish in the quadratic approximation to Redfield theory, for which kct was set equal to zero. The coherences (1,2+2) and (2, 1+1), though involved in the coherence “kinetics” do not contribute to the density matrix trace that yields the signal. All other contributions were neglected. The dashed horizontal lines in the diagrams (Figure 5) indicate the stage in the evolution where a spontaneous process of coherence or population transfer might occur. Such a change may alter not only the detected frequency but also the polarization condition which is the main point when it comes to evaluating the anisotropy. For this reason the time ordered interaction associated with each diagram is indicated below it in Figure 5. Only those terms with an even number of molecular frame transition dipole directions, such as 1111, 1212, 1221, etc., survive the orientation average 27. All such possible changes are included in the simulation and weighted according to the foregoing kinetic model. The diagrams that are moved to the right in Figure 5, describe processes undergone by some molecules in the ensemble that are involved in the kinetics but that do not contribute to the signal because of the vanishing orientation average in the isotropic sample. Of course all these processes are incorporated into the quantum mechanical trace over the bath and system variables that define the measured signal through the diagrams in Figure 5.
In order to assess the relative importance of population and coherence transfer in the pump-probe signal and anisotropy decay, model simulations were carried out based on the diagrams in Figure 5. The various contributions to the anisotropy, free of any complications due to overlapping signals, are presented in Figure 6a. In the absence of population and coherence transfer (Figure 5, diagrams without horizontal dotted lines) the anisotropy is 0.4 when probed at the new absorption region, which contains no (12) coherence diagrams, and 0.37 when probed at the bleach and stimulated emission part of the signal. The drop from 0.4 is caused by the signal difference between having (1+2,1) or (2,0) coherences in the detection period. Coherence decay during T (Figure 5, diagrams 4a,b) causes a small but rapid drop of anisotropy probed at the bleaching region, after which the anisotropy stays constant (Figure 6a, 2): the rate of this decay is determined by the dephasing time of the coherent superposition of the two modes. The response functions used in the simulation incorporated the conventional relaxation parameters36 in the form of those worked out explicitely for anharmonic oscillators39. The autocorrelation function was chosen as in Equation (3) while the cross correlation was defined 39 as: f σ2 where f is a correlation coefficient which could range from +1 to −1. The pure dephasing time calculated from a simulation of diagram 4a of Figure 5 for two equal frequency oscillators was found to vary from 0.6 to 0.2 ps as f varied from +1 to −1 assuming that σ is determined by the autocorrelation. In the former case the decay is exponential and the latter is a mean decay time of a non-exponential function. Thus, the total coherence loss for DGdm+ in D2O, including the T1 process, must occur within a range 0.5 ps (for f=1) to 0.2 ps (for f=−1). The experimental anisotropy decay time is within this range of coherence loss, but as seen from Figure 6a (1 and 2) this mechanism can never account for the decay of the anisotropy to 0.1. When coherence transfer dominates all other processes during the detection time t (Figure 5, diagrams with dotted line during t period only) the anisotropies for both probe frequencies undergo a fast decay to become almost equal at r = 0.2 (Figure 6a, 3 and 4). In order to influence the signal during t, coherence transfer must compete effectively with the total dephasing rate of the coherences generated by the probe pulse in the absence of coherence transfer. For DGdm+ in D2O coherence decay in the excited state manifold has the form exp [−γt −Δ2t2/2 −3t/2T1] which has a mean decay rate of 2.22 ps−1.
Figure 6. Contributions to the pump-probe signal.

Simulated pump-probe spectra (XXXX tensor) and anisotropy for a probe at 1603 cm−1 for bleaching (curves with squares) and at 1569 cm−1 for excited state absorption (smooth curves) transients. Two equivalent oscillators were used with central frequencies ω1=1603.9 cm−1 and ω2=ω1−Δω and population lifetimes and transfer as for DGdm+ in D2O. The auto and cross-correlation functions, determining the dephasing of the transitions and the T interval coherence transfer, were those for DGdm+ in D2O from Table 2. There is no rotational diffusion.
a) Parameters: Δ=∞, Δc=0 cm−1 and Δω=0 cm−1. 1 and 2: population exchange and all coherence transfers set equal to zero; 3 and 4: population exchange and coherence transfers during T set equal to zero and the coherence transfer rate during t set equal to infinity; 5 and 6: population exchange and coherence transfers during T are incorporated and coherence transfer rate during t set equal to zero; 7 and 8: all processes incorporated with coherence transfer during t set equal to infinity; 9 and 10: coherence transfer during T was incorporated; no population exchange or dephasing during T, and no coherence transfer during t;
b) Parameters: Δ=20 cm−1, Δc=3 cm−1 and Δω=10 cm−1. 1 and 2: population exchange and all coherence transfers set equal to zero; 3 and 4: population exchange and coherence transfers during T set equal to zero and the coherence transfer rate during t set equal to infinity; 5 and 6: population exchange and coherence transfers during T are incorporated and coherence transfer rate during t set equal to zero; 7 and 8: all processes incorporated with coherence transfer during t set equal to infinity; 9 and 10: coherence transfer during T was incorporated; no population exchange or dephasing during T, and no coherence transfer during t;
c) Parameters: Δ=20 cm−1, Δc=3 cm−1, Δω=10 cm−1 and T=0. red: no population exchange and no coherence transfer, black: coherence transfer rate during t set equal to infinity.
d) Ratio of signal magnitudes of excited state absorption and bleach versus T for experimental (1) and simulated results with parameters Δ=20 cm−1, Δc=3 cm−1 and Δω=10 cm−1: (2) inclusion of 0.95ps population exchange and coherence transfer during T but no coherence transfer during t; (3) same conditions as for (2) but infinitely fast coherence transfer during t.
We now consider transfer in the T range, without any coherence transfer during t, (Figure 5, diagrams with dotted line during T period only) where both population exchange and coherence transfer occur with the same rate constants during T as predicted by Equation 6. The coherence transfer competes unfavorably with the dephasing of the coherence during T but in any event has a negligible effect on the anisotropy: without pure dephasing and with inclusion only infinitely fast coherence transfer during T, the anisotropy does not decay (Figure 6a, 9 and 10). The population transfer during the T interval diminishes the anisotropy from 0.4 to 0.1 (Figure 6a, 5 and 6) because of the rotation of the transition dipole moment in the molecular frame by 90° and results in stochastic averaging of the dipole in the mean plane of DGdm+. Inclusion of both population exchange and coherence transfer during both the T and t periods also predicts that the anisotropy will decay to values close to 0.1 with a very slight dependence on the probed frequency (Figure 6a, 7 and 8).
Figure 7.

Experimental and fit of pump-probe spectra (a, dashed and solid curves) and anisotropy (b, circles and solid curve) for DGdmCl in D2O. Diagrams from Figure 5 and parameters from Table 2 were used in simulation.
The overlapping of the excited state absorption with the bleaching part decreases the anisotropy from the values presented in Figure 6a for the degenerate states and discussed for the ideal conditions used in the previous paragraph. This effect is seen from the simulated anisotropy of two independent transitions with spectroscopic parameters close to those of DGdm+ in D2O (see Table 2) having diagonal anharmonicities of 20 cm−1 and an off-diagonal anharmonicity of 3 cm−1. As a result of the spectral overlap the initial anisotropies (T=0) in the bleach and excited state regions are reduced to 0.36 and 0.38 respectively. Dephasing further reduces the values to 0.365 and 0.32 as shown in Figure 6b (1 and 2). When coherence transfer is introduced in the t period, the T=0 anisotropy is 0.29. Although dephasing during T causes this anisotropy to drop as shown on (Figure 6b, 3 and 4) it is strongly dependent on probe frequency. When only population exchange occurs during T, the anisotropy decays to 0.1 independently of the probed frequency (Figure 6b, 5 and 6), regardless of whether coherence transfer is occurring during T. In the absence of pure dephasing during T the anisotropy oscillates at the frequency difference of transitions and does not depend on the rate of coherence transfer (Figure 6b, 9 and 10). Pure dephasing of the coherence in T damps the oscillation which is not seen in the experiment. When both population exchange and coherence transfer are present during both T and t, the anisotropy decays to about the same value of 0.1 for the bleach and excited state absorption (Figure 6b, 7 and 8). However, the excited state absorption anisotropy decays more rapidly. This prediction of a faster anisotropy decay when the new absorption region is probed is not in accord with the experiment for DGdm+ in D2O. The experimental anisotropy at the new absorption begins from values close to 0.4 and is higher during whole T period than the anisotropy measured at the bleaching region. This result implies that, for this example, coherence transfer during t does not compete effectively with coherence loss. The simulated pump-probe spectra for parallel polarization (Figure 6c) predicts that the ratio of the new absorption to the bleaching peak amplitude becomes significantly smaller (Figure 6d) when coherence transfer is influencing the decay during t. Figure 6d also includes the experimental ratio which is close to the value with no coherence transfer during t, but with population exchange and coherence transfer during T with equal rates according to Equation 6. The conditions prevailing for the rate of dephasing during the T period makes it impossible to see the oscillations of the (12) coherences which form a negligible contribution to the anisotropy signal. Although we have not explored different conditions experimentally it might be that slowing down the frequency fluctuations by significant decrease in temperature could result in a more significant contribution of coherence transfer during t period.
Table 2.
Parameters used for simulation of polarization dependent pump-probe spectra of guanidinium ion in D2O and D-glycerol/D2O.
| Parameters | Solvent | ||||
|---|---|---|---|---|---|
| D2O | 59% glycerol | 81% glycerol | 100% glycerol | ||
| ω1, cm−1 | 1593.1 | 1588.1 | 1586.2 | 1584.7 | |
| ω2, cm−1 | 1603.9 | 1602.4 | 1601.5 | 1600.5 | |
| Δ1, cm−1 | 20 | 18 | 18 | 18 | |
| Δ2, cm−1 | 20 | 12 | 12 | 14 | |
| Δc, cm−1 | 3 | 2 | 2 | 6 | |
| D||, D⊥, *10−10 s−1 | 3.3 | 0.33 | 0.23 | 0.0033 | |
| μ1/μ2 | 1 | 0.931 | 0.932 | 0.924 | |
| k, ps−1 | 0.53 | 0.71 | 0.59 | 0.45 | |
| kct, ps−1 (upper limit) | 0.33 | 0.33 | 0.33 | 0.25 | |
| 〈δωi(τ) δωi (0)〉 | γ, ps−1 | 0.9 | 0.98 | 0.95 | 0.86 |
| σ, ps−1 | 1.1 | 0.83 | 0.79 | 0.77 | |
| 〈δωi(τ) δωj (0)〉 | σ, ps−1 | 1.1 | 0.83 | 0.79 | 0.77 |
Rotational diffusion
The role of rotational diffusion was considered by treating DGdm+ as an approximate oblate diffuser. The anisotropy of an oblate symmetric diffuser is expected to exhibit two relaxation times that depend on the rotational diffusion constants D|| and D⊥. The initial anisotropy amplitude of the component decaying with time constant (4D|| + 2D⊥)−1 is predicted to be 0.3 and that for the component decaying with time constant is 0.1 46. The ultrafast vibrational energy transfer between the perpendicularly polarized components, as discussed above, averages out the transition dipole orientation in the molecular plane much faster than the overall rotation around the symmetry axis. This averaging implies that the effect of overall rotation can only be sensed during the decay of the anisotropy from 0.1 to zero, and that only D⊥ is measured. Concentration and viscosity dependences of this decay component of the anisotropy are shown in Table 1. The time constant of 4.1 ps for 0.1 M solutions in D2O are in good agreement with NMR data (4.38 ps) obtained for 2 M HGdmBr 47. For the ten times more viscous 59% D-glycerol/D2O mixture and high concentrations of DGdm+ in D2O the slow decay component was too long to be measured confidently. The slow rotational relaxation time is predicted to be:
| (8) |
where T is a temperature (295 K), kB is Boltzmann constant, η, V, and λ are the viscosity (1.192*10−3 kg·m−1sec−1 for D2O), the volume of the molecule (70.6*10−30 m3), and friction coefficient for out of plane rotation. The viscosity of water is not changed by adding 1 M HGdmCl 48. For slip boundary conditions, λ = 1.407 49, 50 so that τS = 4.85 ps, which is reasonable agreement with the experiment. For stick boundary conditions, involving rotation of the solute with its solvation shell, λ=8.0931 and τS = 27.9 ps, which is not close to the experimental results in Table 1 and much longer than could be measured given that the populations last for only a few picoseconds.
In order to assess the remote possibility that a fast rotation might contribute to the diminishment of the anisotropy from 0.4 to 0.1 the rotational diffusion of the HGdm+ ion was also estimated from classical MD simulations using the GROMOS96 force field (G45a1) and SPC water. The partial charges for HGdm+ were known 51 and the box consisted of HGdmCl and 891 water molecules was used. Two hundred configurations with minimized energy were randomly chosen and the trajectory of an angular deviation for a selected CN bond was obtained over a time range of 20 ps for each of the starting configurations. In the simulation the anisotropy dropped from 0.4 to 0.32 in the first 250 fs as a result of residual fast inertial motions. This type of behavior is common in classical MD simulations of anisotropy 52–54. The bulk of the HGdm+ anisotropy decayed with a single exponential time constant of 4.4 ps. This classical simulation of the rotational diffusion does not evidence a sub-picosecond decay of the anisotropy to a level of 0.1 as seen in the experiment and predicts that D⊥ and D|| are similar in magnitude since all the anisotropy is lost in a single exponential decay.
Pump-probe simulation
The complete simulation of the frequency resolved pump-probe spectra and anisotropy were performed by including all diagrams in rows 1–5 of Figure 5. The parameters used in the simulation are shown in Table 2 and the simulated spectrum and anisotropy for DGdm+ in D2O are compared with experiment in Figure 7. It is assumed in the simulation that the fluctuations in the two degenerate component transition frequencies were anticorrelated based on the simple idea that a solvent induced splitting of the degeneracy will shift the transitions in opposite directions. Thus the pure dephasing rate of the coherence in this approximation during the T period was chosen as faster (0.2 ps−1) of the possibilities mentioned above. Orientational factors for spherical rotor were incorporated 27. In both solvents the frequencies of two nearly equal frequency modes, ω1 and ω2, were chosen as ω2-ω1=10.8 cm−1 for D2O, 14.3 cm−1 in 59%D-glycerol/D2O, 15.3 cm−1 in 81%D-glycerol/D2O and 15.8 cm−1 in neat D-glycerol. For the simulation in D2O the same set of parameters was used for both modes. The rate constants for population and coherence transfer in the T period were found to be 0.53 ps−1 in D2O and 0.71, 0.59 and 0.45 ps−1 for 59%, 81%, and 100% D-glycerol mixtures. The transfer time (1/2k) is between 0.7 and 1.1 ps in different solvents and is 3.5 – 5.5 times longer than the time of coherence loss, 0.2 ps, implying that the effect of coherence transfer during T is negligible. Of course this conclusion is demanded by the magnitudes of the anisotropy, which are not possibly decaying to 0.1 by coherence transfer alone. It should be emphasized that the splitting of the degeneracy affects the r(0) and the apparent decay rates and other parameters in Table 1. The relative insensitivity of the rate constant of the population exchange to solvent suggests that the required forces are arising from OD groups of either D2O or D-glycerol directly H-bonded the ND2 groups of DGdm+. The decrease of the transfer rate with increasing friction suggests that out-of-plane and bending distortions of the ND2 groups of DGdm+, which would be slower at higher friction, are inducing the transfer. The upper limit for the rate constant of coherence transfer during t based on the simulation was estimated as 0.33 ps−1 for all solvents except for neat glycerol where the value was 0.25 ps−1. Simulations with faster coherence transfer (see Figure 6) did not match the experimental anisotropy decays. The fitted dephasing rates of the detected transitions vary from 1.69 to 2.22 ps−1 for different solvents consistent with the coherence transfer during t being negligible. Thus, the ultrafast decay of anisotropy is defined mainly by population exchange during T.
A qualitative picture of the HGdm+ dynamics can be gleaned from the present and previous 55 MD simulations which show the water molecules arranged around the periphery of HGdm+ with the oxygens predominantly pointing inward. Water molecules are repelled from approaching in the perpendicular direction so the HGdm+ acts as if it has a hydrophobic shield protecting the perpendicular direction yet it is hydrophilic on the periphery. This is a well-known property of the HGdm+ ion and endows it with its amphiphilicity. Our MD simulation was used to estimate the residence times of peripheral water configurations in the neighborhood of a particular ND2 group and it was consistently found that configurations are maintained for times exceeding 5 ps with high probability. These observations from the simulation are consistent with the conclusion from the experiments that the observed subpicosecond decay of the anisotropy is much faster than these changes.
CONCLUSION
The nearly degenerate asymmetric stretch vibrations of DGdm+ in D2O and D-glycerol/D2O show an ultrafast (~0.5 ps) and slow (~4 ps) decay of their anisotropy The slow component is attributed to rotational diffusion occurring on a time scale of few picoseconds. The fast component is attributed to exchange of population between the two modes with relaxation rate of about 1 ps. The coherence transfer during both waiting and integrated detection times (T and t respectively) of the pump-probe experiment was systematically evaluated and found to have only a small influence on the decay of the anisotropy signal.
Acknowledgments
This research was supported by grants to RMH from NSF-CHE and NIH- GM12592 with instrumentation supported by NIH-RR001348.
References
- 1.Makhatadze GI, Lopez MM, Richardson JM, Thomas ST. Anion binding to the ubiquitin molecule. Protein Sci. 1998;7(3):689–697. doi: 10.1002/pro.5560070318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishimura C, Uversky VN, Fink AL. Effect of salts on the stability and folding of staphylococcal nuclease. Biochemistry. 2001;40(7):2113–2128. doi: 10.1021/bi000861k. [DOI] [PubMed] [Google Scholar]
- 3.Ramos CHI, Baldwin RL. Sulfate anion stabilization of native ribonuclease A both by anion binding and by the Hofmeister effect. Protein Sci. 2002;11(7):1771–1778. doi: 10.1110/ps.0205902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chi EY, Krishnan S, Randolph TW, Carpenter JF. Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm Res. 2003;20(9):1325–1336. doi: 10.1023/a:1025771421906. [DOI] [PubMed] [Google Scholar]
- 5.Perez-Jimenez R, Godoy-Ruiz R, Ibarra-Molero B, Sanchez-Ruiz JM. The efficiency of different salts to screen charge interactions in proteins: A Hofmeister effect? Biophys J. 2004;86(4):2414–2429. doi: 10.1016/S0006-3495(04)74298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins KD. Ions from the Hofmeister series and osmolytes: effects on proteins in solution and in the crystallization process. Methods. 2004;34(3):300–311. doi: 10.1016/j.ymeth.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 7.Herberhold H, Royer CA, Winter R. Effects of chaotropic and kosmotropic cosolvents on the pressure-induced unfolding and denaturation of proteins: An FT-IR study on staphylococcal nuclease. Biochemistry. 2004;43(12):3336–3345. doi: 10.1021/bi036106z. [DOI] [PubMed] [Google Scholar]
- 8.Zhang YJ, Cremer PS. Interactions between macromolecules and ions: the Hofmeister series. Curr Opin Chem Biol. 2006;10(6):658–663. doi: 10.1016/j.cbpa.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 9.Vrbka L, Jungwirth P, Bauduin P, Touraud D, Kunz W. Specific ion effects at protein surfaces: A molecular dynamics study of bovine pancreatic trypsin inhibitor and horseradish peroxidase in selected salt solutions. J Phys Chem B. 2006;110(13):7036–7043. doi: 10.1021/jp0567624. [DOI] [PubMed] [Google Scholar]
- 10.Bagchi B. Water dynamics in the hydration layer around proteins and micelles. Chem Rev. 2005;105(9):3197–3219. doi: 10.1021/cr020661+. [DOI] [PubMed] [Google Scholar]
- 11.Rajamani S, Truskett TM, Garde S. Hydrophobic hydration from small to large lengthscales: Understanding and manipulating the crossover. Proc Natl Acad Sci U S A. 2005;102(27):9475–9480. doi: 10.1073/pnas.0504089102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bakker HJ. Structural dynamics of aqueous salt solutions. Chem Rev. 2008;108(4):1456–1473. doi: 10.1021/cr0206622. [DOI] [PubMed] [Google Scholar]
- 13.Jackson SE, Elmasry N, Fersht AR. Structure of the Hydrophobic Core in the Transition-State for Folding of Chymotrypsin Inhibitor-2 - a Critical Test of the Protein Engineering Method of Analysis. Biochemistry. 1993;32(42):11270–11278. doi: 10.1021/bi00093a002. [DOI] [PubMed] [Google Scholar]
- 14.Mayo SL, Baldwin RL. Guanidinium Chloride Induction of Partial Unfolding in Amide Proton-Exchange in Rnase-A. Science. 1993;262(5135):873–876. doi: 10.1126/science.8235609. [DOI] [PubMed] [Google Scholar]
- 15.Bai YW, Sosnick TR, Mayne L, Englander SW. Protein-Folding Intermediates - Native-State Hydrogen-Exchange. Science. 1995;269(5221):192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tromp RH, Neilson GW, Soper AK. Water-Structure in Concentrated Lithium-Chloride Solutions. J Chem Phys. 1992;96(11):8460–8469. [Google Scholar]
- 17.Leberman R, Soper AK. Effect of High-Salt Concentrations on Water-Structure. Nature. 1995;378(6555):364–366. doi: 10.1038/378364a0. [DOI] [PubMed] [Google Scholar]
- 18.Bello J, Haas D, Bello HR. Interactions of Protein-Denaturing Salts with Model Amides. Biochemistry. 1966;5(8):2539. doi: 10.1021/bi00872a008. [DOI] [PubMed] [Google Scholar]
- 19.Makhatadze GI. Thermodynamics of protein interactions with urea and guanidinium hydrochloride. J Phys Chem B. 1999;103(23):4781–4785. [Google Scholar]
- 20.Makhatadze GI, Privalov PL. Protein Interactions with Urea and Guanidinium Chloride - a Calorimetric Study. J Mol Biol. 1992;226(2):491–505. doi: 10.1016/0022-2836(92)90963-k. [DOI] [PubMed] [Google Scholar]
- 21.Wetlaufer DB, Coffin RL, Malik SK, Stoller L. Nonpolar Group Participation in Denaturation of Proteins by Urea and Guanidinium Salts. Model Compound Studies. J Am Chem Soc. 1964;86(3):508–514. [Google Scholar]
- 22.Arakawa T, Timasheff SN. Protein Stabilization and Destabilization by Guanidinium Salts. Biochemistry. 1984;23(25):5924–5929. doi: 10.1021/bi00320a005. [DOI] [PubMed] [Google Scholar]
- 23.Scott JN, Nucci NV, Vanderkooi JM. Changes in Water Structure Induced by the Guanidinium Cation and Implications for Protein Denaturation. J Phys Chem A. 2008;112(43):10939–10948. doi: 10.1021/jp8058239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moglich A, Krieger F, Kiefhaber T. Molecular basis for the effect of urea and guanidinium chloride on the dynamics of unfolded polypeptide chains. J Mol Biol. 2005;345(1):153–162. doi: 10.1016/j.jmb.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 25.Uversky VN, Fink AL. Protein misfolding, aggregation, and conformational diseases. Springer Science+Business Media; New York, N.Y.: 2006. [Google Scholar]
- 26.Dobson CM. Protein folding and misfolding. Nature. 2003;426(6968):884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 27.Hochstrasser RM. Two-dimensional IR-spectroscopy: polarization anisotropy effects. Chem Phys. 2001;266(2–3):273–284. [Google Scholar]
- 28.Magalhaes AL, Gomes JANF. Density-Functional Methods for the Study of the Ground-State Vibrations of the Guanidinium Ion. Int J Quantum Chem. 1997;61:725–739. [Google Scholar]
- 29.Drozd M. Molecular structure and infrared spectra of guanidinium cation - A combined theoretical and spectroscopic study. Materials Science and Engineering B-Solid State Materials for Advanced Technology. 2007;136(1):20–28. [Google Scholar]
- 30.Lemmon EW, MOMaDGF . Thermophysical Properties of Fluid Systems. Gaithersburg, MD: NIST Chemistry WebBook, NIST Standard Reference Database Number 69. [Google Scholar]
- 31.Segur JB, Oberstar HE. Viscosity of Glycerol and Its Aqueous Solutions. Industrial and Engineering Chemistry. 1951;43(9):2117–2120. [Google Scholar]
- 32.Kuo CH, Vorobyev DY, Chen JX, Hochstrasser RM. Correlation of the vibrations of the aqueous azide ion with the O-H modes of bound water molecules. J Phys Chem B. 2007;111(50):14028–14033. doi: 10.1021/jp076503+. [DOI] [PubMed] [Google Scholar]
- 33.Wynne K, Hochstrasser RM. The Theory of Ultrafast Vibrational Spectroscopy. Chem Phys. 1995;193(3):211–236. [Google Scholar]
- 34.Frisch MJ, Schlegel GWTHB, Scuseria GE, Robb MA, Montgomery JRCJA, Jr, Vreven T, Kudin KN, Millam JCBJM, Iyengar SS, Tomasi J, Barone V, Cossi BMM, Scalmani G, Rega N, Petersson GA, Hada HNM, Ehara M, Toyota K, Fukuda R, Ishida JHM, Nakajima T, Honda Y, Kitao O, Nakai H, Li MKX, Knox JE, Hratchian HP, Cross JB, Adamo C, Gomperts JJR, Stratmann RE, Yazyev O, Austin AJ, Pomelli RCC, Ochterski JW, Ayala PY, Morokuma K, Salvador GAVP, Dannenberg JJ, Zakrzewski VG, Daniels SDAD, Strain MC, Farkas O, Rabuck DKMAD, Raghavachari K, Foresman JB, Cui JVOQ, Baboul AG, Clifford S, Cioslowski J, Liu BBSG, Liashenko A, Piskorz P, Komaromi I, Fox RLMDJ, Keith T, Al-Laham MA, Peng CY, Challacombe ANM, Gill PMW, Johnson B, Wong WCMW, Gonzalez C, Pople JA. Gaussian 03, Revision B05 . Gaussian, Inc; Pittsburgh PA: 2003. [Google Scholar]
- 35.Sension RJ, Hudson B, Callis PR. Resonance Raman Studies of Guanidinium and Substituted Guanidinium Ions. J Phys Chem. 1990;94(10):4015–4025. [Google Scholar]
- 36.Mukamel S. Principles of nonlinear optical spectroscopy. Oxford University Press; New York: 1995. p. xviii.p. 543. [Google Scholar]
- 37.Herzberg G. Molecular spectra and molecular structure. II Infrared and Raman Spectra of Polyatomic molecules. D. Van Nostrand Company, Inc; Princeton, N.J: 1956. [Google Scholar]
- 38.Califano S. Vibrational states. Wiley; London; New York: 1976. pp. xii–335. [Google Scholar]
- 39.Ge NH, Zanni MT, Hochstrasser RM. Effects of vibrational frequency correlations on two-dimensional infrared spectra. J Phys Chem A. 2002;106(6):962–972. [Google Scholar]
- 40.Vorobyev DY, Kuo C-H, Chen J-X, Kuroda DG, Scott JN, Vanderkooi JM, Hochstrasser RM. 2D IR photon echo spectroscopy of guanidinium chloride. 2009 in preparation. [Google Scholar]
- 41.Sando GM, Zhong Q, Owrutsky JC. Vibrational and rotational dynamics of cyanoferrates in solution. J Chem Phys. 2004;121(5):2158–2168. doi: 10.1063/1.1767072. [DOI] [PubMed] [Google Scholar]
- 42.Tokmakoff A, Fayer MD. Homogeneous Vibrational Dynamics and Inhomogeneous Broadening in Glass-Forming Liquids - Infrared Photon-Echo Experiments from Room-Temperature to 10 K. J Chem Phys. 1995;103(8):2810–2826. [Google Scholar]
- 43.Tokmakoff A, Urdahl RS, Zimdars D, Francis RS, Kwok AS, Fayer MD. Vibrational Spectral Diffusion and Population-Dynamics in a Glass-Forming Liquid - Variable Bandwidth Picosecond Infrared-Spectroscopy. J Chem Phys. 1995;102(10):3919–3931. [Google Scholar]
- 44.Khalil M, Demirdoven N, Tokmakoff A. Vibrational coherence transfer characterized with Fourier-transform 2D IR spectroscopy. J Chem Phys. 2004;121(1):362–373. doi: 10.1063/1.1756870. [DOI] [PubMed] [Google Scholar]
- 45.Redfield AG. Adv Magn Reson. 1965;1:1–32. [Google Scholar]
- 46.Lim M, Hochstrasser RM. Unusual vibrational dynamics of the acetic acid dimer. J Chem Phys. 2001;115(16):7629–7643. [Google Scholar]
- 47.Miyajima K, Yoshida H, Kuroda Y, Nakagaki M. Studies on the Aqueous-Solutions of Guanidinium Salts.13. NMR-Study of the Interactions between Guanidinium Salt and Tetraalkylammonium Salts in Water. Bull Chem Soc Jpn. 1980;53(8):2212–2216. [Google Scholar]
- 48.Perl D, Jacob M, Bano M, Stupak M, Antalik M, Schmid FX. Thermodynamics of a diffusional protein folding reaction. Biophys Chem. 2002;96(2–3):173–190. doi: 10.1016/s0301-4622(02)00024-8. [DOI] [PubMed] [Google Scholar]
- 49.Sension RJ, Repinec ST, Szarka AZ, Hochstrasser RM. Femtosecond Laser Studies of the Cis-Stilbene Photoisomerization Reactions. J Chem Phys. 1993;98(8):6291–6315. [Google Scholar]
- 50.Hu CM, Zwanzig R. Rotational Friction Coefficients for Spheroids with Slipping Boundary-Condition. J Chem Phys. 1974;60(11):4354–4357. [Google Scholar]
- 51.Camilloni C, Rocco AG, Eberini I, Gianazza E, Broglia RA, Tiana G. Urea and guanidinium chloride denature protein L in different ways in molecular dynamics simulations. Biophys J. 2008;94(12):4654–4661. doi: 10.1529/biophysj.107.125799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teleman O, Ahlstrom P. Molecular-Dynamics Simulation of a Small Calcium Complex in Aqueous-Solution. J Am Chem Soc. 1986;108(15):4333–4341. [Google Scholar]
- 53.Impey RW, Madden PA, Mcdonald IR. Spectroscopic and Transport-Properties of Water Model-Calculations and the Interpretation of Experimental Results. Mol Phys. 1982;46(3):513–539. [Google Scholar]
- 54.Leenders R, Vangunsteren WF, Berendsen HJC, Visser AJWG. Molecular-Dynamics Simulations of Oxidized and Reduced Clostridium-Beijerinckii Flavodoxin. Biophys J. 1994;66(3):634–645. doi: 10.1016/s0006-3495(94)80837-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mason PE, Neilson GW, Enderby JE, Saboungi ML, Dempsey CE, MacKerell AD, Brady JW. The structure of aqueous guanidinium chloride solutions. J Am Chem Soc. 2004;126(37):11462–11470. doi: 10.1021/ja040034x. [DOI] [PubMed] [Google Scholar]