

Abstract
Serotonin (5-hydroxytryptamine; 5-HT) has an important role in mood regulation, and its dysfunction in the central nervous system (CNS) is associated with depression. Reports of mood and immune disorder co-morbidities indicate that immune-5-HT interactions may mediate depression present in immune compromised disease states including HIV/AIDS, multiple sclerosis, and Parkinson’s disease. Chemokines, immune proteins that induce chemotaxis and cellular adhesion, and their G-protein coupled receptors (GPCRs) distribute throughout the CNS, regulate neuronal patterning, and mediate neuropathology. The purpose of this study is to investigate the neuroanatomical and neurophysiological relationship between the chemokine fractalkine/CX3CL1 or its receptor CX3CR1 and 5-HT neurons in the rat midbrain raphe nuclei (RN). Immunohistochemistry was used to examine the colocalization of CX3CL1 or CX3CR1 with 5-HT in the RN, and whole-cell patch-clamp recordings in rat brain slices were used to determine the functional impact of CX3CL1 on 5-HT dorsal raphe nucleus (DRN) neurons. Greater than 70% of 5-HT neurons colocalize with CX3CL1 and CX3CR1 in the RN. CX3CL1 localizes as discrete puncta throughout the cytoplasm, whereas CX3CR1 concentrates to the perinuclear region of 5-HT neurons and exhibits microglial expression. CX3CL1 and CX3CR1 also colocalize with one-another on individual RN cells. Electrophysiology studies indicate a CX3CL1-mediated enhancement of spontaneous inhibitory postsynaptic current (sIPSC) amplitude and dose-dependent increase of evoked IPSC (eIPSC) amplitude without affecting eIPSC paired-pulse ratio, a finding observed selectively in 5-HT neurons. CX3CL1’s effect on eIPSC amplitude is blocked by pretreatment with an anti-CX3CL1 neutralizing antibody. Thus, CX3CL1 enhances postsynaptic GABA receptor number or sensitivity on 5-HT DRN neurons under conditions of both spontaneous and synaptically-evoked GABA release. CX3CL1 may indirectly inhibit 5-HT neurotransmission by increasing the sensitivity of 5-HT DRN neurons to GABA inputs. Therapies targeting CX3CL1 may treat serotonin related mood disorders, including depression experienced by patients with compromised immune systems.
Keywords: chemokine, CX3CL1, 5-HT, GABA, electrophysiology, immunohistochemistry
Psychological stress alters immune responses and confers vulnerability to a wide spectrum of autoimmune, cardiovascular, and neurological diseases (Glaser and Kiecolt-Glaser, 2005 review). The neurotransmitter serotonin (5-hydroxytryptamine; 5-HT) has an important role in the neuronal response to stress (Chaouloff, 2000), and dysfunction of 5-HT is characteristically associated with stress-related psychiatric disorders including depression and anxiety (Charney et al., 1990; Meltzer, 1990). Recent reports also describe widespread serotonin interactions with the immune system, including regulation of T cells, delayed-type hypersensitivity responses, and natural immunity, as well as activation of natural killer cells (Mössner and Lesch, 1998 review). Functional studies indicate that 5-HT may modulate cytokine release, including interleukin (IL)-1β, IL-6, IL-8, IL-12, and tumor necrosis factor (TNF)-α (Dürk et al., 2005; Idzko et al., 2004). Chemokines may likewise modulate 5-HT activity, as already reported for the chemokine monocyte chemotactic protein-1, which was shown to enhance the mitogenic effect of 5-HT on vascular smooth muscle (Watanabe et al., 2001). Furthermore, the injection of cytokines elicits a behavioral response referred to as “sickness behavior,” resembling the central symptoms of depression including loss of appetite, social withdrawal, and fatigue (Dantzer and Kelley, 2007). Thus, it appears that chemokine-5-HT interactions may occur in a bi-directional manner.
Chemokines (chemoattractant cytokines) are small (7–11 kD) heparin-binding chemoattractant proteins that mediate cell-to-cell communication to promote chemotaxis and cellular adhesion. They form a specialized subclass of cytokines and appear to be over-expressed in diseases such as meningitis, rheumatoid arthritis, multiple sclerosis, cancer, and HIV encephalopathy (Bajetto et al., 2001 review). The chemokine superfamily is divided into four subclasses, C, CC, CXC, and CX3C, based on the location of the first two cysteine residues in the N-terminus. As the only member of the CX3C family, CX3CL1 is a unique chemokine since it has cysteines separated by 3 amino acids and can exist as either a membrane-anchored protein or a soluble glycoprotein. The cell surface-bound CX3CL1 promotes robust cellular adhesion, whereas the soluble glycoprotein form of the protein serves as a potent chemoattractant for T cells, natural killer cells, and monocytes (Bazan et al., 1997). CX3CL1 mediates leukocyte chemotaxis to inflammatory regions by binding to its GPCR, CX3CR1. CX3CR1 specifically interacts with CX3CL1, unlike other chemokine GPCRs which are less specific and interact with multiple chemokines (Imai et al., 1997; Combadiere et al., 1998; Allen et al., 2007 review).
CX3CL1 and CX3CR1 are constitutively expressed in various non-hematopoietic tissues, including the brain. Recent reports describe the localization of CX3CL1 to neurons and astrocytes, and CX3CR1 to neurons and microglia in the hippocampus, cortex, thalamic nuclei, spinal cord, and dorsal root ganglia (Harrison et al., 1998; Nishiyori et al., 1998; Meucci et al., 2000; Tong et al., 2000; Hughes et al., 2002; Hatori et al., 2002; Verge et al., 2004; Zhuang et al., 2007). Furthermore, there is a unique neuronal-glial interaction between neurons and CX3CR1-expressing microglia under normal and pathological states in the CNS (Harrison et al., 1998).
Although CX3CL1 may induce monocytic infiltrates during CNS inflammation (Chapman et al., 2000), it appears to exert an overall neuroprotective action. Mizuno et al. (2003) reported CX3CL1-mediated inhibition both of neuronal apoptosis and of activated microglial nitric oxide, IL-6, and TNF-α production. Interestingly, CX3CL1 is also up-regulated in HIV encephalitis brain tissue, indicating that its induction may be in response to chronic activation of the neurotoxic glutamate-mediated excitatory neurotransmission by HIV neurotoxins platelet-activating factor and Tat (Tong et al., 2000). In fact, CX3CL1 is a reported inhibitor of glutamate synaptic activity in the hippocampus under basal conditions (Bertollini et al., 2006). The ability of CX3CL1 to modulate glutamate function appears to be the mechanism involved in its neuroprotective actions, and it provides support for a chemokine-neuromodulatory role in the CNS (Adler and Rogers, 2005; Adler et al., 2006; Callewaere et al., 2007; Guyon and Nahon, 2007).
The DRN contains the largest population of forebrain-projecting serotonergic neurons (Jacobs and Azmitia, 1992). Although chemokines are expressed in the brain, little is known about chemokine regulation of 5-HT neuronal functions. An impact of chemokines, including CX3CL1, on the 5-HT system would have implications for stress-induced immunological dysfunction as well as our understanding of anxiety and depressive disorders associated with immune disorders. In the present study, we examined the neuroanatomical relationship between CX3CL1 and the 5-HT system as well as the functional impact of CX3CL1 on 5-HT neurons using whole-cell patch-clamp recordings in an in vitro brain slice preparation in rats.
Experimental Procedures
Animals
Male Sprague-Dawley rats (Taconic Farms, Germantown, NY), 10 weeks of age (for immunohistochemistry) and 4–5 weeks of age (for electrophysiology) were housed 2 per cage on a 12-h light schedule (lights on at 07:00 AM) in a temperature-controlled (20 C) colony room. Rats were given access to standard rat chow and water ad libitum. Animal protocols were approved by the Temple University Institutional Animal Care and Use Committee and were conducted in accordance to the NIH Guide for the Care and Use of Laboratory Animals.
Antibodies
Details regarding the primary and secondary antibodies used in our immunohistochemistry experiments are provided in Table 1.
Table 1.
Description of antibodies used for immunohistochemistry
| Primary antibody |
Secondary antibody |
||||
|---|---|---|---|---|---|
| Antibody | Species | Clonality | Dilution | Catalog# | Antibody, Species, Dilution |
| Anti-CX3CL1 | Goat | Polyclonal | 1:100 | SC7225 | Alexa Fluor 647, donkey anti-goat, 1:200 |
| Anti-CX3CR1 | Rabbit | Polyclonal | 1:100 | TP501 | Alexa Fluor 647 and 488, donkey anti-rabbit, 1:200 |
| Anti-TPH | Mouse | Monoclonal | 1:500 | T0678 | Alexa Fluor 488, donkey anti-mouse, 1:200 |
| Anti-NeuN | Mouse | Monoclonal | 1:100 | MAB377 | AMCA, donkey anti-mouse, 1:100 |
| Anti-CD11b | Mouse | Monoclonal | 1:100 | CBL1512F | FITC-conjugated primary antibody, 1:100 |
SC, Santa Cruz; TP, Torrey Pines; T, Sigma; MAB and CBL, Millipore.
The polyclonal goat CX3CL1 antibody obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) was directed against a synthetic peptide corresponding to 20 amino acids in the C-terminal domain of CX3CL1 of human origin. Western blot analysis detected a single band with a molecular size of approximately 95 kD, which was consistent with the previously described size of glycosylated CX3CL1 (Muehlhoefer et al., 2000). Preabsorption of the CX3CL1 antibody with a tenfold excess of the immunogenic peptide (sc-7225P, Santa Cruz Biotechnology, Inc.) eliminated any specific staining (data not shown), confirming preabsorption controls for this antibody in non-neuronal cells (Habasque et al., 2003). CX3CL1 detection on immune cells by immunohistochemistry was also confirmed by single cell RT-PCR (Nanki et al., 2002). Furthermore, the Santa Cruz antibody directed against the C-terminus of CX3CL1 offered identical staining patterns in brain tissue as an antibody directed against the N-terminus of CX3CL1 in hippocampal, spinal cord, and dorsal root ganglia neurons (Hughes et al., 2002; Verge et al., 2004; Lindia et al., 2005; Zhuang et al., 2007).
A polyclonal rabbit CX3CR1 antibody raised against E. coli expressed rat CX3CR1 mapping to amino acids 2–22 in the N-terminal domain was obtained from Torrey Pines Biolabs (East Orange, NJ). Western blot analysis revealed a specific band at approximately 40 kD corresponding to the molecular weight of CX3CR1 (Meucci et al., 2000; Zhuang et al., 2007). Reported preabsorption studies (Meucci et al., 2000; Moon et al., 2006; Furuichi et al., 2006) determined the specificity of CX3CR1 immunohistochemical staining, which were in agreement with our preabsorption experiments using a tenfold excess of immunogenic peptide applied to brain slices (data not shown). Staining patterns with the CX3CR1 antibody from Torrey Pines Biolabs corresponded with in situ hybridization studies for the mRNA of CX3CR1 in the rat spinal cord and dorsal root ganglia (Verge et al., 2004). The staining of brain sections with this antibody produced a pattern of CX3CR1 immunoreactivity consistent with previous neuronal and microglial immunohistochemical labeling in the hippocampus, cortex, thalamic nuclei, spinal cord, and dorsal root ganglia with the same (Verge et al., 2004; Zhuang et al., 2007) or different CX3CR1 antibodies (Hughes et al., 2002).
The staining pattern and specificity of the tryptophan hydroxylase (TPH), NeuN, and CD11b antibodies used in our studies are well established in the rat brain. When brain tissue was stained with these antibodies (listed in Table 1), it produced a pattern that was identical to previously published reports for TPH (Azmitia et al., 1993; Commons and Valentino, 2002), NeuN (Lindia et al., 2005; Karuppagounder et al., 2007) and CD11b (Karuppagounder, 2007). Furthermore, additional tests for secondary antibody specificity were conducted on brain slices with omission of primary antibodies, and no specific staining was detected (data not shown).
Immunohistochemistry
(Antibody details in Antibodies section)
Rats were deeply anesthetized with pentobarbital (60 mg/kg, i.p.) and transcardially perfused with saline and 4% paraformaldehyde. Brains were extracted and cryoprotected in a 20% sucrose solution, frozen at −80 °C, and sectioned coronally (30 μm) by cryostat. Midbrain slices including the DRN and median raphe nucleus (MRN) were preincubated with blocking solution containing 3% normal donkey serum and 0.5% Triton X-100 in phosphate buffered saline for 30 min.
To analyze colocalization of CX3CL1 or CX3CR1 with serotonergic neurons, midbrain slices were initially incubated with either goat anti-CX3CL1 antibody (1:100; Santa Cruz Biotechnology, Inc.) or rabbit anti-CX3CR1 antibody (1:100; Torrey Pines Biolabs) overnight at 4 °C. Sections were incubated in mouse anti-TPH antibody (1:500; Sigma-Aldrich) overnight at 4 °C. Subsequently, immunohistochemical labeling was detected with an Alexa 647-conjugated donkey anti-goat or anti-rabbit secondary antibody (1:200; Molecular Probes, Eugene, OR) and an Alexa 488-conjugated donkey anti-mouse secondary antibody (1:200; Molecular Probes) for 1 h at room temperature in the dark.
CX3CR1 localization to neurons and glia was determined by sequential incubation of brain slices with rabbit anti-CX3CR1 antibody (1:100; Torrey Pines Biolabs), mouse anti-NeuN antibody (1:100; Chemicon, Temecula, CA), and mouse anti-CD11b FITC-conjugated antibody (1:100; Chemicon). Antibody incubations were conducted overnight at 4 °C, and slices were kept in the dark following the addition of the mouse anti-CD11b FITC-conjugated antibody. Immunohistochemical labeling was visualized with an Alexa 647-conjugated donkey anti-rabbit secondary antibody (1:200; Molecular Probes) to label CX3CR1 and AMCA-conjugated donkey anti-mouse secondary antibody (1:100; Jackson ImmunoResearch, West Grove PA) to label NeuN for 1 h at room temperature in the dark.
CX3CL1 and CX3CR1 colocalization in the raphe nuclei was examined by brain slice incubation with antibodies directed towards these proteins (described above). The incubations were conducted overnight at 4 °C. Next, brain sections were placed in a cocktail of Alexa Fluor 647-conjugated donkey anti-goat secondary antibody (1:200; Molecular Probes) to detect CX3CL1, and Alexa Fluor 488-conjugated donkey anti-rabbit secondary antibody (1:200; Molecular Probes) to label CX3CR1, for 1 h at room temperature in the dark.
For all immunohistochemical experiments, brain sections were rinsed with phosphate buffer solution (3 × 10 min) between incubations. Sections were mounted on Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA) and coverslipped with Prolong Gold Antifade Reagent (Molecular Probes) or Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Labs, Burlingame, CA).
Fluorescence Microscopy
Fluorescent images of TPH, CX3CL1, or CX3CR1 were captured with a Leica DMIRE TCS SL-Conformation confocal microscope using a 40X oil immersion objective and Leica operating software (Leica Microsystems, Exton, PA). The laser power and emission filters were adjusted for both the red and green fluorophors to ensure minimal possibility of a false positive result. Fluorescent photomicrographs were produced from six stacks with four-point line averaging. The image format was 1285 by 1285 pixels, and the scan speed was 400 image-lines/s. Images were also captured with a Nikon E800 fluorescent microscope (Nikon Instruments, Melville, NY), using a Retiga EXi Fast 1394 digital camera and QCapture Suite imaging software (Quantitative Imaging Corp., Surrey, BC, Canada).
Quantification Analysis
The Vectashield mounting medium containing DAPI, which detects nuclear DNA, was applied to slices double-labeled with TPH and CX3CL1 or CX3CR1 antibodies to simplify the localization and quantification of single-labeled (SL) and double-labeled (DL) cells. Individual TPH (green), CX3CL1 or CX3CR1 (red), and DAPI (blue) fluorescent photomicrographs were captured by digital camera and QCapture imaging (Fluorescence Microscopy section). All DAPI-labeled cells expressing TPH were counted in the DRN (dorsomedial (DM), lateral wing (LW), and ventromedial (VM) subdivisions) and MRN and subsequently classified as “total TPH-labeled” cells. The individual green, red, and blue images were also merged using Photoshop software. Cells (DAPI-positive, blue) expressing both TPH (green) and CX3CL1 or CX3CR1 (red) were counted and classified as “colocalized” cells. For quantification purposes, the term “colocalized” identifies cells containing green and red labeling when the immunolabeling was detected in separate cellular compartments, i.e. outer membrane vs. cytoplasm. Also, it is important to note that the inclusive term “colocalization” does not rule out the expression of chemokine proteins in terminals synapsing onto, and thus associated with, the cell body or cellular process. “Single-labeled” TPH neurons were determined by subtracting “colocalized” from “total TPH-labeled” cells. The percent of CX3CL1 or CX3CR1 colocalization with TPH was determined by dividing the number of “colocalized” neurons by the “total TPH-labeled” neurons.
Slice Preparation for Electrophysiology
Rats were rapidly decapitated, and the head placed in ice cold artificial cerebrospinal fluid (ACSF) in which sucrose (248 mM) was substituted for NaCl. The brain was rapidly removed and trimmed to isolate the brainstem region. Slices 200 μm thick were cut throughout the rostro-caudal extent of the midbrain RN using a Vibratome 3000 Plus (Vibratome, St. Louis, MO) and placed in a holding vial containing ACSF with l-tryptophan (50 μM) at 35 °C bubbled with 95% O2/5% CO2 for 1 h. Slices were then maintained in ACSF at room temperature bubbled with 95% O2/5% CO2. The composition of the ACSF was (mM), NaCl 124, KCl 2.5, NaH2PO4 2, CaCl2 2.5, MgSO4 2, Dextrose 10 and NaHCO3 26.
Electrophysiological Recordings
Slices were transferred to a recording chamber (Warner Instruments, Hamden, CT) and continuously perfused with ACSF at 1.5–2.0 ml/min at 32–34 °C maintained by an in-line solution heater (TC-324, Warner Instruments). Raphe neurons were visualized using a Nikon E600 upright microscope fitted with a 40X water-immersion objective, differential interference contrast, and infrared filter (Optical Apparatus, Ardmore, PA). The image from the microscope was enhanced using a CCD camera and displayed on a computer monitor. Whole-cell recording pipettes were fashioned on a P-97 micropipette puller (Sutter Instruments, Novato, CA) using borosilicate glass capillary tubing (1.2 mm OD, 0.69 mm ID; Warner Instruments). The resistance of the electrodes was 4–8 MΩ when filled with an intracellular solution of (in mM) Kgluconate 70, KCl 70, NaCl 2, EGTA 4, HEPES 10, MgATP 4, Na2GTP 0.3, 0.1% Biocytin, pH 7.3 (for IPSC experiments) or an intracellular solution of (in mM) Kgluconate 130, NaCl 5, MgCl2 1, EGTA 0.02, HEPES 10, sodium phosphocreatinine 10, MgATP 2, Na2GTP 0.5, 0.1% Biocytin, pH 7.3 (for membrane characteristic experiments).
Recordings were conducted in cells located in the VM subdivision of the DRN at mid-caudal levels (corresponding to −7.56 to −8.28 mm caudal to bregma in Paxinos and Watson (2005)) containing dense clusters of 5-HT neurons. A visualized cell was approached with the electrode to establish a gigaohm seal, and the cell membrane was subsequently ruptured to obtain a whole-cell recording configuration using a HEKA patch clamp EPC-10 amplifier (HEKA Elecktronik, Pfalz, Germany). Series resistance was monitored throughout the experiment. If the series resistance became unstable or exceeded four times the electrode resistance, the cell was discarded. When the whole-cell configuration was obtained in a stable cell, the cell membrane potential and input resistance (calculated by the voltage response to −300 pA current injections) were recorded in current-clamp mode (I = 0 pA).
sIPSC recordings were made in voltage clamp mode (Vm = −70 mV). Signals were stored on-line using Pulse software, filtered at 1 kHz, and digitized at 10 kHz. The liquid junction potential was approximately 9 mV between the pipette solution, and the ACSF and was not subtracted from the data obtained.
For eIPSC experiments, tungsten stimulating electrodes (World Precision Instruments, Sarasota, FL) were placed dorsolateral (100–200 μm distance) to the recorded cell, and stimuli were delivered with an IsoFlex stimulus isolator (A.M.P.I., Jerusalem, Israel). For each cell, a stimulus-response curve was generated, and the stimulus intensity producing a half-maximal response was then used for paired-pulse experiments. Paired-pulses were delivered with a 50 ms inter-pulse interval and a 10 s interval between pairs.
Experimental Protocols
Baseline membrane potential was initially recorded for 5 min to ensure cell stability. CX3CL1 was then added to the perfusion bath and recorded for 10 min or until a change in the resting membrane potential (drug effect) was observed. If a drug effect was present, the cell was recorded from for an additional 5 min following membrane stabilization its new baseline potential. The drug was subsequently removed from the perfusion bath during a washout phase to determine if the cell would return to its initial membrane potential.
GABA-mediated sIPSC recordings were isolated by addition of the non-N-methyl D-aspartate (NMDA) receptor antagonist 6,7-dinitroquinoxaline-2,3(1H, 4H)-dione (DNQX; 20 μM) and were recorded for 6 min. CX3CL1 (10 nM) was then added to the perfusion bath, and sIPSC recordings were obtained for 9 min. For stimulation experiments, baseline paired-pulse data were collected for 10–20 min. CX3CL1 (1.0 or 10 nM) was then added to the perfusion bath, and paired-pulse data was collected for an additional 10–20 min. The specificity of CX3CL1’s effects (10 nM) on eIPSCs was tested by preincubating brain slices for 1 h with an anti-CX3CL1 neutralizing antibody (2 μg/ml) before beginning the eIPSC recordings. In some cells (both sIPSC and eIPSC experiments), GABAA receptor-mediation of IPSC recordings was verified at the end of the experiment by addition of a GABAA receptor antagonist, bicuculline (20 μM). Under these conditions, bicuculline eliminated all sIPSC/eIPSC activity (see Figs. 3–5).
The dose range of CX3CL1 used in these studies (1.0–10 nM) is 10–100 times the reported kD of CX3CL1 for its receptor CX3CR1 (Imai et al., 1997). Previous studies of CX3CL1 using in vitro bioassays and electrophysiological techniques have also employed similar doses. For example, CX3CR1 dose-dependently stimulates calcium mobilization in CX3CR1-expressing cells with an EC50 of 2 nM (Imai et al., 1997). CX3CL1 also dose-dependently reduces glutamate-induced excitotoxicity in hippocampal neurons with an EC50 of 0.7 nM (Limatola et al., 2005). Finally, CX3CL1 dose-dependently inhibits field excitatory postsynaptic potentials (Bertollini et al., 2006) and evoked EPSC amplitude (Ragozzino et al., 2006) in hippocampal neurons with an EC50 of 0.7 and 1.0 nM, respectively.
Immunohistochemistry of the recorded cell
Following electrophysiology studies, standard immunofluorescence procedures were used to visualize the filled cell and neurotransmitter content (Beck et al., 2004). Slices were post-fixed in 4% paraformaldehyde overnight. Sections were incubated in phosphate buffered saline containing 0.5% Triton and bovine serum albumin for 30 min. Sections were then incubated in mouse anti-TPH antibody (1:500; Sigma-Aldrich, St. Louis, MO) overnight at 4 °C. Subsequently, immunohistochemical labeling was visualized using an Alexa 647- conjugated donkey anti-mouse secondary antibody (1:200; Molecular Probes) for 1 h at room temperature. Biocytin in the recorded cell was visualized using a streptavidin-conjugated Alexa 488 secondary antibody (1:200; Molecular Probes) for 1 h at room temperature.
Drugs
Most chemicals for making the ACSF and electrolyte solutions as well as bicuculline were obtained from Sigma-Aldrich. DNQX was purchased from Tocris (Ellisville, MO) and dissolved in dimethyl sulfoxide (final dimethyl sulfoxide concentration in bath 0.015%). CX3CL1 (recombinant rat CX3CL1/Fractalkine carrier free) was obtained from R&D Systems (Minneapolis, MN), dissolved in dH20 at 10 μM and stored at 4 °C for up to 1-week following rehydration. The anti-CX3CL1 neutralizing antibody was obtained from R&D Systems.
Data Analysis
The resting membrane potential and input resistance was recorded from baseline, drug, and washout phases. The maximum drug steady-state value was reported as the drug effect for each cell. Input resistance was calculated from voltage responses to periodic (every 30 s) intracellular current pulse injections (−300 pA). Input resistance was calculated based on the average of three measured voltage responses taken at baseline or when the drug effect had reached steady-state. Membrane potential and voltage responses to current pulses were measured using pClamp 9.0 software (Molecular Devices, Sunnyvale, CA).
MiniAnalysis software (Synaptosoft, Inc., Decatur, GA) was used to analyze sIPSC and eIPSC events. For sIPSC recordings, noise analysis was conducted for each cell and amplitude detection thresholds were set to exceed noise values. Events were automatically selected, analyzed for double peaks, and subsequently visually inspected and confirmed. Event amplitude histograms were generated and compared to the noise histogram to ensure that they did not overlap. Holding current was monitored, and synaptic activity was analyzed for frequency, amplitude, rise time (calculated from 10–90% of peak amplitude) and decay time (calculated by averaging 200 randomly selected events and fitting a double exponential function from 10–90% of the decay phase). The double exponential function for the decay phase generated an initial fast component and a subsequent slow component of the decay phase.
Data were collected in 1-min bins during the 9 min period following drug application, taking into account a 2-min lag time from drug addition to initial drug effect due to recording chamber volume and perfusion rate. The maximum post-drug steady-state value (1-min bin) was reported as the drug effect for each cell and typically occurred 6–9 min following drug application. Group data are reported as mean ± SEM, with the exception of sIPSC rise time which was not normally distributed and was therefore reported as median ± SEM. eIPSC amplitude was calculated by subtracting the peak current from that obtained during a 5 ms window immediately preceding the stimulus artifact. Baseline eIPSC amplitude was averaged from at least 60 consecutive trials calculated over a minimum of 10 min before drug application. Stimuli were delivered in pairs and paired-pulse ratio (PPR) was calculated as the amplitude of the second eIPSC divided by the amplitude of the first eIPSC. CX3CL1’s effects were analyzed from the average of at least 60 consecutive trials following drug application.
Statistics
All data were prescreened for normal distribution prior to selection of appropriate parametric or non-parametric tests. Statistics were analyzed using SigmaStat 3.11 software (Systat, San Jose, CA).
For immunohistochemical studies, percent colocalization of TPH-CX3CL1 and TPH-CX3CR1 was analyzed by two-way repeated measures ANOVA followed by post-hoc Student-Newman-Keuls tests when appropriate for pairwise comparisons. For electrophysiological studies, the effects of CX3CL1 on membrane potential and input resistance was analyzed by paired Student’s t-test. Holding current or synaptic event characteristics of sIPSC recordings were compared between 5-HT and non-5-HT cells by unpaired Student’s t-test or Mann-Whitney Rank Sum test for non-normally distributed data. CX3CL1’s impact on holding current, sIPSC characteristics, eIPSC amplitude and PPR was analyzed by comparing pre- and post-CX3CL1 values using paired Student’s t-test or Wilcoxon Signed Rank Test for non-normally distributed data. The frequency and amplitude of IPSC events in individual cells were also illustrated as cumulative probability histograms for inter-event interval and amplitude. Baseline vs. drug treatments were also compared by the Kolmogorov-Smirnov test (analyzed with MiniAnalysis software). Unpaired Student’s t-test was used to compare the effects of different concentrations of CX3CL1 on eIPSC amplitude and PPR.
Results
CX3CL1 and CX3CR1 are Expressed on TPH-Positive Neurons and on the Same Cells in the DRN
Fluorescent photomicrographs of TPH and CX3CL1 or TPH and CX3CR1 containing cells in the dorsomedial (DM) and ventromedial (VM) subdivisions of the DRN are shown in Fig. 1A–F. Individual panels of TPH-immunoreactivity in green and CX3CL1- (panels A–C) or CX3CR1- (panels D–F) immunoreactivity in red indicate a generalized colocalization of 5-HT neurons with CX3CL1 or CX3CR1 in the DRN (Fig. 1 shows colocalization in DM-DRN (panels A, D) and VM-DRN (panel B, E); Table 2 and Fig. 2 present data from all DRN subdivisions including the lateral wing (LW) subdivision). A limited number of 5-HT neurons not expressing CX3CL1 or CX3CR1 were detected as green-only labeling (Fig. 1A, B, D green arrows). Interestingly, several cell bodies with similar morphology to TPH-positive neurons were detected as single-labeled for CX3CL1 or CX3CR1, suggesting that CX3CL1 and CX3CR1 may be present on other non 5-HT neurons or cells in the raphe nuclei (Fig. 1B, D, E red arrows).
Figure 1.

CX3CL1 or CX3CR1 colocalization with TPH in the DRN. Fluorescent photomicrographs of TPH with CX3CL1 (A–C) or CX3CR1 (D–F) containing cells in the DM (A, D) and VM (B, E) subdivisions of the DRN. The upper left panels show TPH-immunoreactivity in green, the upper right panels depict either CX3CL1- or CX3CR1-immunoreactivity in red, and the large panels show the merge image. Colocalization of CX3CL1 or CX3CR1 in serotonergic neurons is present throughout the DM- and VM-DRN (yellow arrows; A, B, D, E). Individual cells are also labeled only with TPH (green arrows; A, B, D), CX3CL1 (red arrows; B), or CX3CR1 (red arrows; D, E). Panels C and F show magnified images of CX3CL1 or CX3CR1 with TPH in the DRN. CX3CL1 localizes as discrete puncta within the cytoplasm and processes of TPH-positive neurons (white arrows, C merge), whereas CX3CR1 is expressed in a perinuclear pattern in TPH-positive neurons (white arrows, F merge) and other cells (dotted white arrows, F merge). Panel G depicts DRN cells co-expressing CX3CL1 and CX3CR1 (white arrows, merge). Panel H shows CX3CR1 expressing microglia (CD11b in green; white arrows) and neurons (anti-NeuN in blue; dotted white arrows) in DRN sections. Schematics from Paxinos and Watson (2005) are included to indicate the panel locations in the DRN. Scale Bars = 25μm (A, B, D, E); 10 μm (H), 5 μm (C, F, G).
Table 2.
Quantification of CX3CL1 and CX3CR1 localization with serotonergic neurons in the raphe nuclei
| Raphe nuclei subdivisions | Total TPH | TPH-CX3CL1 DL | TPH SL | Total TPH | TPH-CX3CR1 DL | TPH SL |
|---|---|---|---|---|---|---|
| DRN-DM | 23.8+3.1 | 22.5+3.4 | 1.2+0.5 | 29.2+2.4 | 28.0+2.7 | 1.2+0.6 |
| DRN-LW | 30.0+0.9 | 24.5+1.8 | 5.5+1.0 | 31.8+2.5 | 20.0+2.2 | 2.8+0.5 |
| DRN-VM | 72.5+2.6 | 63.2+4.8 | 9.2+4.1 | 71.8+4.2 | 68.5+4.2 | 3.2+0.2 |
| MRN | 23.8+3.4 | 18.0+3.2 | 5.8+1.4 | 28.5+3.2 | 26.0+2.4 | 2.5+0.9 |
TPH-CX3CL1 and TPH-CX3CR1 colocalization studies show data presented as mean number of cells ±SEM for four rats.
TPH, tryptophan hydroxylase; DRN, dorsal raphe nucleus; MRN, median raphe nucleus; DM, dorsomedial; LW, lateral wing; VM, ventromedial; DL, double-labeled; SL, single-labeled.
Figure 2.

Quantified colocalization of TPH with CX3CL1 or CX3CR1 in the raphe nuclei. Panels A and B show CX3CL1 and CX3CR1 expression in over 70% of serotonergic neurons throughout the DRN subdivisions, DM, LW, VM, and the MRN. Panel C indicates the rostrocaudal level (−8.00) and delineates the raphe subdivisions in which colocalization was quantified in a representative TPH-immunoreactive section of the raphe nuclei. * = p < 0.05 and ** = p < 0.01 by post-hoc Student-Newman-Keuls test. All values are mean ± SEM. Dorsal raphe nucleus (DRN); median raphe nucleus (MRN); dorsomedial (DM); lateral wing (LW); ventromedial (VM).
The individual images in Fig. 1C, F are representations of TPH and CX3CL1 or TPH and CX3CR1 localization detected in the RN by confocal microscopy. Fig. 1C depicts a magnified image of 5-HT neurons co-expressing CX3CL1 (white arrows). CX3CL1 appears to be distributed as discrete puncta within the cytoplasm and processes of these neurons. In Fig. 1F, the subcellular distribution of CX3CR1 localized to the perinuclear membrane of 5-HT neurons (white arrows) and other cells (dotted white arrow). Also, fluorescent photomicrographs of cells labeled for CX3CL1-C3XCR1 are shown in Fig. 1G. Individual panels of CX3CL1-immunoreactivity in red and CX3CR1-immunoreactivity in green were captured and merged. The yellow/orange labeled cells in the merge image indicate that these neurons are double-labeled (white arrows).
Localization of CX3CR1 to Neurons as well as Microglia in the Raphe Nuclei
Many reports have demonstrated CX3CR1 localization to microglia in the brain (Nishiyori et al., 1998; Harrison et al., 1998). Recent studies, as well as our own CX3CR1 colocalization data with TPH, indicate that CX3CR1 may be expressed by neurons as well (Tong et al., 2000; Meucci et al., 2000; Hughes et al., 2002). To further investigate CX3CR1 neuronal and microglial localization, sections were incubated with antibodies to CX3CR1, CD11b to label microglia, and NeuN to label neuronal nuclei. The antibodies were visualized as red (CX3CR1-IR), green (CD11b-IR), and blue (NeuN-IR), respectively (Antibodies section, Table 1). Triple-labeling indicates that CX3CR1 exhibits a dual neuronal and microglial expression profile. In Fig. 1H, CX3CR1 is present on neurons (dotted white arrows) as well as microglia (solid white arrows) within the RN. The neuronal localization of CX3CR1 appears to be more prominent in the perinuclear region, as previously detected in the TPH-CX3CR1 merge image (white arrows, Fig. 1F). A neuronal distribution of CX3CR1 was also observed in other brain regions examined, including the CA1 hippocampal pyramidal cell layer, cingulate cortex, and striatum (data not shown).
Quantification of CX3CL1 and CX3CR1 Colocalization to 5-HT Neurons in the Raphe Nuclei
“Total-TPH” labeled as well as TPH-CX3CL1 or TPH-CX3CR1 “colocalized” neurons were quantified in the DRN (DM, VM, and LW subdivisions) and MRN in four rats (Methods). Quantification data shown in Table 2 for CX3CL1 and CX3CR1 were consistent across the different animals as indicated by small standard error of mean (SEM) values. Immunohistochemical analysis revealed a greater than 70% colocalization of CX3CL1 or CX3CR1 in 5-HT neurons in the RN. In Fig. 2A, all DRN subdivisions showed a high degree of CX3CL1 or CX3CR1 colocalization with TPH. There was a significant main effect of DRN subdivision (F(2,12) = 7.80, p < 0.01) but no effect of antibody and no significant subdivision x antibody interaction. Fig. 2B indicates the percent colocalization of CX3CL1 or CX3CR1 to 5-HT neurons in the DRN and MRN. There were significant main effects of raphe division [F(1,6) = 18.77, p < 0.01], antibody [F(1,6) = 6.60, p < 0.05] and a significant interaction between these factors [F(1,6) = 7.76, p < 0.05]. The percent colocalization of CX3CR1 with TPH remained greater than 90% throughout the DRN and MRN. The percent colocalization of its ligand, CX3CL1, with TPH was significantly higher in the DRN (87.0%) than in the MRN (75.3%) (p < 0.01) by post-hoc Student-Newman-Keuls test (Fig. 2B). In the MRN, CX3CL1 also displayed a reduced colocalization with TPH as compared to CX3CR1 (p < 0.05).
Basal IPSC Characteristics and Holding Current
Holding current (pA) as well as sIPSC characteristics were examined, including frequency (Hz), amplitude (pA), rise 10–90% (ms), fast decay (ms) and slow decay (ms). Table 3 shows baseline sIPSC characteristics from 5-HT and non-5-HT DRN neurons. The control sIPSC amplitude of non-5-HT neurons was significantly greater than the control sIPSC amplitude for 5-HT neurons (p < 0.05). These data are consistent with earlier findings (Kirby et al., 2008), which suggest enhanced non-5-HT neuronal postsynaptic GABA receptor number or sensitivity when compared to 5-HT neurons. In contrast, under conditions of synaptic activation no statistical difference was detected between 5-HT and non-5-HT neurons comparing baseline eIPSC amplitude and PPR characteristics, indicating that this difference in postsynaptic GABA receptors between 5-HT and non-5-HT cells exists only under basal conditions (see below). Other sIPSC characteristics or holding current values did not differ between the 5-HT and non-5-HT cell types, suggesting that the resting membrane potential and presynaptic GABA release are similar in the two cell populations in the basal state.
Table 3.
sIPSC characteristics and holding current in 5-HT and non-5-HT neurons
| A 5-HT | ||||||
|---|---|---|---|---|---|---|
| Treatment | Frequency (Hz) | Amplitude (pA) | Rise 10–90% (ms) | Fast decay (ms) | Slow decay (ms) | Current (pA) |
| Control | 4.9±0.5 | 12.9±1.0 | 1.9±0.2 | 5.1±1.5 | 23.1±9.5 | −32.9±7.6 |
| + CX3CL1 | 5.5±0.6 | 16.4±1.7* | 2.0±0.1 | 5.2±0.8 | 21.8±7.1 | −47.0±15.0 |
| B Non-5-HT | ||||||
|---|---|---|---|---|---|---|
| Treatment | Frequency (Hz) | Amplitude (pA) | Rise 10–90% (ms) | Fast decay (ms) | Slow decay (ms) | Current (pA) |
| Control | 4.6±0.7 | 20.2±2.6** | 1.4±0.2 | 6.3±3.5 | 31.1±20.3 | −37.1±8.4 |
| + CX3CL1 | 4.0±0.3 | 18.8±3.1 | 2.0±0.4* | 5.0±0.3 | 35.2±21.0 | −30.6±13.5 |
sIPSC characteristics and holding current determined before (control) and after CX3CL1 (10 nM) application are presented for 5-HT (A; n=9) and non-5-HT (B; n=5) cells.
All values are mean±SEM, except for rise 10–90% which is median±SEM.
P<0.05 by paired Student’s t-test comparing control vs. CX3CL1 treatment groups.
P<0.01 by unpaired Student’s t-test comparing 5-HT vs. non-5-HT neurons.
CX3CL1 Has No Effect on Membrane Characteristics of 5-HT Neurons
The resting membrane potential was recorded under current clamp conditions (I = 0 pA) in 5-HT DRN neurons. CX3CL1 (10 nM) failed to demonstrate a statistically significant impact on the resting membrane potential of serotonergic neurons (N = 11) (data not shown). Baseline recordings, −53.2 ± 1.9 mV, remained unchanged following 10 nM CX3CL1 application, −51.9 ± 2.5 mV. Consistent with this finding, the holding current measured in voltage-clamp experiments also remained unchanged by CX3CL1 (see Table 3). CX3CL1 lacked a significant effect on cellular input resistance as well, 266.7 ± 44.2 MΩ at baseline and 275.0 ± 49.9 MΩ following CX3CL1 application, in the recorded neurons (data not shown). Thus, CX3CL1 does not exhibit effects on membrane potential or cellular input resistance in 5-HT DRN neurons.
CX3CL1 Increases Spontaneous GABA Synaptic Activity Selectively in 5-HT Neurons
sIPSCs were recorded under voltage-clamp conditions (Vm = −70 mV) in 5-HT and non-5-HT neurons in the VM subdivision of the DRN. Table 3 summarizes CX3CL1’s effects on sIPSC characteristics in 5-HT and non 5-HT neurons.
CX3CL1’s actions on sIPSCs in a recorded 5-HT neuron are depicted in Fig. 3A. The recorded cell was identified as serotonergic since it expressed TPH (Fig. 3B merge, white arrow). In this cell, CX3CL1 (10 nM) stimulates sIPSC amplitude from 15.4 pA to 18.8 pA and does not change sIPSC frequency. Application of bicuculline (20 μM), completely eliminates the recorded sIPSCs, confirming that these events are mediated by the GABAA receptor. The increase in sIPSC amplitude was statistically significant (z-score = 3.29, p < 0.05, Kolmogorov-Smirnov two sample test) as indicated by a rightward shift on the cumulative histogram (Fig. 3C’). However, the inter-event interval cumulative histogram for baseline and CX3CL1 treatment was not different (Fig. 3C), indicating no difference in sIPSC frequency.
Figure 3.

CX3CL1 stimulates sIPSC amplitude selectively in 5-HT DRN neurons. CX3CL1 (10 nM) enhances baseline sIPSC amplitude from 15.5 to 18.9 pA without affecting sIPSC frequency in a recorded 5-HT neuron, and bicuculline (20 μM) eliminates all sIPSC events (A, B white arrow). CX3CL1 significantly shifts the cumulative histogram for amplitude to the right, but does not impact the inter-event interval (C′ vs. C). Group data demonstrate a CX3CL1 selective increase in sIPSC amplitude (D′) with no change in sIPSC frequency (D) at 5-HT DRN neurons. * = p < 0.05 by paired Student’s t-test, # = p <0.05 by the Kolmogorov-Smirnov two sample test. Group data are represented as mean ± SEM.
Group analysis of recorded serotonergic neurons (N = 9) reveals that CX3CL1 significantly increases sIPSC amplitude (Fig. 3D’, p < 0.05; Table 3) without impacting sIPSC frequency (Fig. 3D; Table 3), suggesting an enhanced postsynaptic expression or sensitivity of the GABAA receptor.
CX3CL1’s effects on a recorded non-5-HT neuron are depicted in Fig. 4. The recorded cell was identified as non-5-HT since it did not label with TPH (Fig. 4B merge, white arrow). In this non-5-HT cell, CX3CL1 (10 nM) does not change baseline sIPSC amplitude or frequency. Application of bicuculline (20 μM) completely eliminates the recorded sIPSCs, confirming that these events are mediated by the GABAA receptor. The inter-event interval and sIPSC amplitude cumulative histograms for baseline and CX3CL1 treatment were not different (Fig. 4C, C’). Therefore, in contrast to 5-HT neurons, non-5-HT neurons (N = 5) do not exhibit changes in sIPSC frequency or amplitude following CX3CL1 application (Fig. 4D, D’).
Figure 4.

CX3CL1 does not affect sIPSC frequency or amplitude in non-5-HT DRN neurons. CX3CL1 (10 nM) fails to modulate sIPSC frequency or amplitude in a recorded non-5-HT neuron, and bicuculline (20 μM) eliminates all sIPSC events (A, B white arrow). CX3CL1 does not impact the cumulative histogram for inter-event interval (C) or amplitude (C′). Group data show no effect of CX3CL1 on either sIPSC frequency (D) or amplitude (D′) parameters in non-5-HT DRN neurons. Group data are represented as mean ± SEM.
Thus, CX3CL1 (10 nM) shows unique actions on 5-HT vs. non-5-HT neurons in the DRN since it selectively enhances postsynaptic GABA receptor number or sensitivity in 5-HT neurons.
CX3CL1 Increases Evoked GABA Synaptic Activity at 5-HT Neurons
Additionally, we examined eIPSCs under voltage-clamp conditions (Vm = −70 mV) in 5-HT DRN neurons to complement recordings of spontaneous GABA synaptic activity with recordings under conditions of synaptic activation.
A representative recording of a 5-HT neuron shows that CX3CL1 application stimulates baseline eIPSC amplitude from 241.4 pA to 359.1 pA but does not alter baseline PPR (Fig. 5A, B). The addition of bicuculline (20 μM) completely eliminates recorded eIPSC events, thus confirming that the eIPSC recordings are mediated by the GABAA receptor (Fig. 5A).
Figure 5.

CX3CL1 selectively enhances eIPSC amplitude without modulating PPR in 5-HT DRN neurons. Panels A and B are representative traces (averaged from 60 events) showing eIPSC amplitude (A) and PPR (B) chart recordings before and after CX3CL1 (A, B) and bicuculline (A) treatment. CX3CL1 (10 nM) stimulates eIPSC amplitude (A, A′) and does not change baseline PPR (B, B′) in 5-HT neurons, and bicuculline (20 μM) completely eliminates all eIPSC events (A). * = p < 0.05 by paired Student’s t-test. Group data are represented as mean ± SEM.
In serotonergic neurons (N = 6), the application of CX3CL1 (10 nM) significantly enhances eIPSC amplitude (Fig. 5A’, p < 0.05) with no effect on PPR (Fig. 5B’). CX3CL1’s ability to selectively increase eIPSC amplitude but not PPR in 5-HT DRN neurons suggests that its effects are mediated by an enhanced postsynaptic GABA receptor number or sensitivity. These results suggest CX3CL1 enhancement of postsynaptic GABA receptors on 5-HT neurons under conditions of spontaneous as well as synaptically-evoked GABA release.
CX3CL1’s Concentration-Dependent Effect on Evoked GABA Synaptic Activity
The concentration-dependent actions of CX3CL1 on evoked GABA synaptic activity in 5-HT neurons were examined using 1.0 and 10 nM doses of CX3CL1. The 1.0 nM application of CX3CL1 had no impact on eIPSC amplitude in 5-HT DRN neurons (N = 6). In contrast, 10 nM of CX3CL1 significantly increased eIPSC amplitude above predrug baseline (p < 0.05, N = 6). The PPR was not affected by either dose of CX3CL1. Fig. 6 summarizes the concentration-dependent nature of CX3CL1’s actions on eIPSC amplitude and PPR in 5-HT DRN neurons.
Figure 6.
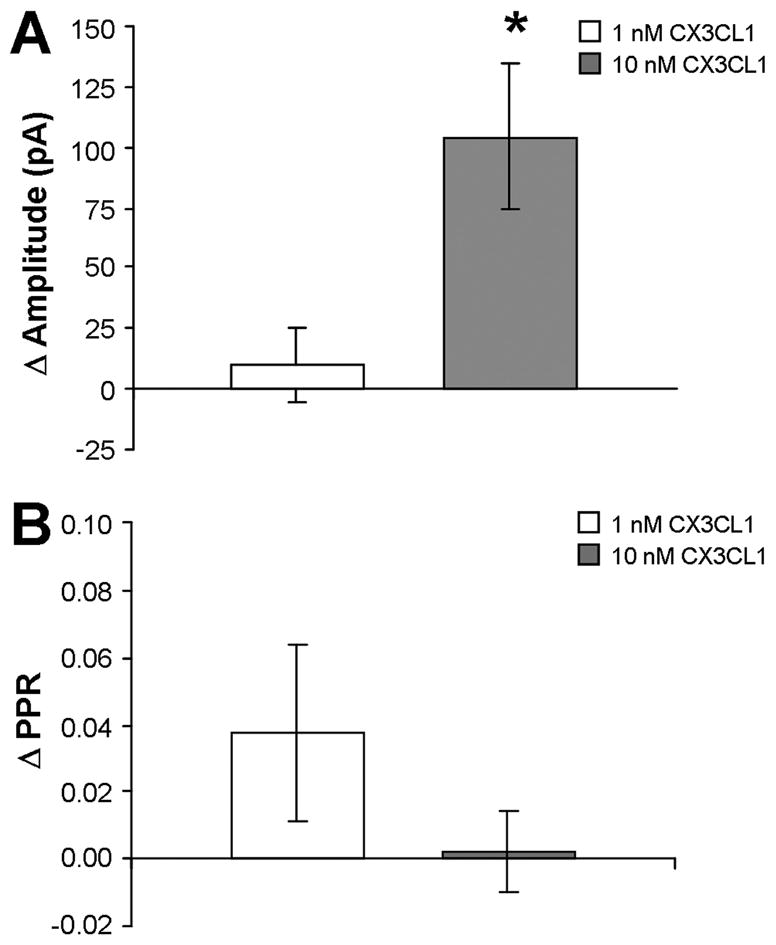
CX3CL1’s concentration-dependent elevation of eIPSC amplitude with no effect on PPR in 5-HT DRN neurons. CX3CL1 enhances eIPSC amplitude at 10 nM but not at 1.0 nM (A). PPR is not changed by the 10 nM or 1.0 nM concentration of CX3CL1 (B). * = p < 0.05 by unpaired Student’s t-test. All data are represented as mean ± SEM.
An Anti-CX3CL1 Neutralizing Antibody Inhibits CX3CL1’s Effects on Evoked GABA Synaptic Activity in 5-HT Neurons
In order to investigate the specificity of the observed actions of CX3CL1 on eIPSC amplitude in 5-HT DRN neurons, we utilized an anti-CX3CL1 neutralizing antibody to prevent CX3CL1 from binding to and activating its receptor CX3CR1. In Fig. 7A, the increase in eIPSC amplitude normally produced by 10 nM CX3CL1 (see Fig. 5A, A’) was eliminated by preincubation of the brain slice with the anti-CX3CL1 neutralizing antibody. Furthermore, CX3CL1’s lack of effect on PPR in 5-HT neurons was unchanged following application of the anti-CX3CL1 neutralizing antibody (Fig. 7B ). Thus, CX3CL1-CX3CR1 interactions appear necessary to enhance presynaptic GABA receptor number or sensitivity on 5-HT neurons.
Figure 7.

Anti-CX3CL1 neutralizing antibody eliminates CX3CL1’s effects on evoked GABA synaptic activity in 5-HT DRN neurons. The increase in eIPSC amplitude produced by CX3CL1 (see Fig. 5) was blocked by CX3CL1-neutralizing antibody (2 μg/ml) pretreatment (A). Statistical analysis was performed by paired Student’s t-test. Group data are represented as mean ± SEM.
Discussion
These data provide evidence for anatomical and functional interactions of CX3CL1 with 5-HT neurons in the RN. Immunohistochemical data show extensive (>70%) colocalization of CX3CL1 or its receptor with 5-HT RN neurons. Electrophysiology studies indicate that CX3CL1 increases postsynaptic GABA receptor number or sensitivity selectively at 5-HT neurons under conditions of spontaneous and synaptically-evoked GABA release. This enhancement of postsynaptic GABA receptors would be predicted to reduce 5-HT neuronal excitability. Furthermore, we show that CX3CL1’s effects in 5-HT neurons are dose-dependent and appear to be specific since pretreatment with an anti-CX3CL1 neutralizing antibody eliminates CX3CL1’s effect on synaptically-evoked activity. In non-5-HT neurons, CX3CL1 does not impact the parameters examined, suggesting that this chemokine selectively targets 5-HT neurons in the DRN.
Immunohistochemical studies show CX3CL1 colocalization both to 5-HT neurons and to CX3CR1-expressing neurons. These localization profiles indicate that CX3CL1 may have different functional roles in the DRN: neuromodulator vs. autoregulator. The localization of CX3CL1 within the cytoplasm of 5-HT neurons suggests that it may be co-released with serotonin, potentially modulating 5-HT neurotransmitter release upon neuronal activation. It is important to note, however, that the limitations of light microscopy leave open the alternative possibility that CX3CL1 is instead located in terminals synapsing onto 5-HT neurons. This alternative still implies interactions between CX3CL1-containing cells and 5-HT neurons. However, the interaction would be via a different mechanism than if the chemokine was co-expressed in individual 5-HT neurons. Colocalization, co-release and functional interactions as described with various neuropeptide and neurotransmitter systems (Zupanc, 1996; Hokfelt et al., 2000), is likely to occur with CX3CL1 and 5-HT, either directly via cellular colocalization, indirectly via terminal synapses, or perhaps a combination of both mechanisms.
In addition to a potential neuromodulatory role, the colocalization of CX3CL1 and CX3CR1 to individual DRN neurons suggests that CX3CL1 can be released from these neurons, possibly interacting directly with its receptor expressed on the same cell. CX3CL1 may thus function through an autoregulatory mechanism to modulate neuronal activity and/or excitability. Interestingly, the subcellular localization profile of CX3CL1 and CX3CR1 are distinct. CX3CL1 localizes as cell-filled puncta whereas its receptor localizes in a perinuclear pattern within neurons. This further suggests synthesis, as well as potential storage and release, of CX3CL1 in 5-HT neurons. In contrast, CX3CR1 may require cellular activation for receptor mobilization to the cellular membrane in order for CX3CL1-CX3CR1 interaction to occur. Future studies investigating CX3CR1 trafficking and distribution under basal conditions, in activated/CX3CL1-bound states following exogenous CX3CL1 administration, in disease states, or following immune challenges that stimulate endogenous CX3CL1 release are necessary to completely examine the mechanism(s) governing CX3CL1 and CX3CR1 interactions with 5-HT neurons and other DRN cells.
CX3CL1 and CX3CR1 are expressed on neurons and glia, and a unique neural-glial interaction for neuronally derived CX3CL1 and CX3CR1-expressing microglia under normal and disease states within the CNS may exist (Harrison et al., 1998). Ré and Przedborski (2006) reported CX3CL1-mediated neuroprotective effects on hippocampal neurons via inhibition of microglial activation. CX3CL1 may activate CX3CR1 on microglia (Harrison et al., 1998; Nishiyori et al., 1998) to directly prevent microglial activation under neuroinflammatory conditions. Inhibition of microglial activation in neurodegenerative diseases, such as Parkinson’s disease, via the application of various pharmacological agents targeting CNS inflammation, appears to be neuroprotective in animal models, and these drug therapies are currently undergoing preclinical trials (Wu et al., 2002; Gao et al., 2003 review).
Within the 5-HT system, triple-labeling of CX3CR1 with NeuN and CD11b, to label neuronal nuclei and microglial cells respectively, confirms a dual neuronal and microglial localization profile for CX3CR1 in the DRN. Endogenous CX3CL1 released from 5-HT neurons may interact with neuronal or microglial expressed CX3CR1 to modulate neurotransmission, microglial activation, and receptor trafficking. Exogenous administration of CX3CL1 agonists may also represent novel pharmacological treatments to modify 5-HT neurotransmission or microglial activation during neuroinflammatory responses. CX3CL1’s ability to provide neuroprotection implies that it may serve to enhance 5-HT levels within the brain. Enhancement of 5-HT levels via CX3CL1 may improve the mood of individuals afflicted with depression or mood changes associated with impaired immunity. However, because our electrophysiology data indicate enhanced GABAergic inhibition of 5-HT neurons by CX3CL1 administration, the degree to which 5-HT can be enhanced under these circumstances appears to depend on the relative balance between CX3CL1-induced neuroprotection and GABAergic inhibition at 5-HT neurons.
Previous reports using electron microscopy demonstrate localization of chemokines in dense-core vesicles in vasopressin-containing nerve terminals in the rat posterior pituitary (Callewaere et al., 2007). These data provide support for the proposed release of chemokines from nerve terminals (de Jong et al., 2005), and their ability to act via autocrine and paracrine mechanisms in the CNS (Callewaere et al., 2006; Meucci et al., 2000). Previous reports indicate that CX3CL1 inhibits glutamate synaptic activity postsynaptically at hippocampal neurons (Ragozzino et al., 2006). Our data indicate additional postsynaptic effects of CX3CL1: increasing GABA receptors on 5-HT DRN neurons.
Another chemokine, stromal-cell derived factor-1α/CXCL12, has also been similarly examined with respect to the 5-HT RN system in our laboratory. Like the CX3CL1 and CX3CR1 findings, CXCL12 and its receptor CXCR4 show extensive colocalization profiles with TPH in the DRN and MRN. Functionally, both chemokines CX3CL1 and CXCL12 also interact with the 5-HT system via a GABA-mediated mechanism. Yet, these two chemokines differ in their intrinsic GABA synaptic mechanism of action. As already discussed, CX3CL1 activates GABA synaptic activity at the postsynaptic level, however, we have reported stimulatory presynaptic actions of CXCL12 on GABA release (Heinisch and Kirby, 2007). Interestingly, CXCL12 also produces a membrane depolarization in 5-HT neurons, an effect mediated indirectly by glutamate afferents, whereas CX3CL1 does not exhibit any effect on this parameter. Thus, although the chemokine family is comprised of a large variety of small chemotactic proteins, it appears that two structurally distinct chemokines, CX3CL1 and CXCL12, share common colocalization profiles with the 5-HT system as well as common functional actions on GABA synaptic activity on 5-HT neurons within the RN.
Future studies will be needed to elucidate the mechanism whereby CX3CL1 postsynaptically activates GABAA receptors at 5-HT neurons. Because we employed a chloride-based intracellular solution, we cannot rule out the possibility that our observed changes in sIPSC amplitude are mediated by CX3CL1-induced changes in Cl- driving force across the membrane (Wagner et al., 1997). However, there are examples in the literature of extracellular signals regulating GABAA receptor trafficking and inhibitory synaptic signaling. These include insulin, epileptic activity and brain-derived neurotrophic factor (Wan et al., 1997; Nusser et al., 1998; Brunig et al., 2001; Mizoguchi et al., 2003; Wang et al., 2003; Elmariah et al., 2004; Jovanovic et al., 2004). Insulin, for example, induces a rapid translocation of the GABAA receptor from the intracellular compartment to the plasma membrane and enhances synaptic currents via phosphorylation of the 2 receptor subunit by a downstream serine/threonine kinase, Akt (Wan et al., 1997; Wang et al., 2003). It is possible that CX3CL1 similarly stimulates postsynaptic expression of GABA receptors on 5-HT neurons. Alternatively, CX3CL1 could enhance the sensitivity of postsynaptic receptors to GABA via intracellular mechanisms such as more efficient coupling of the receptor for its G-protein.
Modulation of GABA synaptic activity at 5-HT neurons by CX3CL1, and by additional members of the chemokine family, such as CXCL12, may be clinically significant in the pathophysiology and treatment of 5-HT related mood disorders, including depression experienced by immunocompromised individuals. CX3CL1 and other chemokines may also represent a new class of CNS neuromodulators, impacting brain function via their effects on traditional neurotransmitter and neuropeptide systems, including the 5-HT system (Adler and Rogers, 2005; Adler et al., 2006; Callewaere et al., 2007; Rostene et al., 2007). Thus, our study provides additional support for the general hypothesis that chemokines, in addition to traditional neurotransmitters and neuropeptides, represent a novel neuromodulatory system in the CNS.
Acknowledgments
We would like to thank Dr. Patrick Piggot, members of his laboratory, and Emily Freeman-Daniels for their technical assistance. We also thank Dr. Mary Barbe and Dr. Martin Adler for their helpful advice and guidance in interpreting our anatomical data and examining our research report. This work was supported by an NIH grant DA 20126 given to Dr. Kirby and collaborative grants from NIH (DA 06650, M. Adler, PI) and the Pennsylvania Dept. of Health as well as an NIH Center grant (DA 13429). S. Heinisch was supported by a Pennsylvania Health Research Formula Fund Grant.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- CNS
central nervous system
- DAPI
4′,6-diamidino-2-phenylindole
- DNQX
dinitroquinoxaline-2,3 (1H, 4H)-dione
- DRN
6,7- dorsal raphe nucleus
- DM
dorsomedial
- DL
double-labeled
- eIPSC
evoked inhibitory postsynaptic current
- GPCR
G-protein coupled receptor
- 5-HT
5-hydroxytryptamine
- IL
interleukin
- IPSC
IPSC, inhibitory postsynaptic current
- LW
lateral wing
- MRN
median raphe nucleus
- NMDA
non-N-methyl D-aspartate
- PPR
paired-pulse ratio
- RN
raphe nuclei
- SL
single-labeled
- sIPSC
spontaneous inhibitory postsynaptic current
- SEM
standard error of the mean
- SDF
stromal cell-derived factor
- TNF
tumor necrosis factor
- TPH
tryptophan hydroxylase
- VM
ventromedial
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler MW, Geller EB, Chen X, Rogers TJ. Viewing chemokines as a third major system of communication in the brain. AAPS J. 2006;7:865–870. doi: 10.1208/aapsj070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler MW, Rogers TJ. Are chemokines the third major system in the brain? J Leukoc Biol. 2005;78:1204–1209. doi: 10.1189/jlb.0405222. [DOI] [PubMed] [Google Scholar]
- Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- Azmitia EC, Liao B, Chen YS. Increase of tryptophan hydroxylase enzyme protein by dexamethasone in adrenalectomized rat midbrain. J Neurosci. 1993;13:5041–5055. doi: 10.1523/JNEUROSCI.13-12-05041.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G. Chemokines and their receptors in the central nervous system. Front Neuroendocrinol. 2001;22:147–184. doi: 10.1006/frne.2001.0214. [DOI] [PubMed] [Google Scholar]
- Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- Beck SG, Pan YZ, Akanwa AC, Kirby LG. Median and dorsal raphe neurons are not electrophysiologically identical. J Neurophysiol. 2004;91:994–1005. doi: 10.1152/jn.00744.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertollini C, Ragozzino D, Gross C, Limatola C, Eusebi F. Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacology. 2006;51:816–821. doi: 10.1016/j.neuropharm.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. Eur J Neurosci. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Callewaere C, Banisadr G, Desarmenien MG, Mechighel P, Kitabgi P, Rostene WH, Melik PS. The chemokine SDF-1/CXCL12 modulates the firing pattern of vasopressin neurons and counteracts induced vasopressin release through CXCR4. Proc Natl Acad Sci USA. 2006;103:8221–8226. doi: 10.1073/pnas.0602620103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaere C, Banisadr G, Rostene W, Parsadaniantz SM. Chemokines and chemokine receptors in the brain: implication in neuroendocrine regulation. J Mol Endocrinol. 2007;38:355–363. doi: 10.1677/JME-06-0035. [DOI] [PubMed] [Google Scholar]
- Chaouloff F. Serotonin, Stress, and Corticoids. J Psychopharm. 2000;14:139–151. doi: 10.1177/026988110001400203. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores KE, Gohil J, Berkhout TA, Patel L, Green P, Macphee CH, Stewart BR. The role of fractalkine in the recruitment of monocytes to the endothelium. Eur J Pharmacol. 2000;392:189–195. doi: 10.1016/s0014-2999(00)00117-5. [DOI] [PubMed] [Google Scholar]
- Charney DS, Woods SW, Krystal JH, Heninger GR. Serotonin function and human anxiety disorders. Ann NY Acad Sci. 1990;600:558–572. doi: 10.1111/j.1749-6632.1990.tb16910.x. [DOI] [PubMed] [Google Scholar]
- Combadiere C, Salzwedel K, Smith ED, Tiffany HL, Berger EA, Murphy PM. Identification of CX3CR1. A chemotactic receptor for the human CX3C chemokine fractalkine and a fusion coreceptor for HIV-1. J Biol Chem. 1998;273:23799–23804. doi: 10.1074/jbc.273.37.23799. [DOI] [PubMed] [Google Scholar]
- Commons KG, Valentino RJ. Cellular basis for the effects of substance P in the periaqueductal gray and dorsal raphe nucleus. J Comp Neurol. 2002;447:82–97. doi: 10.1002/cne.10228. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21:153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong EK, Dijkstra IM, Hensens M, Brouwer N, van Amerongen M, Liem RS, Boddeke HW, Biber K. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci. 2005;25:7548–7557. doi: 10.1523/JNEUROSCI.1019-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürk T, Panther E, Müller T, Sorichter S, Ferrari D, Pizzirani C, Di Virgilio F, Myrtek D, Norgauer J, Idzko M. 5-Hydroxytryptamine modulates cytokine and chemokine production in LPS-primed human monocytes via stimulation of different 5-HTR subtypes. Int Immunol. 2005;17:599–606. doi: 10.1093/intimm/dxh242. [DOI] [PubMed] [Google Scholar]
- Elmariah SB, Crumling MA, Parsons TD, Balice-Gordon RJ. Postsynaptic TrkB-mediated signaling modulates excitatory and inhibitory neurotransmitter receptor clustering at hippocampal synapses. J Neurosci. 2004;24:2380–2393. doi: 10.1523/JNEUROSCI.4112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi K, Gao JL, Murphy PM. Chemokine receptor CX3CR1 regulates renal interstitial fibrosis after ischemia-reperfusion injury. Am J Pathol. 2006;169:372–387. doi: 10.2353/ajpath.2006.060043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Novel anti-inflammatory therapy for Parkinson’s disease. Trends Pharmacol Sci. 2003;24:395–401. doi: 10.1016/S0165-6147(03)00176-7. [DOI] [PubMed] [Google Scholar]
- Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- Guyon A, Nahon JL. Multiple actions of the chemokine stromal cell-derived factor-1 on neuronal activity. J Mol Endocrinol. 2007;38:365–376. doi: 10.1677/JME-06-0013. [DOI] [PubMed] [Google Scholar]
- Habasque C, Satie AP, Aubry F, Jégou B, Samson M. Expression of fractalkine in the rat testis: molecular cloning of a novel alternative transcript of its gene that is differentially regulated by pro-inflammatory cytokines. Mol Hum Reprod. 2003;9:449–455. doi: 10.1093/molehr/gag059. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. J Neurosci Res. 2002;69:418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- Heinisch S, Kirby LG. Serotonin-chemokine interactions in the raphe nuclei: anatomical and electrophysiological studies. Soc Neurosci Abs. 2007:873.7. [Google Scholar]
- Hokfelt T, Broberger C, Xu ZQ, Sergeyev V, Ubink R, Diez M. Neuropeptides--an overview. Neuropharmacology. 2000;39:1337–1356. doi: 10.1016/s0028-3908(00)00010-1. [DOI] [PubMed] [Google Scholar]
- Hughes PM, Botham MS, Frentzel S, Mir A, Perry VH. Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia. 2002;37:314–327. [PubMed] [Google Scholar]
- Idzko M, Panther E, Stratz C, Müller T, Bayer H, Zissel G, Dürk T, Sorichter S, Di Virgilio F, Geissler M, Fiebich B, Herouy Y, Elsner P, Norgauer J, Ferrari D. The serotoninergic receptors of human dendritic cells: identification and coupling to cytokine release. J Immunol. 2004;172:6011–6019. doi: 10.4049/jimmunol.172.10.6011. [DOI] [PubMed] [Google Scholar]
- Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, Azmitia EC. Structure and function of the brain serotonin system. Physiol Rev. 1992;72:165–231. doi: 10.1152/physrev.1992.72.1.165. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J Neurosci. 2004;24:522–530. doi: 10.1523/JNEUROSCI.3606-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Shi Q, Xu H, Gibson GE. Changes in inflammatory processes associated with selective vulnerability following mild impairment of oxidative metabolism. Neurobiol Dis. 2007;26:353–362. doi: 10.1016/j.nbd.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby LG, Freeman-Daniels EL, Lemos JC, Nunan JD, Lamy C, Akanwa A, Beck SG. Corticotropin-releasing factor increases GABA synaptic activity and induces inward current in 5-hydroxytryptamine dorsal raphe neurons. J Neurosci. 2008;28:12927–12937. doi: 10.1523/JNEUROSCI.2887-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limatola C, Lauro C, Catalano M, Ciotti MT, Bertollini C, Di Angelantonio S, Ragozzino D, Eusebi F. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J Neuroimmunol. 2005;166:19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Lindia JA, McGowan E, Jochnowitz N, Abbadie C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain. 2005;6:434–438. doi: 10.1016/j.jpain.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Meltzer HY. Role of serotonin in depression. Ann NY Acad Sci. 1990;600:486–499. doi: 10.1111/j.1749-6632.1990.tb16904.x. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci USA. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi Y, Kanematsu T, Hirata M, Nabekura J. A rapid increase in the total number of cell surface functional GABAA receptors induced by brain-derived neurotrophic factor in rat visual cortex. J Biol Chem. 2003;278:44097–44102. doi: 10.1074/jbc.M305872200. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- Moon SO, Kim W, Sung MJ, Lee S, Kang KP, Kim DH, Lee SY, So JN, Park SK. Resveratrol suppresses tumor necrosis factor-alpha-induced fractalkine expression in endothelial cells. Mol Pharmacol. 2006;70:112–119. doi: 10.1124/mol.106.022392. [DOI] [PubMed] [Google Scholar]
- Mössner R, Lesch KP. Role of serotonin in the immune system and in neuroimmune interactions. Brain Behav Immun. 1998;12:249–271. doi: 10.1006/brbi.1998.0532. [DOI] [PubMed] [Google Scholar]
- Muehlhoefer A, Saubermann LJ, Gu X, Luedtke-Heckenkamp K, Xavier R, Blumberg RS, Podolsky DK, MacDermott RP, Reinecker HC. Fractalkine is an epithelial and endothelial cell-derived chemoattractant for intraepithelial lymphocytes in the small intestinal mucosa. J Immunol. 2000;164:3368–3376. doi: 10.4049/jimmunol.164.6.3368. [DOI] [PubMed] [Google Scholar]
- Nanki T, Imai T, Nagasaka K, Urasaki Y, Nonomura Y, Taniguchi K, Hayashida K, Hasegawa J, Yoshie O, Miyasaka N. Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheum. 2002;46:2878–2883. doi: 10.1002/art.10622. [DOI] [PubMed] [Google Scholar]
- Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A, Satoh M. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 1998;429:167–172. doi: 10.1016/s0014-5793(98)00583-3. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Hajos N, Somogyi P, Mody I. Increased number of synaptic GABA(A) receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998;395:172–177. doi: 10.1038/25999. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Amsterdam: Elsevier Academic Press; 2005. [Google Scholar]
- Ragozzino D, Di AS, Trettel F, Bertollini C, Maggi L, Gross C, Charo IF, Limatola C, Eusebi F. Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. J Neurosci. 2006;26:10488–10498. doi: 10.1523/JNEUROSCI.3192-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ré DB, Przedborski S. Fractalkine: moving from chemotaxis to neuroprotection. Nat Neurosci. 2006;9:859–861. doi: 10.1038/nn0706-859. [DOI] [PubMed] [Google Scholar]
- Rostène W, Kitabgi P, Parsadaniantz SM. Chemokines: a new class of neuromodulator? Nat Rev Neurosci. 2007;8:895–903. doi: 10.1038/nrn2255. [DOI] [PubMed] [Google Scholar]
- Tong N, Perry SW, Zhang Q, James HJ, Guo H, Brooks A, Bal H, Kinnear SA, Fine S, Epstein LG, Dairaghi D, Schall TJ, Gendelman HE, Dewhurst S, Sharer LR, Gelbard HA. Neuronal fractalkine expression in HIV-1 encephalitis: roles for macrophage recruitment and neuroprotection in the central nervous system. J Immunol. 2000;164:1333–1339. doi: 10.4049/jimmunol.164.3.1333. [DOI] [PubMed] [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. 2004;20:1150–1160. doi: 10.1111/j.1460-9568.2004.03593.x. [DOI] [PubMed] [Google Scholar]
- Wagner S, Castel M, Gainer H, Yarom Y. GABA in the mammalian suprachiasmatic nucleus and its role in diurnal rhythmicity. Nature. 1997;387:598–603. doi: 10.1038/42468. [DOI] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/s0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Pakala R, Katagiri T, Benedict CR. Monocyte chemotactic protein 1 amplifies serotonin-induced vascular smooth muscle cell proliferation. J Vasc Res. 2001;38:341–349. doi: 10.1159/000051065. [DOI] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21:642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupanc GK. Peptidergic transmission: from morphological correlates to functional implications. Micron. 1996;27:35–91. doi: 10.1016/0968-4328(95)00028-3. [DOI] [PubMed] [Google Scholar]