

Abstract
We have been investigating the functional consequences of rare disease-associated amino acid substitutions in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). Mutations of the arginine residue at codon 1070 have been associated with different disease consequences R1070P and R1070Q have “severe” pancreatic insufficient cystic fibrosis (CF) and R1070W have “mild” pancreatic sufficient CF or congenital bilateral absence of the vas deferens. Intriguingly, CFTR bearing each of these mutations is functional when expressed in non-polarized cells. To determine whether R1070 mutations cause disease by affecting CFTR localization, we created polarized MDCK cell lines that express either wild-type or mutant CFTR from the same genomic integration site. Confocal microscopy and biotinylation studies revealed that R1070P was not inserted into the apical membrane, R1070W was inserted at levels reduced from wild-type while R1070Q was present in the apical membrane at levels comparable to wild-type. The abnormal localization of CFTR bearing R1070P and R1070W was consistent with deleterious consequences in patients however the profile of CFTR R1070Q was inconsistent with a “severe” phenotype. Re-analysis of 16 patients with the R1070Q mutation revealed that 11 carried an in cis nonsense mutation, S466X. All 11 patients carrying the complex allele R1070Q-S466X had severe disease, while 4 of 5 patients with R1070Q had “mild” disease, thereby reconciling the apparent discrepancy between the functional studies of R1070Q and the phenotype of patients bearing this mutation. Our results emphasize that localization studies in relevant model systems are needed for the interpretation of the disease-causing potential of rare missense mutations.
Keywords: cystic fibrosis, CF, CFTR, polarized cells, complex allele, recombinase-mediated integration
INTRODUCTION
Cystic fibrosis (CF; MIM# 219700) is the most common autosomal recessive disorder in Caucasians, affecting approximately one in every 3000 births (Hamosh et al., 1997). CF is caused by dysfunction of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR; MIM# 602421), a cAMP-induced chloride channel located in the apical membrane of exocrine epithelial cells. Mutations in CFTR disrupt epithelial salt and water transport, causing tissue dehydration and accumulation of thick mucus in pancreatic, bronchial, and vas deferens ducts, leading to obstruction and inflammation of ducts as well as eventual lung failure such that the median survival of CF patients is about 36 years (Welsh et al., 2001; Cystic Fibrosis Foundation, 2006). Over 1,300 putative disease-causing mutations are reported in the Cystic Fibrosis Mutation Database and half (647) are rare mutations predicted to substitute a single amino acid (http://www.genet.sickkids.on.ca/cftr). The consequence of most of these missense mutations upon CFTR function is unknown.
CFTR consists of two membrane spanning domains, each with six transmembrane segments, two nucleotide binding domains (NBDs), and a regulatory region with multiple sites for phosphorylation by protein kinases A and C (Gadsby et al., 2006). The N- and C-termini contain motifs for protein interactions that facilitate CFTR trafficking (Naren et al., 1997; Moyer et al., 1999). While functions have been assigned to the aforementioned domains, the four cytoplasmic loops of CFTR, which make up 16.8% of the protein, are of unclear function. Although the loops were initially proposed to contribute to the CFTR pore, loop mutants generally showed little effect on conduction and permeation (Sheppard and Welsh, 1999). Structural analysis of CFTR indicates that the loops are proximate to NBDs and experimental evidence indicates that the loops couple conformational changes in the NBDs with channel function (Serohijos et al., 2008). Each loop shows considerable amino acid conservation across species with cytoplasmic loop 4 (CL4) being the most highly conserved compared to the rest of CFTR (63% identity compared to the rest of CFTR (34%, unpublished data). Furthermore, a substantial function of the reported amino acid substitutions observed in CF patients occur in the cytoplasmic loops with CL4 having the highest number of putative disease-causing mutations (36 of 68 residues mutated; 11 sites have multiple missense mutations) compared to the other three cytoplasmic loops. Intriguingly, codon 1070 at the center of CL4 has three reported missense mutations that have been associated with different disease severity. Patients with R1070W (c.3208C>T; p.Arg1070Trp) have pancreatic sufficient Cystic Fibrosis (CF) or Congenital Bilateral Absence of the Vas Deference (CBAVD) and a normal life span, whereas patients with R1070Q (c.3209G>A; p.Arg1070Gln) and R1070P (c3209G>C; p.Arg1070Pro) have classic CF with significant clinical features of lung disease, pancreatic insufficiency, and elevated sweat chloride levels (Mickle et al., 2000). Previous studies revealed that the biogenesis and function of CFTR bearing R1070W and R1070P is substantially altered consistent with their pathologic role. However, CFTR with R1070Q had a less severe channel gating abnormality than R1070W and either no defect (Seibert et al., 1996; Mickle et al., 2000) or a milder defect than R1070W upon protein processing(Seibert et al., 1996). These functional studies indicate that the R1070Q mutation should cause a milder, rather than the observed more severe phenotype than R1070W. However, the studies performed to date evaluated mutant CFTR in non-polarized cells. We therefore hypothesized that the severe CF phenotype attributed to R1070Q was due to a localization defect in native epithelial cells.
To determine the properties of CFTR R1070 mutants in polarized cells, a situation that more closely mimics the lung epithelia, we have made use of a recombinase-based integration Flp-In® system to express heterologous CFTR in Madin Darby Canine Kidney (MDCK) cells. The primary advantage of this system is that a series of clones are obtained with integrations of a single copy cDNA in the same genomic site that results in comparable levels of expression of the introduced cDNA by the individual cell lines of many cell lines. Furthermore, MDCK cells have been used extensively to study the localization of proteins, including CFTR, that distribute asymmetrically in polarized cells (Mendes et al., 2005). While CFTR bearing R1070W and R1070P displayed localization defects consistent with their associated phenotypes, R1070Q was difficult to distinguish from wild-type CFTR. These localization studies prompted a search for alternate explanations for the CF phenotype associated with the R1070Q mutation.
MATERIALS AND METHODS
Constructs
The current GenBank reference for CFTR RNA sequence is NM_000492.3. Mutations in CFTR are reported according to the convention used by the CF Mutation Database where the A of the initiation codon is nucleotide 133 (http://www.genet.sickkids.on.ca/cftr) and codon 1 is the initiation codon. Nucleotide numbering and changes at the protein level are also reported in parentheses using the HGVS nomenclature convention with a "c." symbol and “p.” symbol, respectively (www.hgvs.org/mutnomen). The pcDNA5/FRT/GFP-CFTR wild-type and R1070 plasmids were made by removing full length cDNA from the existing peGFP-CFTR (Mickle et al., 2000) plasmids by NheI (5’ end) and EcoRV (3’ end) digestion and ligation into the same sites in the multiple cloning sequence of the pcDNA/FRT plasmid (Invitrogen, Carlsbad, CA). The pcDNA5/FRT/GFP-CFTR plasmids were then sequenced to ensure the presence of the appropriate R1070 mutation prior to their use in the creation of isogenic stable MDCK-CFTR cell lines.
Transfection and clone selection
The MDCK type-II FRT cell line, which contains the Flp recombinase target (FRT) but expresses no endogenous CFTR, was the kind gift of Gregory Germino (Johns Hopkins School of Medicine, Baltimore, MD). Zeocin-resistant parental MDCK cells were cotransfected using 8-ul of Lipofectamine 2000 (Invitrogen #11668-019, Carlsbad, CA) with the appropriate pcDNA5/FRT/GFP-CFTR construct and pOG44 recombinase (Flp-In System kit, Invitrogen #K6010, Carlsbad, CA) at 1:12 or likewise at 1:15 ratios, respectively, that facilitated site-specific integration. Since the recombinase plasmid has no antibiotic resistance and integration of the hygromycin-expressing pcDNA5/FRT/GFP-CFTR plasmid disrupts zeocin expression from the FRT site in the cell line, selection for hygromycin-resistant and zeocin-sensitive cells yielded clones with CFTR cDNA integrated at the FRT site. Hygromycin selection proceeded until clones were at least 3 mm in diameter (3–5 weeks), at which point each clone was picked, grown to cover a 10 cm plate, and tested by PCR to confirm integration. MDCK cells were cultured in DMEM (GIBCO #11965, Carlsbad, CA) with 10% fetal bovine serum (FBS; Gibco #16140-071, Carlsbad, CA) and zeocin (100-ug/ml; Invitrogen #45–0430, Carlsbad, CA) for parental cells or hygromycin (200-ug/ml, Invitrogen #10687-010, Carlsbad, CA) for cells with integrations at the FRT site in a humidified incubator at 37°C in the presence of 5% CO2.
Verification of pcDNA5/FRT/GFP-CFTR integration into the FRT site in MDCK cells
To confirm that full-length CFTR integrated into the FRT site, genomic DNA was extracted from the stable CFTR cell lines and subjected to a PCR-based test displayed in Figure 1. Genomic DNA was isolated from one 10-cm plate for each sample using Puregene kit (Gentra #D5500, Minneapolis, MN); the sample was rehydrated in 50-ul and 1-ul was used for each PCR reaction. Briefly, PCR primers (Invitrogen) were created to amplify the 5’ integration site from pSV40 of the parental cell line to the hygromycin gene of the CFTR stable line (699 bp, 5’F CCTAACTGACACACATTCCACA and 5’R TCAGCGAGAGCCTGACCTAT) and, likewise, to confirm integration at the 3’ end of CFTR (1379 bp, 3’F AGCATTTGCTGATTGCACAG and 3’R CCGTAATGGGATAGGTCACG). Presence of the parental cell line was detected using the 5’F pSV40 and 3’R lacZeo primers that produce a unique 745 bp product. To ensure that full-length 5.4 kb CFTR cDNA was present, the overlapping primers were used. Specifically, exons 1–7 amplified a 1023 bp region (AGAGGTCGCCTCTGGAAAAG and TGCTCCAAGAGAGTCATACC); exons 4–12 amplified 1297 bp (CACATTGGAATGCAGATGAG and GTGTTAAAACATCTAGGT ATC); exons 11–16 amplified 1381 bp (CATCTCCAAGTTTGCAGAGA and GTCAAATATGGTAAGAGGC); exons 15–21 amplified 1087 bp (CACCTATGTCAACCCTCAAC and CATCTGCAACTTTCCATATTTC); and exons 19–24 amplified 933 bp (GAATGGCCAACTCTCGAAAG and CCATGAGCAAATGTCCCATG).
Figure 1. Integration of full-length CFTR into FRT site of MDCK-FRT cells.
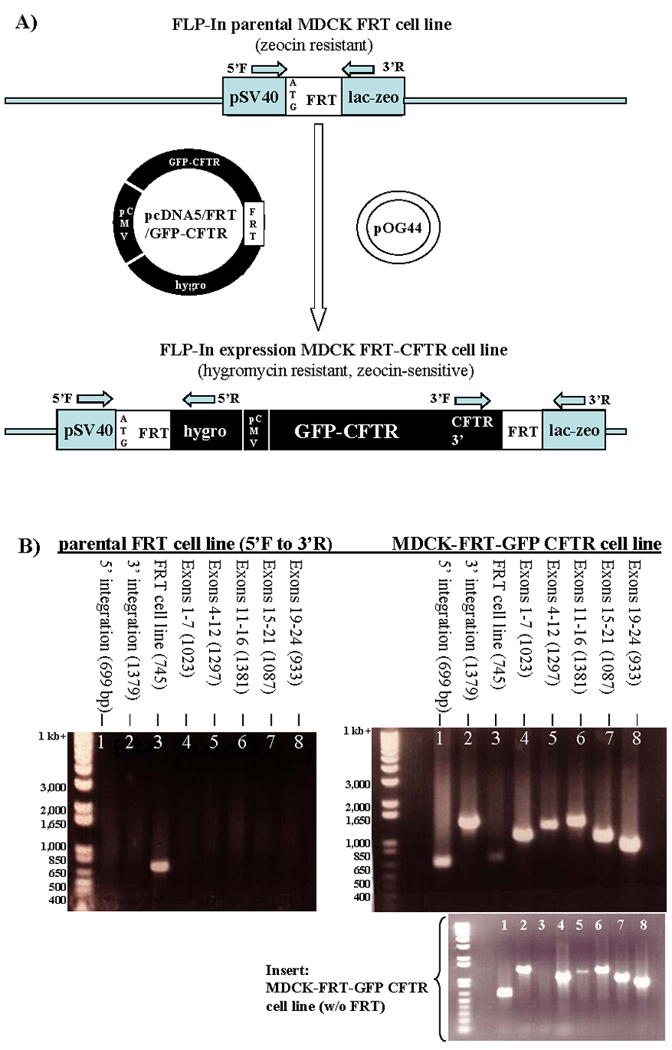
A) Diagrams of the components in the integration site of the MDCK-FRT cell line and of the integration site after recombination with the FRT sequence in the pcDNA/FRT/GFP-CFTR plasmid. The relative locations of DNA primers used to verify integration are shown. pSV40: simian virus 40 promoter; hygro, hygromycin-resistance gene; LacZeo, β-lactamase zeomycin fusion protein; pCMV, cytomegalovirus promoter. B) Confirmation of GFP-CFTR cDNA integration by PCR analysis. Photographs of agarose gels with amplified DNA stained with ethidium bromide and visualized by UV trans-illumination. Gel on the left shows amplification of genomic DNA from the parental MDCK-FRT cell line. As expected, a 745 bp product is amplified using the 5’ F and 3’ R flanking the FRT site (lane 3). Gel on the right shows amplification of genomic DNA from a hygromycin-resistant, zeocin-sensitive cell line. Amplification of 699 bp and 1379 bp products using primers 5’ F and 5’ R (lane 1) and 3’ F and 3’ R (lane 2) confirms integration of the cDNA into the FRT site. The panel below is an example of genomic DNA from a cell line with integration of full-length CFTR cDNA with no residual parental MDCK-FRT cells. Water controls were negative but are not shown.
Immunostaining and confocal microscopy
Cells were plated on filters (Falcon #353090, Franklin Lakes, NJ) or glass coverslips at >50% density, cultured for 72 hr with daily media changes then fixed in 4% paraformaldehyde and either treated with wheat germ agglutinin (WGA, Molecular Probes #W-849, Carlsbad, CA) to stain the membrane surface (5-ul per sample, 1 hr at room temperature followed by washes with PBS) or permeabilized with 0.25% Triton-X for immunostaining. Non-specific binding sites were blocked with 2.5% normal goat serum (Invitrogen #16201, Carlsbad, CA). The cells were incubated for 1.5 hours with ZO-1 (polyclonal anti-rabbit, Zymed #61–7300, San Francisco, CA, 1:50 dilution in goat serum blocking solution) or Na+/K+ ATP-ase (monoclonal anti-mouse, Upstate #05–369, Lake Placid, NY, 1:50), washed with PBS, and, thereafter, incubated for 30 minutes with rabbit antiserum (Sigma #C2306, St. Louis, MO) or mouse antiserum (Sigma #C2181, St. Louis, MO). Filters were mounted face up on slides, layered with DAPI (Sigma #D9542, St. Louis, MO) and antifade solution (Invitrogen #S36936, Carlsbad, NY, 1:30), overlaid with a coverslip, and sealed with nail polish. The specimens were visualized on a Zeiss 510 confocal microscope (Johns Hopkins School of Medicine Microscope Facility). At least 2 clones of each construct was evaluated.
Surface biotinylation and Western blotting
Surface biotinylation of CFTR at the plasma membrane has been described previously (Cheng et al., 2004). Briefly, the cell-surface proteins were labeled with cell-impermeable EZ-LINK sulfo-NHS-LC-biotin (Pierce #21335, Rockford, IL) at 4°C for 30 min, washed for 30 min with glycine in phosphate buffered saline containing calcium and magnesium (PBS; Gibco #14040, Carlsbad, CA) to remove excess biotin, and lysed for 20 min with lysis buffer (1% Nonidet P-40, 150 mM NaCl, 50 mM Tris-HCl at pH 7.5) containing protease inhibitors (Roche #10481700, Indianapolis, IN). The lysate samples were separated into the “total fraction” and the “apical fraction”. Biotinylated proteins in the apical fraction were extracted by incubation with NeutrAvidin beads (Pierce #53151, Rockford, IL) at 4°C for 2 hr or overnight, washing with lysis solution (as above but without protease inhibitors) and eluting with 2X Laemmli sample buffer (Biorad #161–0737, Hercules, CA) containing 100-mM dithothreitol (DTT) at 42°C for 30 min. Lysates of cells expressing wild-type and each R1070 mutant were obtained by treating 10 cm plates with lysis solution and with Laemmli sample buffer plus DTT as described above. Samples were subjected to SDS-PAGE using a Tris-HCl 5% gel to detect GFP-CFTR (Biorad #161–1213, Hercules, CA) or a Tris-HCl 4–15% gel to detect beta-actin (Biorad #161–1158, Hercules, CA) and transferred to a nitrocellulose membrane (Biorad #162–0174, Hercules, CA) by the semi-dry transfer method (Mickle et al., 2000). After blocking in 5% milk with PBS-tween (1:1000) overnight, GFP-CFTR was detected using an anti-rabbit GFP antibody (Invitrogen #A11122, Carlsbad, CA, 1:4,000, in 5% milk with PBS-tween) and subsequently rabbit antiserum (Amersham Biosciences #NA934V, Piscataway, NJ, 1:10,000). Actin (42 kDa), used as the loading control, was detected with anti-mouse beta-actin (Abcam #ab6276-100, Cambridge, MA, 1:10,000) and subsequently mouse antiserum (Amersham Biosciences #NA931V, Piscataway, NJ, 1:10,000). The blots were developed via ECL-Plus (Amersham Biosciences #RPN2108, Piscataway, NJ). All studies were repeated a minimum of 3 times.
Collection of the clinical data
The corresponding authors of papers reporting patients with R1070 mutations were contacted (Audrézet et al., 1993; Bienvenu et al., 1997; Casals et al., 2000; Casals et al., 1995; Chillón et al., 1995; Choi et al., 2001; Claustres et al., 2000; Cotten et al., 1996; Dayangac et al., 2004; Estivill et al., 1997; Feldmann et al., 2003; Ferec et al., 1995; Gervais et al., 1996; Groman et al., 2002; Jezequel et al., 2000; Jezequel et al., 1995; Le Lannou et al., 1995; McGinniss et al., 2005; Mercier et al., 1994; Mercier et al., 1993; Mickle et al., 2000; Osborne et al., 1993; Radivojevic et al., 2004; Savov et al., 1994; Seibert et al., 1996; Shrimpton et al., 1997; de la Taille et al., 1998; Wang et al., 2002). Detailed clinical information was received from Milan Macek Jr. (Charles University Hospital Motol, Prague, Czech Republic), Teresa Casals (Center of Genetic Diagnostics and Molecular Medicine, Barcelona, Spain), and Thierry Bienvenu (Cochin Hospital, Paris, France), and Claude Ferec (University of Brest, Brest, France). The Cystic Fibrosis Foundation (Washington, DC) maintains a registry database, which was accessed by way of application process that corresponded with HIPAA requirements to obtain clinical information on R1070 patients in the United States.
RESULTS
Wild-type CFTR is localized to apical membranes of MDCK cells
To determine the effect of R1070 mutations on CFTR localization, we expressed heterologous CFTR (wild-type or one of 3 CFTR mutants—R1070P, R1070Q, or R1070W) from an FRT integration site in MDCK type II cell lines (Figure 1A). PCR amplification confirmed orientation of integration and presence of residual parental cells (Figure 1B). Presence of full length CFTR cDNA was verified with overlapping primers; exons 1–7 (1023 bp, lane 4), exons 4–12 (1297 bp, lane 5), exons 11–16 (1381 bp, lane 6), exons 15–21 (1087 bp, lane 7), and exons 19–24 (933 bp lane 8). As seen in lane 3 of Figure 1B, amplification of a faint 745 bp fragment indicated presence of parental MDCK-FRT cells. This outcome occurred in about two-thirds of all clones; in the remainder, there was no evidence of residual parental cells (see insert). We observed no difference in our studies between clones that had residual parental cells and those that had no parental cells.
Cells with stable expression of wild-type CFTR were immunostained to assess polarization and location of CFTR relative to the proteins that localize to the tight junction, basolateral membrane, and the cell surface. The zona occludens 1 (ZO-1) antibody selectively stains the tight junctions, which are located proximal to the apical surface providing delineation of the boundary between apical and lateral membranes. The Na+/K+ ATP-ase antibody stains this basolateral transporter and indicates lateral cell boundaries and portions of the basal membrane. Wheat germ agglutinin (WGA) is a carbohydrate-binding protein dye that recognizes sialic acid and N-acetylglucosaminyl sugars that are highly prevalent on the plasma membrane. When applied to non-permeabilized cells, it delineates the apical surface thus establishing whether a protein such as GFP-CFTR is located on the apical membrane when the yellow composite signal is observed or subapically when the red WGA signal and the green GFP signal are separate.
Confocal microscopy indicated that the wild-type CFTR was primarily localized to apical membranes. Immunostaining for ZO-1 distinguished protein of tight junctions as expected for polarized cells and, in each cell, CFTR was localized at the level of ZO-1 suggesting the presence within or very near the apical membrane (Figure 2, wild-type, left column). Immunostaining for Na+/K+ ATP-ase distinguished the basolateral membranes as expected and wild-type CFTR was not localized to these basolateral membranes (Figure 2, wild-type, middle column). Likewise, staining with the apical surface dye shows overlap (yellow) of the green GFP-CFTR and the red WGA dye (Figure 2, wild-type, right column). The presence of wild-type CFTR at the apical surface of MDCK cells is consistent with its location in polarized cells in vivo indicating that these cells are suitable for analyzing the consequences of mutations in terms of CFTR localization.
Figure 2. CFTR R1070P and R1070W show different cellular distribution patterns in polarized epithelial cells, whereas CFTR R1070Q shows an apical localization pattern similar to wild-type CFTR.
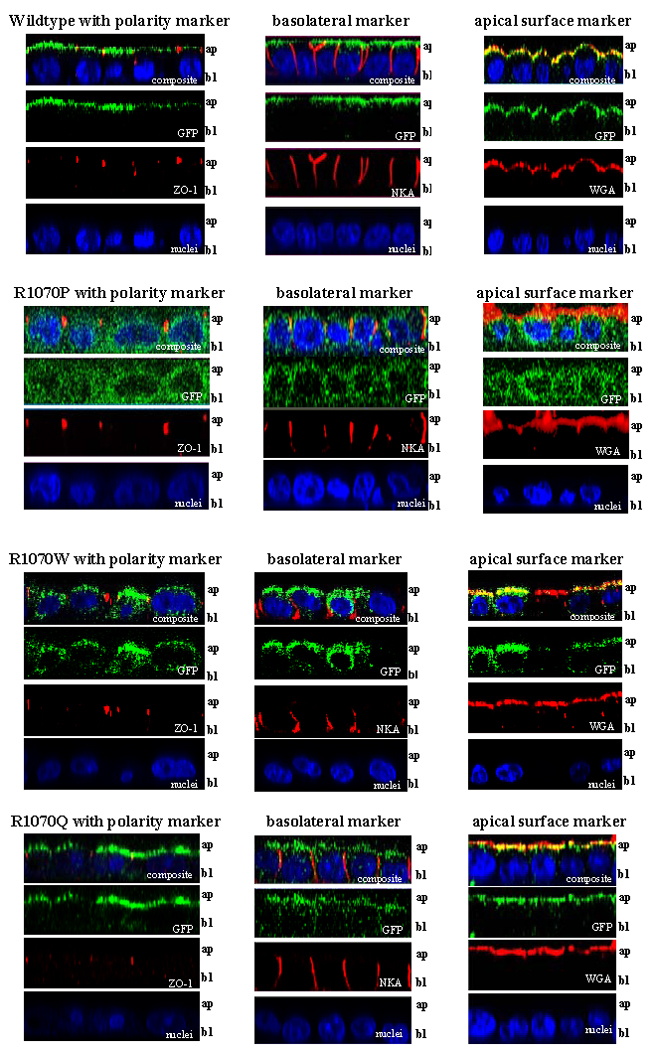
Confocal microscopy images (63x magnification) of immunostained MDCK-FRT-GFP-CFTR cells imaged in the xz-plane (scanned apical to basal membrane), with top row in each panel representing the composite scan and the panels below showing each channel separately (from top: composite, green, red, and blue). Each panel shows MDCK-FRT cells expressing either wild-type, R1070P, R1070W, or R1070Q CFTR. The location of CFTR is detected by green signal emanating from the green fluorescence protein (GFP) fused to the N-terminus of CFTR. Cells were immunostained with one of three markers: ZO-1 (red, left column) to indicate polarity, Na+/K+ ATPase (red, middle column) to show the basolateral membrane, or wheat germ agglutinin (WGA, red, right column) to delineate the apical surface. All cells were counterstained with DAPI (blue) to detect the nuclei. An occasional individual red signal within the cell indicated nonspecific background staining.
Proline (P) and tryptophan (W) substitutions of arginine at codon 1070 alter localization while CFTR bearing a glutamine (Q) substitution is found in apical membranes
CFTR bearing R1070P, a mutation which is associated with classic CF, did not reach—or was not appreciably retained at—the cell surface, instead localizing throughout the cytoplasm. The cytoplasmic distribution seen with R1070P was similar to that of CFTR bearing the common delta-F508 mutation (p.F508fdel; data not shown) that does not reach the apical membrane due to CFTR misfolding and is degraded (Cheng et al., 1990; Kartner et al., 1992; Denning et al., 1992). Immunostaining with ZO-1 (Figure 2, R1070P, left column), Na+/K+ ATP-ase (Figure 2, R1070P, middle column), and staining of the surface apical membrane with WGA dye (Figure 2, R1070P, right column) showed that the CFTR R1070P protein does not localize to the apical membrane. CFTR bearing R1070W, a mutation associated with mild disease, localized to the apical surface of MDCK cells (see overlap with apical surface marker, Figure 2, R1070W, left column) but was also present in other locations in the cell. The absence of co-localization with Na+/K+ ATP-ase basolateral marker is consistent with presence of CFTR R1070W in the cytoplasm. On the other hand, CFTR bearing R1070Q localized primarily to the apical membrane of MDCK cells. Staining R1070Q cells with the apical surface dye, WGA, revealed yellow signal (Figure 2, R1070Q, right column) indicating overlap of green GFP-CFTR and red WGA signals at the apical membrane. Green signal was observed in other locations in the cell (Figure 2, R1070Q, left column) and co-localization of this signal with Na+/K+ ATP-ase (Figure 2, R1070Q, right column) suggests that a minor fraction of R1070Q is present in the basolateral membranes. Overall, CFTR R1070Q is preferentially located at the apical membrane, a pattern very similar to that observed for wild-type CFTR.
Biochemical assays of wild-type CFTR and R1070 mutants concur with localization in polarized cells
To verify the results of the confocal studies, we performed Western blotting to assess the degree of CFTR maturation and also performed biotinylation studies to determine if CFTR is inserted into the apical cell membrane. The glycosylation status of CFTR is used to assess its maturity, whereby fully glycosylated protein that is properly folded and able to exit the Golgi is present as “C band” and biosynthetic defects in CFTR present as incompletely glycosylated protein is present as “B band” or “A band” (Cheng et al., 1990). Wild-type CFTR expressed in MDCK cells is present primarily as mature C band (Figure 3A, lane 1). Some immature “B band” protein is present likely due to the inability of the cells to completely process all of the protein expressed by the heterologous CFTR cDNA, as noted in other cell lines (Seibert et al., 1996; Mickle et al., 2000). Almost all of the wild-type CFTR is present in the apical membrane as biotinylated, mature C band protein (Figure 3B, lane 1). CFTR bearing R1070Q is also processed to mature band C protein (Figure 3A, lane 2) that is accessible to biotinylation at the apical membrane (Figure 3B, lane 3). Thus, wild-type and R1070Q CFTR are processed to fully mature protein that is inserted into the apical membrane, consistent with the localization studies above.
Figure 3. CFTR bearing mutants in R1070 show different levels of insertion into the apical membrane of polarized epithelial cells.
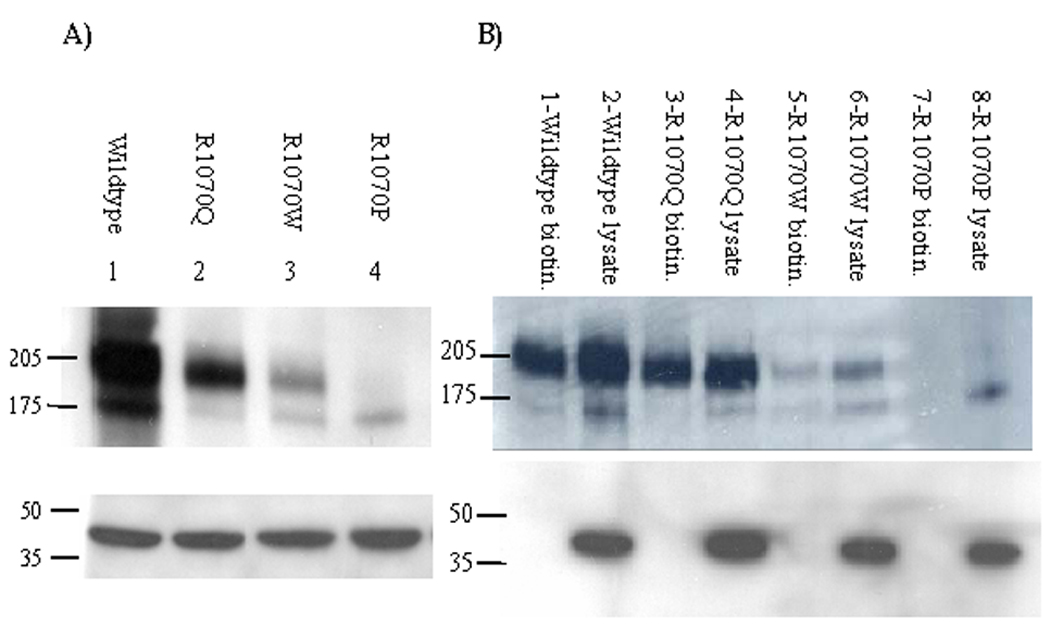
A) Western blotting of MDCK cell lysates expressing wild-type, R1070P, R1070W, or R1070 CFTR. CFTR is detected by immunoblotting against GFP that is fused to the N-terminus of CFTR. Wild-type CFTR (lane 1) displays bands of molecular masses 205 kDa and 175 kDa consistent with fully glycosylated “C band” (175 kDa plus 30 kDa for GFP) and partially glycosylated “B band” protein (130 kDa plus 30 kDa). CFTR R1070Q (lane 2) has a pattern similar to wild-type, primarily C band with some B band visible. The quantity of CFTR R1070Q mutant protein is slightly lower than wild-type CFTR (note actin control loading in lower panel). CFTR R1070W has a lower amount of stable protein expressed and is readily visible as both C and B bands (lane 3). CFTR R1070P has minimal stable protein and is present only as B band (lane 4). An equal volume of each lysate was immunoblotted with actin (42 kDa) to confirm equal loading (bottom panel). B) Western blotting of the apical biotinylated fraction (odd numbered lanes) or total lysates (even numbered lanes) of MDCK cells expressing wild-type, R1070Q, R1070W, or R1070P CFTR. Wild-type CFTR (lanes 1–2) and CFTR R1070Q (lanes 3–4) have a 205 kDa band in the biotinylated fraction that is consistent with the presence of mature protein in the apical membrane. The fraction of R1070Q inserted into the apical membrane is similar to wild-type. CFTR R1070W (lanes 5–6) has a faint 205 kDa band and a very faint 175 kDa band suggesting that mature and some immature protein is inserted in the apical membrane. CFTR R1070P (lanes 7–8) has no bands visible in the biotinylated fraction and only a 175 kDa band is seen in the total lysate indicating that this mutant protein is not present in the apical membrane. Lower panel is an immunoblot of each apical fraction and each total lysate after incubation with an actin antibody. A 42 kDa band is present only in the lanes with total lysate, as expected for a protein that is exclusively intracellular.
CFTR with R1070W was present in both mature (C band) and immature (B band) forms (Figure 3A, lane 3), although the ratio of C band to B band and the ratio of C band to actin control (Figure 3A, lane 3, lower panel) was lower than seen for wild-type CFTR. Biotinylation revealed that CFTR R1070W is inserted into the apical membrane, albeit at very low levels when compared to wild-type (Figure 3B, lane 5). These results indicate that R1070W is able to process into mature apically localized protein but with decreased efficiency compared to wild-type. As shown by confocal microscopy, these biochemical assays are consistent with only a fraction of R1070W at the apical membrane. There was a small amount of B band in the apical biotinylated fraction of R1070W, implying that some immature CFTR protein bearing this mutation may be inserted into the apical membrane. Similarly, a minor fraction of immature wild-type CFTR was biotinylated (Figure 3B, lane 1). Finally, Western blotting revealed that R1070P is present only as partially glycosylated immature B band protein (Figure 3A, lane 4) that is not detectable in the biotinylated apical membrane fraction. These results show that the R1070P mutation causes CFTR to be improperly processed such that it remains within the cell and does not reach the apical membrane, consistent with confocal microscopy studies showing R1070P only in the cytoplasm.
Altered function of CFTR bearing R1070W and R1070P is consistent with phenotype of patients carrying these mutations
A worldwide survey identified 29 patients who carried the R1070W mutation (of whom 24 had detailed clinical information); 26 patients with R1070Q (of whom 16 had detailed data), and 2 patients with R1070P (one of which had detailed data). For each patient with detailed clinical information, we selected those that had a known CFTR mutation in their other gene, pancreatic status (sufficient or insufficient) reported by authors or determined as insufficient if the patient was taking pancreatic enzymes, lung disease (defined by the U.S. CF Foundation (CFF) criteria), fertility status in males (defined as subfertile when diagnosed with congenital bilateral absence of the vas deferens (CBAVD)), and results of the sweat chloride concentration test (defined as per CFF criteria). Complete data collected for the patients is available in Supplementary Table S1.
Out of 24 patients who carry the R1070W mutation, 16 had F508del (c.1521_1523delCTT; p.Phe508del) on the other allele and all were diagnosed with either pancreatic sufficient CF or CBAVD (Table 1). Pancreatic status was unavailable for 6 of the 16 patients with the R1070W and F508del genotype. Four of these 6 patients were diagnosed as adults and presumably were not tested for gastrointestinal (GI) symptoms. For the remaining 2 individuals, we were unable to determine whether the report intended to declare no pancreatic disease and thus pancreatic sufficiency or that the test was not performed due to absence of GI symptoms. As for the remaining R1070W patients (8 of 24 with detailed clinical information), 5 individuals carried a CFTR mutation associated with pancreatic insufficiency (2869insG (c.2737insG; p.Tyr913fs), G542X (c.1624G4T, p.Gly542X), R668C-K710X (c.[2002C>T, c.2128A>T; p.[Arg668Cys,Lys710X]), N1303K (c.3909C>G, p.Asn1303Lys), and S1235R (c.3705T>G; p.Ser1235Arg). Four of these 5 patients had pancreatic sufficient CF or CBAVD. The one remaining individual with the R1070W/R668C-K710X genotype had pancreatic insufficient CF. Overall, the R1070W mutation in combination with a CFTR mutation associated with pancreatic insufficiency was associated with either pancreatic sufficient CF or CBAVD, indicating that this mutation generally conferred mild disease.
Table 1.
Summarized clinical information on R1070 patients.
| patients' mutation | R1070W | R1070P | R1070Q | R1070Q in cis S466X |
|---|---|---|---|---|
| number of patients** | 24 | 2 | 5 | 11 |
| 2nd mutation: | ||||
| dF508 | 16 | 1 | 0 | 7 |
| other | 8 | 1 | 5 | 4 |
| disease diagnosis: | ||||
| CBAVD (infertility) | 15 | 0 | 3 | 0 |
| nonclassic CF | 9 | 1 | 1 | 0 |
| classic CF | 1 | 1 | 1 | 11 |
| pancreatic status: | ||||
| sufficient | 9 | 0 | 1 | 0 |
| insufficient | 4** | 1 | 1 | 10 |
| not reported | 11* | 1 | 3* | 1 |
| sweat chloride levels: | ||||
| normal or low | 12 | 0 | 1 | 0 |
| elevated >60 mmol/L | 4 | 1 | 1 | 10 |
| not reported | 8* | 1 | 3* | 1 |
These patients are adults diagnosed with infertility who were then found to have CF mutations.
One patient has classic CF; the other 3 have normal sweat chloride levels and high FVC values. Additional information on all patients appears in supplementary materials.
Only 2 patients were found to carry the R1070P mutation and only one of them has detailed clinical information, therefore only limited conclusions can be drawn. One patient is known to have F508del on the other allele and was diagnosed with CF but no additional information is available. The other patient carries 2143delT (c.2011delT) on the other allele and, although initially diagnosed with “mild” CF (nasal polyps, elevated sweat chloride levels, mild lung disease, late childhood diagnosis), developed classic disease with age, including obstructive lung disease and pancreatic insufficiency. Based on the latter case, the R1070P mutation is considered to be associated with severe disease.
CFTR R1070Q with an in cis nonsense mutation, S466X (c.1397C>G; p.Ser466X), is associated with severe CF
Previous studies have reported that patients with R1070Q have classic CF; however, studies shown here indicate that CFTR R1070Q functions and localizes like wild-type CFTR suggesting that an alternative mechanism exists to create the severe phenotype associated with the R1070Q mutation. A literature review of patients carrying R1070Q uncovered a report that briefly mentioned two Serbian patients with classic CF who carried the R1070Q mutation in cis with S466X, both having the F508del mutation on the other CFTR gene (Radivojevic et al., 2004). We found 14 R1070Q patients with detailed clinical information (16 patients in total when including those of the Radivojevic group) and sequencing of CFTR exon 10 showed that 11 of 16 carried an in cis S466X mutation (Table 1). Seven of these 11 R1070Q-S466X patients had F508del as the other allele and the remaining 4 had a variety of CF alleles in trans, one each of N1303K, 621+1G>T (c.489+1G>T), 711+3A>G (c.579+3A>G), and R1070Q-S466X (Supplementary Table S1). Regardless of the CF mutation on the other CFTR gene, all 11 R1070Q-S466X patients had pancreatic insufficient CF. Of the five patients with R1070Q only, 3 had CBAVD and 2 were diagnosed as CF. Of those with CBAVD, one patient carried a mutation known to cause pancreatic insufficient CF (S549N (c.1646G>A; p.Ser549Asn)), a second patient carried a mutation associated with CBAVD (D1152H (c.3454G>C, p.Asp1152His)), and the third carried a mutation of unknown disease association (F1337V(c.4009T>G; Phe1337Val)). Of the 2 patients diagnosed with CF, a female patient with the E822X (c.2464G>T; p.Glu822X) mutation in her other CFTR gene had pancreatic insufficient CF and a patient carrying 2789+5G>A (c.2657+5G>A) had pancreatic sufficient CF. The E822X mutation has been associated with pancreatic insufficient CF while 2789+5G>T is a pancreatic sufficient mutation (http://www.genet.sickkids.on.ca/cftr/). In summary, R1070Q alone appears to be able to confer mild disease (i.e., CBAVD) in some cases when paired with a known “severe” CF mutation, while the presence of the in cis S466X mutation was consistently associated with pancreatic insufficient CF.
DISCUSSION
In the present study, we focused on a specific residue, arginine at codon 1070 (R1070), at the center of the largest cluster of evolutionarily conserved amino acids in cytoplasmic loop 4 of CFTR. This residue is of interest because mutations of R1070 are associated with disease phenotypes varying from male infertility to pancreatic insufficient cystic fibrosis (Mickle et al., 2000). Although prior studies identified functional abnormalities in CFTR bearing R1070 mutations, there was an incomplete correlation with phenotype, especially for the glutamine (Q) substitution. Prior studies had evaluated the effect of R1070Q in non-polarized cells (Cotten et al., 1996; Seibert et al., 1996; Mickle et al., 2000). Use of site-specific integration in the current study via the Flp-In® system enabled uniform expression of CFTR bearing different R1070 mutations in a polarized cell line that closely resembles the epithelial cells in which CFTR normally resides. We had hypothesized that the severe CF phenotype associated with the R1070Q mutation was caused by abnormal trafficking of CFTR to apical membranes. However, CFTR bearing R1070Q localizes to apical membranes in a manner that is qualitatively similar to wild-type CFTR. This result led to new theories explaining the phenotype associated with R1070Q that ranged from underestimation of the chloride transport defect to lack of interaction with the macromolecular complex at the apical membrane.
Before embarking on more detailed analysis of R1070Q CFTR properties, the clinical features of patients bearing this mutation were revisited to confirm its propensity to cause disease. Additionally, other deleterious variants that may exist in the CFTR gene bearing R1070Q were considered. A literature review uncovered a report that the nonsense mutation S466X had been found in cis with R1070Q in two patients (Radivojevic et al., 2004). Demonstration that S466X correlated with the CF phenotype in 11 patients combined with the known severe functional consequences of nonsense mutations (nonsense mediated RNA decay or, less commonly, protein truncation) indicated that S466X, rather than R1070Q, was responsible for the observed severe phenotype. Thus, functional studies were informative for all three R1070 mutations, whereby R1070P and R1070W revealed processing defects that are consistent with their role in disease, while the properties of CFTR bearing R1070Q provoked a re-evaluation of the established genotype/phenotype relationship that led to a more plausible explanation for the pathology observed in patients with a glutamine substitution at codon 1070.
The results obtained for the proline substitution, the rarest of the three R1070 mutants, point to the importance of studying protein expression in polarized cells as well as non-polarized cells. Observed in only two patients to date, this mutation causes classic CF and, although it was partly functional in non-polarized cells, R1070P did not localize to the cell surface in polarized MDCK cells. Certain cell types, such as non-polarized HEK-293, permit mutant CFTR access to the membrane at sufficient quantity to allow functional studies (e.g., whole cell patch clamping to assess channel function (Mickle et al., 2000). However, non-polarized cell systems do not mimic the lung epithelium, which is composed of cells with a distinct apical and basal domain. Since R1070P was not detectable on the apical membrane of FRT-MDCK cells by biotinylation studies but was present on the membrane of non-polarized HEK-293 cells (Mickle et al., 2000), the localization machinery for R1070P is apparently different in polarized and non-polarized cells. Therefore, predictions regarding localization in polarized epithelia in vivo drawn from studies using non-polarized cells should be considered carefully and applied with caution to clinical situations.
Although CFTR R1070W is primarily associated with male infertility (CBAVD), previously published reports and the current study showed that the tryptophan mutation consistently has decreased protein expression levels and incomplete apical localization. CFTR R1070W transiently expressed in non-polarized HEK-293 cells (Mickle et al., 2000), COS-1 cells (Seibert et al., 1996), and the stable expression in isogenic MDCK cell lines shown here all demonstrated lower levels than wild-type CFTR. Thus, one could propose that the presence of reduced amounts of functional protein at the apical membrane may create an abnormal phenotype, albeit one that is moderate compared to those with complete loss of CFTR. The loss of complete apical localization for CFTR R1070W may be due to alteration in the rate of membrane recycling, as has previously been observed with N287Y, a CFTR cytoplasmic loop 2 mutant (Silvis et al., 2003). Additional studies would be necessary to test this concept.
The presence of nonsense mutation S466X in CFTR genes bearing R1070Q provides a parsimonious explanation for the CF phenotype in patients with this combination. However, the 5 patients carrying R1070Q alone manifested phenotypes ranging from male infertility to pancreatic insufficient CF. While it is possible that the more severe phenotype was due to other environmental factors, genetic modifiers, or erroneous information, the abnormal clinical features in the remaining 4 patients indicate that R1070Q has some deleterious effect upon CFTR function. This conclusion is consistent with the observation that CFTR R1070Q has a reduced steady-state protein level compared to CFTR wild-type in MDCK cells. On the other hand, studies in non-polarized COS-1 cells showed wild-type-like levels of R1070Q protein yet generation of only half the chloride efflux of wild-type cells; additionally, R1070Q had normal levels of conductance in CHO cells but a decreased open probability (Seibert et al., 1996). One or more of these moderate dysfunctions caused by R1070Q may explain why it is associated with disease of varying severity when appearing alone.
The combination of R1070Q and S466X adds to the growing list of complex alleles reported in CFTR (Claustres et al., 2000). Ascertainment of the status each in cis variant is often critical for accurate prediction of functional consequences. For example, R117H occurs with two variants in intron 8 (5T and 7T) that alter the splicing efficiency of exon 9 (Chu et al., 1993) consequently, the phenotypes associated with the R117H and 5T combination are different than when R117H occurs with 7T (Kiesewetter et al., 1993). Similarly, interpretation of the clinical spectrum associated with R1070Q also requires determination of the presence or absence of S466X. The current study points to the availability and ease of use of the stable MDCK-FRT cell line system to test the functional consequences of any missense mutation in CFTR. Our results emphasize that a thorough review of the literature and clinical observations of all available rare mutation patients is imperative in situations where functional studies and phenotype are inconsistent. These steps will facilitate accurate translation of genetic data to patient care.
Supplementary Material
Acknowledgments
We thank Drs Susan Michaelis, Greg Germino and Carol Machamer for critical evaluation of this work. We also thank Dr. Germino for the kind gift of the MDCK-FRT parental cells, Dr. Tim Hefferon for CFTR amino acid alignments, Dr. Milan Macek for facilitating clinical analysis, Dr. Claude Ferec for mutation analysis and Andrea Lopez for technical assistance. This work was funded by grants from the Cystic Fibrosis Foundation Research Development Program and the National Institutes of Health (DK44003 to GRC and HL47122 to WBG).
References
- Audrézet MP, Novelli G, Mercier B, Sangiuolo F, Maceratesi P, Férec C, Dallapiccola B. Identification of three novel cystic fibrosis mutations in a sample of Italian cystic fibrosis patients. Hum Hered. 1993;43:295–300. doi: 10.1159/000154147. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Adjiman M, Thiounn N, Jeanpierre M, Hubert D, Lepercq J, Francoual C. Molecular diagnosis of congenital bilateral absence of the vas deferens: analyses of the CFTR gene in 64 French patients. Annales de Genetique. 1997;40:5–9. [PubMed] [Google Scholar]
- Casals T, Bassas L, Egozcue S, Ramos MD, Gimenez J, Segura A, Garcia F, Carrera M, Larriba S, Sarquella J, Estivill X. Heterogeneity for mutations in the CFTR gene and clinical correlations in patients with congenital absence of the vas deferens. Hum Reprod. 2000;15:1476–1483. doi: 10.1093/humrep/15.7.1476. [DOI] [PubMed] [Google Scholar]
- Casals T, Bassas L, Ruiz-Romero J, Chillón M, Giménez J, Ramos MD, Tapia G, Naraez H, Nunes V, Estivill X. Extensive analysis of 40 infertile patients with congenital absence of the vas deferens: in 50% of cases only one CFTR allele could be detected. Hum Genet. 1995;95:205–211. doi: 10.1007/BF00209403. [DOI] [PubMed] [Google Scholar]
- Cheng J, Wang H, Guggino WB. Modulation of mature cystic fibrosis transmembrane regulator protein by the PDZ domain protein CAL. J Biol Chem. 2004;279:1892–1898. doi: 10.1074/jbc.M308640200. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Chillón M, Casals T, Mercier B, Bassas L, Lissens W, Silber S, Romey M-C, Ruiz-Romero BS, Verlingue C, Claustres M, Nunes V, Férec C, Estivill X. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332:1475–1480. doi: 10.1056/NEJM199506013322204. [DOI] [PubMed] [Google Scholar]
- Choi JY, Muallem D, Kiselyov K, Lee MG, Thomas PJ, Muallem S. Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature. 2001;410:94–97. doi: 10.1038/35065099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C-S, Trapnell BC, Curristin S, Cutting GR, Crystal RG. Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nature Genet. 1993;3:151–156. doi: 10.1038/ng0293-151. [DOI] [PubMed] [Google Scholar]
- Claustres M, Guittard C, Bozon D, Chevalier F, Verlingue C, Ferec C, Girodon E, Cazeneuve C, Bienvenu T, Lalau G, Dumur V, Feldmann D, Bieth E, Blayau M, Clavel C, Creveaux I, Malinge MC, Monnier N, Malzac P, Mittre H, Chomel JC, Bonnefont JP, Iron A, Chery M, Georges MD. Spectrum of CFTR mutations in cystic fibrosis and in congenital absence of the vas deferens in France. Hum Mutat. 2000;16:143–156. doi: 10.1002/1098-1004(200008)16:2<143::AID-HUMU7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Cotten JF, Ostedgaard LS, Carson MR, Welsh MJ. Effect of cystic fibrosis-associated mutations in the fourth intracellular loop of cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271:21279–21284. doi: 10.1074/jbc.271.35.21279. [DOI] [PubMed] [Google Scholar]
- Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry Annual Data Report 2006. 2006 [Google Scholar]
- Dayangac D, Erdem H, Yilmaz E, Sahin A, Sohn C, Ozguc M, Dork T. Mutations of the CFTR gene in Turkish patients with congenital bilateral absence of the vas deferens. Hum Reprod. 2004;19:1094–1100. doi: 10.1093/humrep/deh223. [DOI] [PubMed] [Google Scholar]
- de la Taille A, Rigot JM, Mahe P, Vankemmel O, Gervais R, Dumur V, Lemairtre L. Correlation between genito-urinary anomalies, semen analysis and CFTR genotype in patients with congenital bilateral absence of the vas deferens. British Journal of Urology`. 1998;81:614–619. doi: 10.1046/j.1464-410x.1998.00589.x. [DOI] [PubMed] [Google Scholar]
- Denning GM, Ostedgaard LS, Welsh MJ. Abnormal localization of cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia. J Cell Biol. 1992;118:551–559. doi: 10.1083/jcb.118.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estivill X, Bancells C, Ramos C. Georgraphic distribution and regional origin of 272 cystic fibrosis mutations in european populations. The Biomed CF Mutation Analysis Consortium. Hum Mutat. 1997;10:135–154. doi: 10.1002/(SICI)1098-1004(1997)10:2<135::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Feldmann D, Couderc R, Audrezet MP, Ferec C, Bienvenu T, Desgeorges M, Claustres M, Mittre H, Blayau M, Bozon D, Malinge MC, Monnier N, Bonnefont JP, Iron A, Bieth E, Dumur V, Clavel C, Cazeneuve C, Girodon E. CFTR genotypes in patients with normal or borderline sweat chloride levels. Hum Mutat. 2003;22:340. doi: 10.1002/humu.9183. [DOI] [PubMed] [Google Scholar]
- Ferec C, Verlingue C, Parent P, Morin JF, Codet JP, Tault G, Dagorne M. Neonatal screening for cystic fibrosis: result of a pilot study using both immunoreactive trypsinogen and cystic fibrosis gene mutation analyses. Hum Genet. 1995;96:542–548. doi: 10.1007/BF00197409. [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais R, Dumur V, Letombe B, Larde A, Rigot JM, Roussel P, Lafitte JJ. Hypofertility with thick cervical mucus: another mild form of cystic fibrosis? JAMA. 1996;276:1638. [PubMed] [Google Scholar]
- Groman JD, Meyer ME, Wilmott RW, Zeitlin PL, Cutting GR. Variant cystic fibrosis phenotypes in the absence of CFTR mutations. N Engl J Med. 2002;347:401–407. doi: 10.1056/NEJMoa011899. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Fitzsimmons SC, Macek M, Jr, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in African-Americans and Caucasians. 1997 doi: 10.1016/s0022-3476(98)70441-x. [DOI] [PubMed] [Google Scholar]
- Jezequel P, Dorval I, Fergelot P, Chauvel B, LeTreut A, Legall JY, Le Lannou DS. Structural analysis of CFTR gene in congenital bilateral absence of vas deferens. Clin Chem. 1995;41:833–835. [PubMed] [Google Scholar]
- Jezequel P, Dubourg C, Le Lannou D, Odent S, Le Gall JY, Blayau M, Le Treut A, David V. Molecular screening of the CFTR gene in men with anomalies of the vas deferens: identification of three novel mutations. Mol Hum Reprod. 2000;6:1063–1067. doi: 10.1093/molehr/6.12.1063. [DOI] [PubMed] [Google Scholar]
- Kartner N, Augustinas O, Jensen TJ, Naismith AL, Riordan JR. Mislocalization of deltaF508 CFTR in cystic fibrosis sweat gland. Nature Genet. 1992;1:321–327. doi: 10.1038/ng0892-321. [DOI] [PubMed] [Google Scholar]
- Kiesewetter S, Macek M, Jr, Davis C, Curristin SM, Chu C-S, Graham C, Shrimpton AE, Cashman SM, Tsui LC, Mickle J, Amos J, Highsmith WE, Jr, Shuber A, Witt DR, Crystal RG, Cutting GR. A mutation in the cystic fibrosis transmembrane conductance regulator gene produces different phenotypes depending on chromosomal background. Nature Genet. 1993;5:274–278. doi: 10.1038/ng1193-274. [DOI] [PubMed] [Google Scholar]
- Le Lannou DS, Jezequel P, Blayau M, Dorval I, Lemoine P, Dabadie A, Roussey M, Le Marec B, Legall JY. Obstructive azoospermia with agenesis of vas deferens or with bronchiectasia (Young's syndrome): a genetic approach. Hum Repro. 1995;10:338–341. doi: 10.1093/oxfordjournals.humrep.a135939. [DOI] [PubMed] [Google Scholar]
- McGinniss MJ, Chen C, Redman JB, Buller A, Quan F, Peng M, Giusti R, Hantash FM, Huang D, Sun W, Strom CM. Extensive sequencing of the CFTR gene: lessons learned from the first 157 patient samples. Hum Genet. 2005;118:331–338. doi: 10.1007/s00439-005-0065-1. [DOI] [PubMed] [Google Scholar]
- Mendes F, Wakefield J, Bachhuber T, Barroso M, Bebok Z, Penque D, Kunzelmann K, Amaral MD. Establishment and characterization of a novel polarized MDCK epithelial cellular model for CFTR studies. Cell Physiol Biochem. 2005;16:281–290. doi: 10.1159/000089857. [DOI] [PubMed] [Google Scholar]
- Mercier B, Lissens W, Novelli G, Djieva L, De Arce M, Kapranov N, Canki-Klain N. A cluster of cystic fibrosis mutations in exon 17b of the CFTR gene: a site for rare mutations. J Med Genet. 1994;31:731–734. doi: 10.1136/jmg.31.9.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier B, Lissens W, Novelli G, Kalydjieva L, De Arce M, Kapranov N, Klain NC, Lenoir G, Chauveau P, Lenaerts C, Rault G, Cashman S, Sangiuolo F, Audrézet MP, Dallapiccola B, Guillermit H, Bonduelle M, Liebaers I, Quéré I, Verlingue C, Férec C. Identification of eight novel mutations in a collaborative analysis of a part of the second transmembrane domain of the CFTR gene. Genomics. 1993;16:296–297. doi: 10.1006/geno.1993.1183. [DOI] [PubMed] [Google Scholar]
- Mickle JE, Milewski MI, Macek M, Jr, Cutting GR. Effects of cystic fibrosis and congenital bilateral absence of the vas deferens-associated mutations on cystic fibrosis transmembrane conductance regulator-mediated regulation of separate channels. Am J Hum Genet. 2000;66:1485–1495. doi: 10.1086/302893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer BD, Denton J, Karlson KH, Reynolds D, Wang S, Mickle JE, Milewski M, Cutting GR, Guggino WB, Li M, Stanton BA. A PDZ-interacting domain in CFTR is an apical membrane polarization signal. J Clin Invest. 1999;104:1353–1361. doi: 10.1172/JCI7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naren AP, Nelson DJ, Xie W, Jovov B, Pevsner J, Bennett MK, Benos DJ, Quick MW, Kirk KL. Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature. 1997;390:302–305. doi: 10.1038/36882. [DOI] [PubMed] [Google Scholar]
- Osborne LR, Lynch M, Middleton PG, Alton EWFW, Geddes DM, Pryor JP, Hodson ME, Santis GK. Nasal epithelial ion transport and genetic analysis of infertile men with congenital bilateral absence of the vas deferens. Hum Mol Genet. 1993;2(10):1605–1609. doi: 10.1093/hmg/2.10.1605. [DOI] [PubMed] [Google Scholar]
- Radivojevic D, Djurisic M, Lalic T, Guc-Scekic M, Savic J, Minic P, Antoniadi T, Tzetis M, Kanavakis E. Spectrum of cystic fibrosis mutations in Serbia and Montenegro and strategy for prenatal diagnosis. Genet Test. 2004;8:276–280. doi: 10.1089/gte.2004.8.276. [DOI] [PubMed] [Google Scholar]
- Savov A, Mercier B, Kalaydijeva L, Férec C. Identification of six novel mutations in the CFTR gene of patients from Bulgaria by screening the twenty seven exons and exon/intron boundaries using DGGE and direct DNA sequencing. Hum Mol Genet. 1994;3:57–60. doi: 10.1093/hmg/3.1.57. [DOI] [PubMed] [Google Scholar]
- Seibert FS, Linsdell P, Loo TW, Hanrahan JW, Clarke DM, Riordan JR. Disease-associated mutations in the fourth cytoplasmic loop of cystic fibrosis transmembrane conductance regulator compromise biosynthetic processing and chloride channel activity. J Biol Chem. 1996;271:15139–15145. doi: 10.1074/jbc.271.25.15139. [DOI] [PubMed] [Google Scholar]
- Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci U S A. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- Shrimpton AE, Borowitz DC, Swender P. Cystic Fibrosis mutation frequencies in upstate New York. Hum Mutat. 1997;10:436–442. doi: 10.1002/(SICI)1098-1004(1997)10:6<436::AID-HUMU4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Silvis MR, Picciano JA, Bertrand C, Weixel K, Bridges RJ, Bradbury NA. A mutation in the cystic fibrosis transmembrane conductance regulator generates a novel internalization sequence and enhances endocytic rates. J Biol Chem. 2003;278:11554–11560. doi: 10.1074/jbc.M212843200. [DOI] [PubMed] [Google Scholar]
- Wang Z, Milunsky J, Yamin M, Maher T, Oates R, Milunsky A. Analysis by mass spectrometry of 100 cystic fibrosis gene mutations in 92 patients with congenital bilateral absence of the vas deferens. Hum Reprod. 2002;17:2066–2072. doi: 10.1093/humrep/17.8.2066. [DOI] [PubMed] [Google Scholar]
- Welsh MJ, Ramsey BW, Accurso FJ, Cutting GR. Cystic Fibrosis. In: Scriver CR, Beaudet AL, Valle D, Sly WS, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, Inc; 2001. pp. 5121–5188. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.