

Abstract
Globoid cell leukodystrophy (GLD) or Krabbe disease is a neurodegenerative disorder caused by the deficiency of the lysosomal enzyme galactocerebrosidase (GALC). GALC deficiency results in a progressive demyelination of the central and peripheral nervous systems. Inflammatory cells and increased levels of cytokines and chemokines are present in the CNS of GLD mice and may play a significant role in the pathogenesis of the disease. In this study we evaluate the effect of non-steroidal anti-inflammatory drugs, such as indomethacin and ibuprofen, and minocycline, a tetracycline analog with neuroprotective and anti-apoptotic properties, on the progression of the disease using a transgenic mouse model of GLD. Real-time quantitative PCR was used to analyze the expression of several markers of the immune/inflammatory response. IL-6, TNF-α, MIP-1β, MCP-1, iNOS/NOS2, CD11b, CD68, CD4 and CD8 mRNA levels were measured in cortex, cerebellum and spinal cord of untreated and treated affected mice at different ages. In addition, the pharmacological treatments were compared to bone marrow transplantation (BMT). The pharmacological treatments significantly extended the life-span of the treated mice and reduced the levels of several of the immuno-related factors studied. However, BMT produced the most dramatic improvements. In BMT-treated mice, factors in the spinal cord were normalized faster than the cerebellum, with the exception of CD68. There was a decrease in the number of apoptotic cells in the cerebellum of mice receiving anti-inflammatory drugs and BMT. These studies indicate a possible role for combined therapy in the treatment of GLD.
Keywords: globoid cell leukodystrophy, apoptosis, inflammation, bone marrow transplantation, NSAIDs, Krabbe disease
1. INTRODUCTION
Globoid cell leukodystrophy (GLD) or Krabbe disease is an autosomal recessive disorder caused by the deficiency of galactocerebrosidase (GALC) activity, an enzyme required for the catabolism of certain galactolipids found in myelin (reviewed in Wenger et al., 2001; Wenger, 2008). The disease is characterized by microglia activation, astrogliosis, macrophage infiltration and apoptotic death of oligodendrocytes, leading to progressive loss of myelin in the central and peripheral nervous systems. Several well-characterized naturally occurring animal models of this disease are available including the twitcher (twi) mouse (Duchen et al., 1980; Kobayashi et al., 1980), the Cairn and Westhighland white terriers (Fletcher et al., 1966) and the Rhesus monkey (Baskin et al., 1989). In addition, a transgenic (trs) mouse model has been generated in our laboratory by introducing a missense mutation into the GALC gene by homologous recombination (Luzi et al., 2001). The trs mouse model presents with onset of symptoms only slightly delayed compared to the twi mouse (22-24 vs 20 days), and death occurs at an average of 58 days for the trs mice versus 40 days for the twi mice. In addition, the trs mice are larger, produce more pups per litter, and their slightly extended life-span allows a longer time for evaluation of therapeutic effects.
Hematopoietic stem cell transplantation (HSCT) is the only treatment currently available for patients with Krabbe disease. When performed in asymptomatic or mildly affected patients, HSCT slows the progression of the disease (Krivit et al., 1998; Escolar et al., 2005). Animal models have been used to evaluate various therapy options (reviewed in Suzuki et al., 2003), however bone marrow transplantation (BMT) still remains the most effective treatment. In the trs mouse, BMT resulted in a much longer life-span (over one year for some animals) with significant improvements in clinical features as well as in pathological and biochemical parameters (Luzi et al., 2005). However, the animals still die with symptoms of a neurological disorder, indicating that BMT alone is not sufficient to reverse the progression of the disease. Activation of the immune system with inflammatory components has been shown to be present in mice with Krabbe disease (LeVine and Brown, 1997; Wu et al., 2001). The cytokines IL-6 and TNF-α and chemokines MCP-1 and MIP-1β are up-regulated in the CNS of twi mice indicating the possibility that they play a significant role in the demyelinating process characteristic of GLD. We were interested in determining whether anti-inflammatory treatments might be able to affect these markers and slow the progression of the disease. Therefore, trs mice were treated with two known non-steroidal anti-inflammatory drugs (NSAIDs), ibuprofen and indomethacin. In addition, several studies have reported that minocycline, a tetracycline analog, has neuroprotective and anti-apoptotic effects in several neurodegenerative disorders including Huntington’s disease (Chen et al., 2000), amyotrophic lateral sclerosis (Zhu et al., 2002) and multiple sclerosis (Brundula et al., 2002). Since apoptosis has been described in GLD (Taniike et al., 1999; Jatana et al., 2002), the effect of minocycline was also evaluated in our mouse model.
The first step of this project was to determine if any of these treatments could prolong the lives of affected mice. Once a positive effect by these drugs on survival was demonstrated, real-time quantitative RT-PCR was used to analyze several factors involved in the immune/inflammatory response, comparing data from untreated and treated mice. The factors studied were IL-6, a pro-inflammatory cytokine produced by monocytes, macrophages and T-cells, TNF-α, a pro-apoptotic cytokine produced by macrophages, mast cells and NK cells, MIP-1β and MCP-1, two chemokines characterized by chemotactic activity on monocytes and associated with several proinflammatory activities, inducible nitric oxide synthase (iNOS/NOS2), an enzyme involved in the production of toxic radicals with pro-inflammatory activity, CD11b, a marker for microglia and macrophages, CD68, a marker for phagocytosis generally associated with activated macrophages, CD4, a marker for T-helper cells and CD8, a marker for cytotoxic T-cells. For comparison, the same factors were also analyzed in BMT-treated mice. Different CNS regions, including cortex (brain hemispheres without cerebellum), cerebellum and pons and spinal cord were analyzed separately at different stages of the disease.
These studies showed a significant increase in the levels of immune-related factors with progression of the disease, but with some significant regional differences. Some treatments were better than others in partially correcting these abnormalities, however, BMT produced the most dramatic improvements. These studies provide some important clues as to the pathology of GLD and suggest some avenues of treatment to explore.
2. RESULTS
Since a substantial inflammatory component is involved in the pathology of Krabbe disease, several anti-inflammatory and neuroprotective agents were tested for their ability to slow the progression of the disease. Trs mice were treated with indomethacin or ibuprofen, both well-known non-steroidal anti-inflammatory agents, or minocycline, a second generation tetracycline with neuroprotective and anti-apoptotic proprieties (reviewed in Blum et al., 2004). Because of the rapid course of this disease, treatments were started as early as possible. Since these pharmacological agents are excreted into the milk of a lactating mother, the drugs were administered, initially, in the drinking water of the nursing mother. The age for starting the treatments was chosen in an empirical way; several litters were treated with the different pharmacological agents beginning at different ages and survival was recorded. The starting point that produced the longest survival was then selected for further experiments. The treatments were able to prolong the lives of the affected mice as shown in Fig. 1. The average increase in survival was about 10-12 days for all the treatments, with some indomethacin-treated mice living more than 80 days.
FIG. 1.
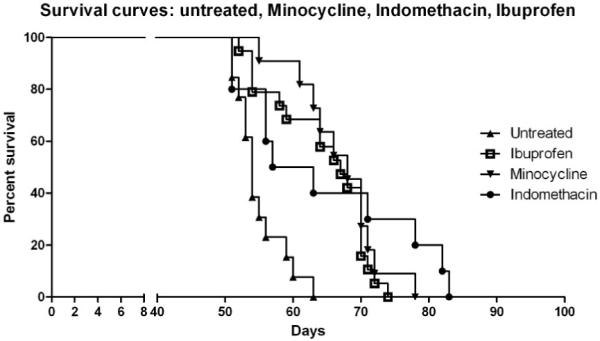
Survival curve for affected trs mice untreated (n= 13) and treated with ibuprofen (n=19), minocycline (n= 11), and indomethacin (n= 10). Treatments were administered, at the beginning, in the drinking water of the nursing mother and then provided for the entire life-span of the mice, starting at PND 5 for minocycline (final concentration 1 mg/ml), PND 10 for ibuprofen (final concentration 0.25 mg/ml) and PND 14 for indomethacin (final concentration 4μg/ml).
After showing that treatments were able to extend the life-span of affected trs mice, we used real-time quantitative RT-PCR to evaluate if the effects of these pharmaceutical treatments could be correlated with a decrease in the levels of pro-inflammatory cytokines, chemokines and markers specific for cell-mediated inflammation. Before analyzing the treated mice, the baseline levels for these factors in normal and untreated affected mice were established. Analysis of the levels of IL-6, TNF-α, MIP-1β, MCP-1, iNOS/NOS2, CD11b, CD68, CD4, CD8 with progression of disease was undertaken in different CNS regions. Cortex, cerebellum and spinal cord from mice at PND 30, PND 40 and terminal stage, were evaluated. The choice of PND 30 as the earliest time point was based on preliminary studies showing that at PND 10 no differences were detected in the levels of IL-6, TNF-α, MIP-1β and MCP-1 between normal and affected mice, while at PND 20 a slight increase in the levels of these factors was detected, mostly in the cerebellum, and to a less degree in the cortex, of affected mice (data not shown).
The fold increase over normal values for IL-6, TNF-α, MIP-1β and MCP-1 is presented in Fig. 2 and for CD11b, CD68, CD4, CD8 in Fig. 3. Please note the scale differences on each graph. In general, there is a progressive increase in mRNA levels for these factors in the cortex and cerebellum, with the cerebellum showing a more pronounced and earlier elevation for several markers.
FIG. 2.
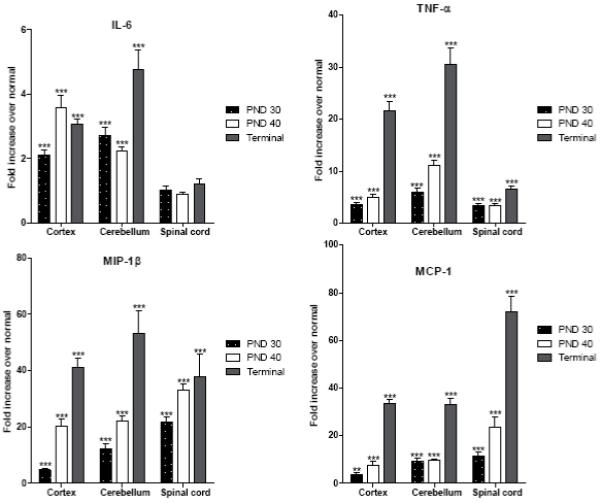
Expression of IL-6, TNF-α, MIP-1β and MCP-1 in the cortex, cerebellum and spinal cord of untreated affected trs mice at PND 30, PND 40 and terminal stage. Levels of mRNA specific for each factor were determined using real-time quantitative RT-PCR as described in Materials and Methods. Levels are presented as the mean ± SEM fold increase in five affected mice over normal mice. All values are corrected for total mRNA content using the housekeeping gene L13. Statistically significant differences in expression levels between normal mice and affected mice at a given time point were determined using the unpaired t test (*, P 0.01-0.05; **, P 0.001-0.01; ***, P < 0.001).
FIG. 3.
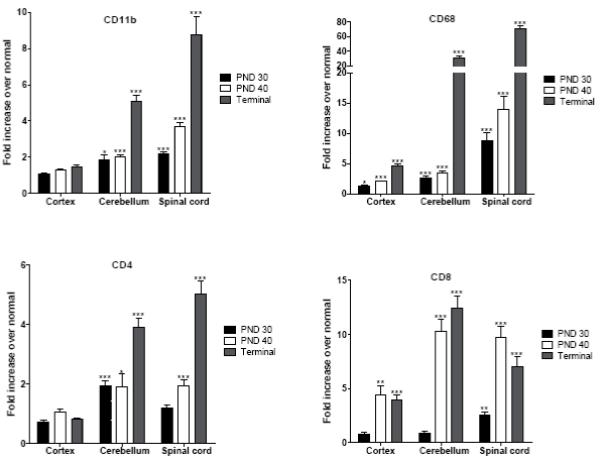
Expression of CD11b, CD68, CD4 and CD8 in the cortex, cerebellum and spinal cord of untreated affected trs mice at PND 30, PND 40 and terminal stage. Levels of mRNA specific for each factor were determined using real-time quantitative RT-PCR as described in Materials and Methods. Levels are presented as the mean ± SEM fold increase in five affected mice over normal mice. All values are corrected for total mRNA content using the housekeeping gene L13. Statistically significant differences in expression levels between normal mice and affected mice at a given time point were determined using the unpaired t test (*, P 0.01-0.05; **, P 0.001-0.01; ***, P < 0.001).
IL-6
IL-6 values in cortex and cerebellum increased to a lesser degree than the other cytokine and chemokines (Fig. 2). At the terminal stage, both in the cortex and in the cerebellum the average increase was only a few folds over normal. Spinal cord values were not elevated at any time.
TNF-α
TNF-α values in cortex and cerebellum were already elevated over normal at PND 30 and increased considerably with age reaching values of more than 20 and 30-fold over normal, in cortex and cerebellum, respectively, at the terminal stage (Fig. 2). In spinal cord the increase over normal was more modest.
MIP-1β
At PND 30, MIP-1β values were significantly elevated over normal especially in the cerebellum and spinal cord. MIP-1β levels continued to rise with disease progression, reaching a 20-fold increase in cortex and cerebellum and 30-fold in spinal cord at PND 40, and even higher levels at the terminal stage (Fig. 2).
MCP-1
While there was only a modest increase in MCP-1 in cortex at PND 30, larger increases were measured in the cerebellum and spinal cord. There was little change at PND 40 in cortex and cerebellum, however spinal cord showed a greater increase. All tissues showed very high levels at the terminal stage (Fig. 2).
i-NOS/NOS2
i-NOS/NOS2 was analyzed in cortex and cerebellum at different ages. No changes were detected between affected and normal mice at any time point studied (not shown).
CD11b
Values for CD11b in the cortex of affected mice were not significantly different from normal at any age (Fig. 3). At PND 30 and 40, significant increases were measured in the cerebellum. The CD11b levels reached 5-fold over normal at the terminal stage. A progressive increase was measured in the spinal cord reaching about 9-fold over normal at the terminal stage.
CD68
This cell marker presented a more significant raise than CD11b (Fig. 3). There was a modest increase in the cortex, however in the cerebellum the levels of CD68 increased slowly at the early stages but then abruptly rose to more than a 30-fold increase over normal at the terminal stage. In the spinal cord, there was a continuous increase from PND 30 that reached 70-fold over normal at the terminal stage.
CD4
Values for CD4 in the cortex of affected mice were not significantly elevated from normal at any age (Fig. 3). In the cerebellum, the CD4 values were only slightly elevated over normal at the earlier ages but increased at the terminal stage. A similar trend was observed in the spinal cord.
CD8
At PND 30, CD8 values were not different from normal in cortex and cerebellum; however there was a significant increase at PND 40 in cortex and especially in cerebellum. These increased values remained relatively stable at the terminal stage. Interestingly in spinal cord there was an earlier increase in CD8 at PND 30 compared to cortex and cerebellum. At PND 40 the value rose even more, but no further increase was detected at the terminal stage.
Treatments
Animals treated with minocycline, indomethacin and ibuprofen were sacrificed at PND 58 and terminal stage. The PND 58 time point was selected since it represents the mean age of death of untreated affected mice. Preliminary studies had shown that at PND 30 there was no significant difference in the factors studied between untreated affected mice and affected mice receiving these three drugs (data not shown). Therefore, values in the cortex and cerebellum of PND 58 treated affected mice and terminal treated mice were both compared to terminal untreated affected mice (average age 58 days) (Fig. 4 and 5). This was done to determine if, at the moribund stage, there was a difference in the measured factors between treated and untreated mice.
FIG. 4.
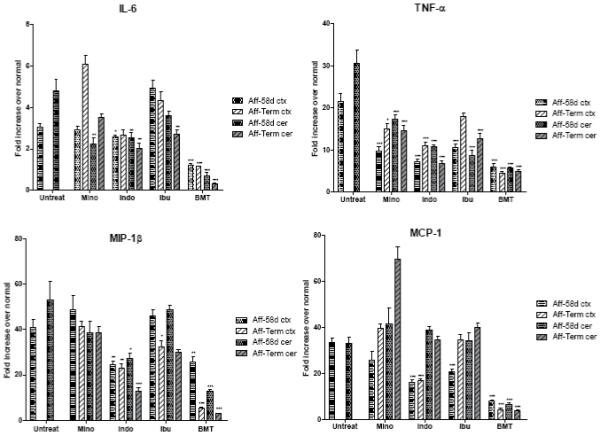
Expression of IL-6, TNF-α, MIP-1β and MCP-1 in the cortex (ctx) and cerebellum (cer) of affected trs mice (Aff) treated with minocycline (Mino), indomethacin (Indo), ibuprofen (Ibu) and BMT at PND 58 (58d) and terminal stage (Term) compared to untreated mice (Untreat) at PND 58. For the untreated mice, PND 58 corresponds to the terminal stage, for minocycline and ibuprofen the terminal stage corresponds to an average age of 70 days, for indomethacin of 67 days and for BMT-treated mice of 228 days. Each time point represent the mean ± SEM fold increase in copies of specific mRNA in the affected treated or untreated mice over the normal mice. For the pharmacological treatments five mice were used for time point, for BMT treatment 3 to 4 mice were used. Statistically significant decreases in expression levels between treated mice and untreated mice were determined by the Mann-Whitney U test (*, P 0.01-0.05; **, P 0.001-0.01; ***, P < 0.001).
FIG. 5.
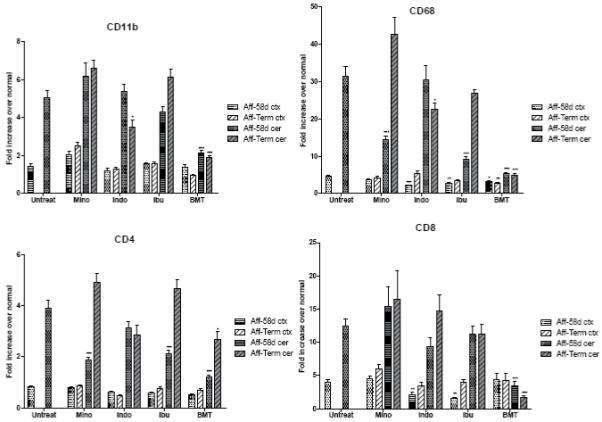
Expression of CD11b, CD68, CD4 and CD8 in the cortex (ctx) and cerebellum (cer) of affected trs mice (Aff) treated with minocycline (Mino), indomethacin (Indo), ibuprofen (Ibu) and BMT at PND 58 (58d) and terminal stage (Term) compared to untreated mice (Untreat) at PND 58 (see legend to Fig. 4).
The mice that received the pharmacological treatments were also compared to mice that received BMT. The mice receiving BMT were sacrificed at PND 58 and the terminal stage (average age 228 days).
Cortex and cerebellum from all mice and, in addition, spinal cord from the BMT-treated mice, were analyzed for IL-6, TNF-α, MIP-1β, MCP-1, CD11b, CD68, CD4, CD8 by real-time quantitative PCR. Data obtained are shown in Fig. 4 and 5 expressed as fold increase over normal, comparing levels present in the untreated affected mice with levels in the treated affected mice. This representation of data allows a direct comparison of the effects of the different treatments.
IL-6
BMT treatment prevented the increase in IL-6 levels seen in untreated affected mice and maintained the level within the normal range in both cortex and cerebellum at PND 58 and terminal stage (Fig. 4). In cortex, minocycline and ibuprofen did not prevent the rise of IL-6. In fact, IL-6 mRNA levels in minocycline-treated mice continued to increase with time and at the terminal stage the values were higher than in untreated affected mice. Treatment with indomethacin, while it did not decrease the high levels of IL-6 in cortex, it did prevent any further rise in IL-6 as the disease progressed. In cerebellum, the levels of IL-6 were slightly lower in all three pharmacologically-treated groups with the IL-6 level in the indomethacin-treated group reduced to about half of that measured in untreated mice.
TNF-α
At PND 58, BMT prevented the increase of the TNF-α levels in both cortex and cerebellum, stabilizing them to about 5-fold over normal compared to 21-fold and 30-fold, respectively, in untreated affected mice (Fig. 4). This 5-fold increase remained about the same in long-living BMT-treated mice. In cortex, the pharmacological treatments were able to stabilize the levels of TNF-α to about half of that measured in untreated mice at PND 58. At the terminal stage, the TNF-α values were increased in all treated mice, however the increase was less pronounced in the indomethacin-treated mice. In cerebellum, all three pharmacological treatments were able to significantly inhibit the increase of TNF-α levels at both time points studied. Again, indomethacin was the most effective, stabilizing TNF-α values from 30-fold over normal to 10-fold at PND 58 and 6-fold over normal at the terminal stage.
MIP-1β
In cortex, the only two treatments able to partially prevent the high levels of MIP-1β seen in untreated affected mice (41-fold over normal) were indomethacin and BMT which reduced the values to about half at PND 58 (Fig. 4). For indomethacin this value remained the same at the terminal stage, while BMT treatment reduced the MIP-1β value to only 5 times over normal in the long-living animals. In cortex, minocycline failed to produce any effect at either time point, while ibuprofen had no effect at PND 58 and only a slight decrease at the terminal stage. In cerebellum, the levels of MIP-1β decreased in BMT-treated mice from 53-fold over normal to 12-fold over normal at PND 58 and only to 3-fold over normal in the long-living mice. Indomethacin treatment halved the value at PND 58 and reduced it even further, to 12-fold over normal at the terminal stage. Minocycline and ibuprofen had no significant effects at both time points studied.
MCP-1
In cortex, BMT treatment prevented the increase of the MCP-1 levels from a 33-fold elevation to 8-fold at PND 58 and to only 4-fold in the long-living mice (Fig. 4). Indomethacin was able to limit the raise of the MCP-1 values to about half at both time points. Minocycline had no significant effect on MCP-1 levels. Ibuprofen was able to partially prevent the MCP-1 increase at PND 58, however it reached the untreated level at the terminal stage. In the cerebellum of BMT-treated mice, MCP-1 levels were 6-fold over normal at PND 58 and only 3-fold over normal at terminal stage compared to 33-fold over normal in untreated affected mice. In cerebellum, the three pharmacological treatments were unable to prevent the increase in the levels of MCP-1 at any time point. In fact, the minocycline-treated mice showed a very significant increase at the terminal stage with values reaching 69-fold over normal.
CD11b
CD11b levels in cortex of untreated affected mice were not significantly different from normal, and treatments did not produce any significant change (Fig. 5). Levels of CD11b in cerebellum of untreated affected mice were 5-fold over normal at PND 58, and the pharmacological treatments failed to lower this level at either time point. Only indomethacin reduced the CD11b level at the terminal stage to 3.5-fold over normal. BMT was successful in stabilizing the CD11b level to only 2-fold over normal at both time points examined.
CD68
In cortex, levels of CD68 were not greatly elevated in untreated affected mice and they were only partially reduced by BMT and certain pharmacological treatments at both time points (Fig. 5). The cerebellum of untreated affected mice shows a very large increase in CD68 levels (31-fold over normal). BMT limited the increase in CD68 mRNAs at PND 58 and terminal stage to only about 5-fold over normal. Of the pharmacological treatments, ibuprofen produced the most pronounced effect on CD68 at PND 58. However, this value goes up to untreated levels at the terminal stage. Minocycline follows the same trend as ibuprofen. Indomethacin does not have a significant effect on the CD68 level at PND58 and produced only a modest decrease at the terminal stage.
CD4
CD4 levels in cortex of untreated affected mice were not significantly different from normal and treatments did not produce any significant change (Fig. 5). However, in the cerebellum CD4 was increased 4-fold. The values of CD4 were normalized at PND 58 by BMT, but in the long-living mice the level increased to about 3-fold over normal. This is the only factor analyzed in the BMT-treated mice that increased from PND 58 to the terminal stage. Minocycline and ibuprofen treatments were able to prevent the increase in CD4 to about half at PND 58 but then the values increased with time to a level higher than measured in untreated affected mice. Indomethacin did not produce any significant effect on CD4 values.
CD8
In cortex, CD8 values were not changed by BMT treatment (Fig. 5). This is the only factor studied for which BMT treatment was not able to prevent an increase to, at least, some degree. Minocycline also did not produce any effect. Indomethacin and ibuprofen were able to inhibit the raise in the CD8 levels at PND 58 by more than 50%, but at the terminal stage CD8 increased to near untreated levels. In the cerebellum of BMT-treated mice, CD8 levels were only 3-fold over normal at PND 58 and normal in the long-living mice compared to 12-fold over normal in untreated affected mice. Minocycline, indomethacin and ibuprofen did not produce any significant effect in the cerebellum.
Spinal Cord Data
The data reported above show that cortex and cerebellum behave differently to the disease process and to treatment, therefore it was of interest to study the spinal cord of BMT-treated mice. Data shown in Fig. 6 compare the fold increase over normal in cortex, cerebellum and spinal cord for all the factors analyzed above.
FIG. 6.

Expression of IL-6, TNF-α, MIP-1β, MCP-1, CD11b, CD68, CD4 and CD8 in cortex, cerebellum and spinal cord of BMT-treated affected trs mice at PND 58 (Aff-58d + BMT) and terminal stage (Aff-Term + BMT) compared to untreated affected at PND 58 (Aff-58d unt). The terminal stage for BMT-treated mice corresponds to an average age of 228 days. Data are presented as fold increase over normal as described in the legend of Fig. 4. Statistically significant decreases in expression levels between treated mice and untreated mice were determined by the Mann-Whitney U test (*, P 0.01-0.05; **, P 0.001-0.01; ***, P < 0.001).
IL-6 values in spinal cord were not different from normal in the untreated affected mice (Fig. 6).
TNF-α values in spinal cord of BMT-treated mice were 2.5 times over normal at PND 58 and were reduced to near normal in the long-living animals (Fig. 6).
MIP-1β level in spinal cord of BMT-treated mice reached only a 5-fold increase over normal at PND 58 and was normalized in the long-living mice (Fig. 6). This effect in spinal cord was more dramatic than the one seen in both cortex and cerebellum, especially at the 58-day time point.
The steep rise in MCP-1 levels seen in the spinal cord of untreated affected mice (72-fold over normal) was prevented by BMT treatment. The value reached only a 7-fold increase over normal at PND 58 and only a 3-fold in the long-living mice, a similar response to that seen in cortex and cerebellum.
CD11b mRNA levels in the spinal cord of BMT-treated mice were around 2-fold over normal at both time points, similar to the cerebellum response (Fig. 6).
CD68 levels were very high in the spinal cord of untreated affected mice (70-fold over normal), higher than in the cerebellum and much higher than in the cortex. Treatment with BMT was able to partially prevent the increase of this factor; CD68 levels reached a 16-fold increase over normal at PND 58 and no further decline was seen with time. These levels, even if lower than in the untreated mice, were still very elevated above the normal value.
CD4 in the spinal cord was 5-fold elevated in untreated mice, however in BMT-treated mice dropped at PND 58 and rose slightly at the terminal stage (Fig. 6). This is similar to the trend seen in cerebellum.
The CD8 level in spinal cord of BMT-treated mice was only 2.5-fold over normal at PND 58 and was normalized in the terminal stage, following the same trend seen in cerebellum (Fig. 6).
Histological detection of apoptotic cells
Since apoptosis is involved in the pathology of Krabbe disease, the brains of trs mice were examined for apoptotic cells. Cryosections of cortex and cerebellum from 58-day-old untreated affected mice, normal mice and mice subjected to different treatments were assayed to detect apoptotic cells. Very few apoptotic cells were detected in cortex and cerebellum from normal mice. Many more apoptotic cells were observed in the cerebellum of affected mice compared to the cortex. In Fig. 7, TUNEL staining in the cerebellar granule cell layer from a normal mouse and untreated affected mouse at PND 56 is shown. In order to have a more quantitative measure, the total number of cells presents in the cerebellar granule cell layer were counted, using high power magnification, in normal and affected mice with and without treatments. Results represented as percentage of granule cells with positive staining are shown in Fig. 8. All the treatments showed a significant decrease of apoptotic cells. BMT treatment reduced the number of apoptotic cells by more than 50%. The most dramatic result was obtained in the mouse treated with both minocycline and BMT. The level of apoptotic cells was close to that seen in unaffected mice. Combining BMT with minocycline seems to have a synergistic effect in decreasing the number of cells undergoing apoptosis in this mouse model.
FIG. 7.
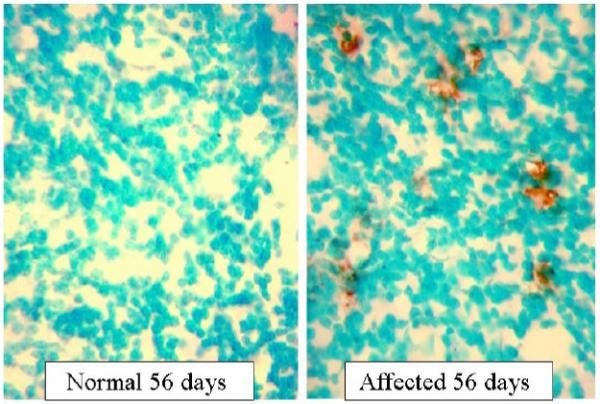
Detection of apoptosis by the TUNEL assay in the cerebellar granule cell layer of a normal trs mouse at PND 56 and an affected trs mouse at PND 56. The brown DAB-stained cells are TUNEL positive cells (original magnification 640X).
FIG. 8.
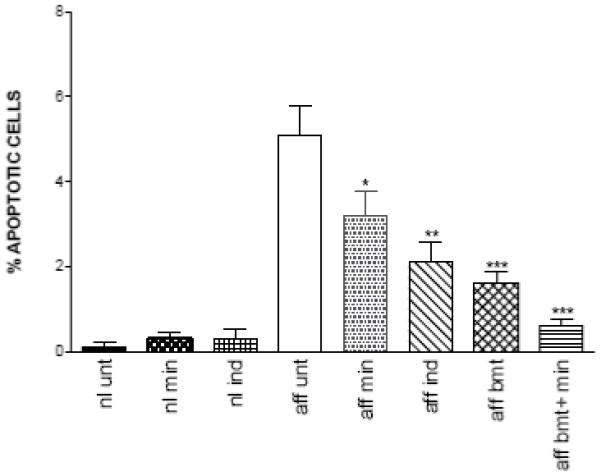
Percent of cells showing positive staining for TUNEL assay in the cerebellar granule cell layer. Cryosections of cerebella of 58-day-old mice: untreated normal (nl unt), normal treated with minocycline (nl min), normal treated with indomethacin (nl ind), untreated affected (aff unt), affected treated with minocycline (aff min), affected treated with indomethacin (aff ind), affected treated with BMT (aff bmt), affected treated with both BMT and minocycline (aff bmt + min). Sections were stained with the ApopTag Plus Peroxidase In Situ Apoptosis Kit (Chemicon) and the total number of cells present in ten representative fields per sections were counted using high power magnification. The cells undergoing apoptosis are represented as percent of tunnel positive cells. Statistically significant decreases in number of apoptotic cells between treated mice and untreated mice were determined using the unpaired t test (*, P 0.01-0.05; **, P 0.001-0.01; ***, P < 0.001).
3. DISCUSSION
Most lysosomal storage disorders characterized by a neurodegenerative course are associated with activation of the immune system and presence of an inflammatory response in the CNS (reviewed in Jeyakumar et al., 2005). Studies in animal models of different LSDs have shown that microglia activation and macrophage recruitment play a key role in the inflammation process by production of pro-inflammatory factors (Wada et al., 2000; Jeyakumar et al., 2003; LeVine and Brown, 1997; Wu et al., 2001; Baudry et al., 2003). Anti-inflammatory therapies have been evaluated in several neurodegenerative disorders. Mouse models of Alzheimer’s and Parkinson’s diseases treated with NSAIDs, such as indomethacin and ibuprofen, showed reduced microglia activation and improved pathology (Netland et al., 1998; Lim et al., 2000; Kurkowska et al., 2002). Treatment of a mouse model of Sandhoff disease with NSAIDs significantly extended the life-span of the mice (Jeyakumar et al., 2004), and treatment of twitcher mice with ibudilast decreased the number of apoptotic oligodendrocytes and reduced demyelination (Kagitani-Shimono et al., 2005).
In this paper we investigated the effects of indomethacin, ibuprofen and minocycline treatments on the trs mouse model of Krabbe disease. All the three pharmacological treatments were able to extend the life-span of the treated mice about 10-12 days from an average of 58 days in the untreated affected mice. This corresponds to an average increase in life expectancy of about 20 %. Some indomethacin-treated mice survived more than 80 days compared to 63 days in the longest living untreated mouse. This prolongation in survival is significant in a mouse model affected with such a rapidly progressing disease. A similar increase in life span was achieved by more drastic therapies, such as direct injection of GALC-expressing AAV particles into the brain of neonatal mice affected with GLD (Rafi et al., 2005; Lin et al., 2005). On the other hand, treatment of twitcher mice with ibudilast resulted in improved pathology but no extension in life-span (Kagitani-Shimono et al., 2005).
Levels of certain pro-inflammatory cytokines and chemokines measured in untreated affected trs mice showed an increase with time and progression of disease. In both cortex and cerebellum the increase in IL-6 was very modest while TNF-α, MIP-1β and MCP-1 values increased very drastically with time, reaching levels ranging from 21 to 70 times over normal at the terminal stage. These high values indicate the presence of a very strong pro-inflammatory response in the latter stages of the disease. The spinal cord showed no changes in IL-6 levels, and a modest increase in TNF-α with time, while MIP-1β and particularly MCP-1 reached extremely elevated levels especially at the terminal stage. Therefore, the induction of the chemokines appears to be independent of the expression of the pro-inflammatory cytokines IL-6 and TNF-α. This very marked increase in chemokines correlates with the very elevated values for CD-68, a marker for activated macrophages and microglia, in spinal cord (70 times over normal at terminal). These data are in agreement with the pathologic findings in affected twitcher mice. The CNS areas that myelinate first, that is spinal cord, followed by cerebellum and then brain hemispheres, are the first to show signs of pathology in affected mice (Taniike and Suzuki, 1994). In our studies, CD-68 increased from cortex to cerebellum to spinal cord at each time point examined, probably reflecting the different degree of macrophage infiltration or microglia activation in the different regions. In addition, the more marked accumulation of inflammatory cells in the spinal cord and cerebellum, compared to cortex, has been previously noted in autoimmune CNS inflammation as well as in the immune clearance of rabies vaccine virus from CNS tissues (Phares et al., 2006; Fabis et al., 2007) and may represent a naturally tighter control of cell invasion into the cortex.
In our model, iNOS/NOS2 levels were not altered by the disease (data not shown). Other authors have reported an increased iNOS/NOS2 signal in brain sections of patients with Krabbe disease using immunohistochemistry, (Giri et al., 2002). Increased iNOS/NOS2 levels have been associated with increased blood-brain barrier (BBB) permeability (Kean et al., 2000). We conducted some studies to evaluate changes in BBB permeability by measuring the CNS uptake of sodium fluorescein, following intravenous injection, in several affected trs mice at the latter stages of the disease (data not shown). These preliminary studies showed no increase in BBB permeability. This is in agreement with previously reported studies (Kondo et al., 1989). The unchanged iNOS/NOS2 levels seen in our studies also correlate with the lack of a compromised BBB.
Currently BMT represents the gold standard for the treatment of Krabbe disease in both humans and animal models. For this reason data obtained from mice treated with BMT are particularly significant. The data reported clearly show that among the studied treatments, BMT was the most effective in preventing the increase in the levels of all the analyzed factors seen in untreated affected mice, in both cortex and cerebellum, even at the early 58-day time point (Fig. 4 and 5). Among the pharmacological treatments, indomethacin was the most effective, however the reduction measured was less than with BMT. Also, all the treatments were able to produce a much more significant inhibition in the rise of the levels of chemokines and cytokines than in the immuno/inflammatory cell markers. In general, the tissues of the cerebellum respond to the treatments better than those of the cortex. The factors analyzed reached much higher values in the cerebellum than cortex of the untreated affected animals, but treatments were able to down regulate cytokines and chemokines more efficiently in cerebellum than cortex, with the exception of MCP-1. The pharmacological treatments were, in fact, more effective in preventing an increase in the levels of IL-6, TNF-α and MIP-1β in the cerebellum than in cortex, but with MCP-1 the opposite effect was noticed (Fig. 4). The treatments were able to inhibit the raise of mRNA for MCP-1 to different degrees in the cortex, but none of them had any effect on its level in the cerebellum.
BMT reduces the levels of the chemokine mRNAs to more or less similar levels in the cortex and cerebellum, but has a considerably greater inhibitory effect in the spinal cord (Fig. 6). Accumulation of CD4, CD8, CD11b and CD68 in the cerebellum and spinal cord is also reduced by bone marrow treatment. However CD68 remains highly elevated in the spinal cord and, to a lesser extent, in the cerebellum. CD68 is a marker for both infiltrating monocytes and microglia and its elevation may have two interpretations in the BMT-treated mice: a positive one, more normalized microglial function correlating with donor-derived cells, and a negative one, persistence of inflammation. It is possible that a high level of CD-68 reflects the greater accumulation of transferred bone marrow-derived cells in cerebellum and particularly in spinal cord. In this case the higher numbers of transplanted cells present in the spinal cord tissues, may have a more profound inhibitory effect on the other inflammatory factors.
On the other hand, since CD68 is a marker associated with phagocytosis, these elevated values might indicate the presence of a significant residual level of damage in the myelin and myelin-forming cells. BMT treatment in trs mice can prolong their life-span up to one year but the mice still die with symptoms of a neurological disorder, despite the fact that the pathology in cortex and cerebellum is greatly improved and psychosine levels in brain are in the normal range (Luzi et al., 2005). The CD68 data reported in this study might indicate that BMT treatment is less effective in correcting certain pathological changes in the spinal cord than in cortex and cerebellum.
In addition, of particular interest are the results of our analyses of mRNAs for the T cell markers CD4 and CD8. CD8, and to a lesser extent, CD4 levels become elevated in the cerebellum of the mice toward the latter stages of the disease, whether treated with anti-inflammatory agents or not. This may be taken as suggesting that there is a T cell component to the disease pathogenesis. However, BMT largely prevents the increase in T cell mRNA levels even in terminally diseased animals. This suggests that the invasion of T cells into the cerebellum of mice developing the disease may be a consequence of the CNS tissue pathology rather than a primary contributor.
Studies of apoptosis show a decrease in the number of cells undergoing apoptosis in the cerebellum of affected mice subjected to these treatments (Fig. 8). BMT was the most effective in reducing apoptotic cells followed by indomethacin and then minocycline. It is interesting to note that the combination of BMT and minocycline showed a synergistic effect in reducing apoptosis (Fig. 8). The apoptosis data correlate also very closely with the reduction of the pro-apoptotic cytokine TNF-α and other factors in cerebellum.
Treatment of GLD mice with anti-inflammatory drugs is not expected to provide a “cure” for the disease since the underlying defect causing the pathology, the GALC deficiency, is not corrected. However our data support the possibility that combining anti-inflammatory treatments with other therapies directed at correcting the GALC deficiency, may result in a better outcome for affected individuals.
4. EXPERIMENTAL PROCEDURE
Animals
Studies were conducted using the trs mouse model previously described (Luzi et al., 2001). Heterozygous mice were mated to obtain affected offspring. The newborn mice were genotyped at 2-3 days of age by analyzing DNA extracted from clipped toes using a PCR-based test (Luzi et al., 2001). Four to six animals for each time point were used. All procedures were conducted according to the guidelines of the Institutional Animal Care and Use Committee.
Pharmacological treatments
Pharmacological treatments were provided, at the beginning, in the drinking water of the nursing mother and then administered for the entire life-span of the mice.
Minocycline (Sigma) was dissolved in the drinking water, containing 5% sucrose, at a final concentration of 1 mg/ml (200 mg/kg/day). The treatment was started at PND 5. Indomethacin (Sigma) was first dissolved in polyethylene glycol/ Tween 20 (95:5 v/v) at a concentration of 0.4 mg/ml and then diluted 1:100 in drinking water to achieve a therapeutic dose of 0.8 mg/kg/day. Treatment was started at PND 14.
Ibuprofen (Sigma) treatment was started at PND 10 using a final concentration of 0.25 mg/ml (50 mg/kg/day) in water.
The untreated mice were given regular water.
Bone marrow transplantation (BMT)
The previously described procedure for BMT was followed (Luzi et al., 2005). Briefly, nine-day-old mice received a lethal total body dose of radiation (9.0 GY), and 24 hrs later were injected intraperitoneally with 3-4×107 bone marrow cells obtained by flushing tibiae and femora of non-carrier donor mice. Before injection, the bone marrow cells were treated with IGF-1 (Sigma) at a final concentration of 50 ng/ml for 30 min at room temperature. For 7 wks after BMT the mice received prophylactic Neomycin (final concentration 0.5 mg/ml) and Polymixin B (final concentration 13 μg/ml) in the drinking water.
Tissue preparation
Treated and untreated mice were sacrificed at different ages including PND 30, PND 40, PND 58 as well as the terminal stage, which is the time at which the animals presented with paralysis of the hind legs. The terminal stage for untreated mice was between 50 and 63 days (average 58 days), for minocycline-treated mice between 71 and 74 days (average 70 days), for indomethacin-treated mice between 64 and 71 days (average 67 days). The ibuprofen-treated mice group at terminal stage was composed by five 70-day-old mice (average 70 days). The terminal stage for BMT-treated trs mice is between 6 and 13 months of age (Luzi et al., 2005). In this study, three long-living transplanted affected mice (173, 216 and 295 days old) were used, while for the 58-day time point, four transplanted affected mice and four transplanted unaffected mice were analyzed. Five to six animals for each time point were used for both untreated and pharmacologically-treated mice, including normal mice subjected to the same treatments as the affected mice.
At the time of sacrifice the mice were subjected to transcardiac perfusion. Briefly, the animals were deeply anesthetized using Pentobarbital sodium (100 μg/g body weight), the left ventricle incannulated and then perfused using phosphate buffered saline (PBS) plus heparin followed by PBS. Brain and spinal cord were removed and the former was separated into cerebral cortex (brain hemispheres without cerebellum) and cerebellum and pons. The tissues were quickly frozen and stored at -80°. Tissues used for the detection of TUNEL assay were prepared by transcardiac perfusion with 4% paraformaldehyde and soaking in 25% sucrose as previously described (Luzi et al., 2005). Frozen sections were then used for apoptosis studies.
Real-time quantitative RT-PCR
RNA was isolated from cortex, cerebellum and spinal cord using the Qiagen RNeasy kit (Qiagen) followed by DNase I treatment (Qiagen). cDNA was synthesized from 2 μg of total RNA using M-MLV reverse transcriptase (Promega) and dT15 primers. Real-time quantitative PCR was performed on equal volumes of cDNA using specific primers and probe sets and the iTaq™ DNA polymerase (IQ™Supermix, Bio-Rad). The probe and primers sets specific for the factors L-13, IL-6, TNF-α, MIP-1β, MCP-1, CD11b, CD4, and CD8 have been previously described (Phares et al., 2006). Probe and primers for CD68 were as follow: probe, CCA GCC CCT CTG AGC ATC TGC CCC A; 5′primer, GTG CTC ATC GCC TTC TGC ATC A; 3′primer, GGC GCT CCT TGG TGG CTT AC. Probe and primers for iNOS/NOS2 have been previously described (Scott et al., 2004). Probes were double labeled at the 5′end with the reporter dye 6-FAM and at the 3′end with the quencher BHQ-1. Real-time quantitative PCR was performed using a Bio-Rad iCycler iQ Real-Time Detection System. Data were calculated based on a threshold cycle (Ct) determined as the PCR cycle at which the fluorescent signal becomes higher than that of the background (cycles 2-10) plus 10 times the SD of the background. The number of copies of specific mRNA in each sample was determined using standard curves obtained by diluting synthetic cDNA standards. Data were expressed as the number of copies of a specific mRNA per copy of the housekeeping mRNA L13 in each particular sample and presented as fold increase in mRNA copy numbers in affected mice over levels in normal mice at the same age/treatment, with all the values normalized to the L13 mRNA content for each sample.
TUNEL assay
To detect apoptotic cells on brain cryosections, the ApopTag Plus Peroxidase In Situ Apoptosis Kit (Chemicon) was used, following the manufacturer’s instructions. Briefly, frozen sections of cortex and cerebellum from treated and untreated trs mice were fixed in 1% paraformaldehyde, permeabilized by treatment with ethanol:acetic acid at -20°, quenched in 30% hydrogen peroxide, and reacted with terminal deoxynucleotidyl transferase and digoxinin deoxynucleotide triphosphates followed by peroxidase conjugated anti-digoxinin antibody and visualized with diaminobenzidine (DAB). The specimens were then counterstained with methyl green and viewed under a light microscope (Olympus BX51). Apoptotic cells, appearing as brown, were counted in the cerebellar granule cell layer. Ten fields per section were counted using high power magnification.
Statistical analyses
Survival curves were analyzed using the log-rank test.
Data obtained by real-time PCR are expressed as the mean ± SEM for each group of mice. Evaluation of the significance of difference between the means of parameters in untreated and treated groups was performed using the unpaired t test or Mann-Whitney U test. Statistical significance for the apoptosis data were evaluated using the unpaired t test and ANOVA with Dunnett’s and Tukey’s multiple comparison tests. In all cases P values < 0.05 were considered significant.
Graphs were plotted and statistics assessed using the program GraphPad Prism 4.0 (GraphPad Software).
ACKNOWLEDGMENTS
We gratefully acknowledge the contributions of Christine M. Brimer and Han Zhi Rao to this research. This work was supported in part by a grant from The National Institutes of Health [DK38795] awarded to D.A.W.
Abbreviations
- GLD
globoid cell leukodystrophy
- GALC
galactocerebrosidase
- BMT
bone marrow transplantation
- HSCT
hematopoietic stem cell transplantation
- twi
twitcher
- trs
transgenic
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Section: Disease-Related Neuroscience
REFERENCES
- Baskin G, Alroy J, Li YT, Dayal Y, Raghavan SS, Sharer L. Galactosylceramide-lipidosis in rhesus monkeys. Lab. Invest. 1989;60:7A. [Google Scholar]
- Baudry M, Yao Y, Simmons D, Liu J, Bi X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp. Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]
- Blum D, Chtarto A, Tenenbaum L, Brotchi J, Levivier M. Clinical potential of minocycline for neurodegenerative disorders. Neurobiol. Dis. 2004;17:359–366. doi: 10.1016/j.nbd.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW. Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain. 2002;125:1297–1308. doi: 10.1093/brain/awf133. [DOI] [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000;6:797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Eicher EM, Jacobs JM, Scaravilli F, Teixeira F. Hereditary leukodystrophy in the mouse: the new mutant twitcher. Brain. 1980;103:695–710. doi: 10.1093/brain/103.3.695. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. New. Engl. J. Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Fabis MJ, Scott GS, Kean RB, Koprowski H, Hooper DC. Loss of blood-brain barrier integrity in the spinal cord is common to experimental allergic encephalomyelitis in knockout mouse models. Proc. Natl. Acad. Sci. USA. 2007;104:5656–5661. doi: 10.1073/pnas.0701252104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher TF, Kurtz HJ, Low DG. Globoid cell leukodystrophy (Krabbe type) in the dog. J. Am. Vet. Med. Assoc. 1966;149:165–172. [PubMed] [Google Scholar]
- Giri S, Jatana M, Rattan R, Won JS, Singh I, Singh AK. Galactosylsphingosine (psychosine)-induced expression of cytokine-mediated inducible nitric oxide synthases via AP-1 and C/EBP: implication for Krabbe disease. FASEB J. 2002;16:661–672. doi: 10.1096/fj.01-0798com. [DOI] [PubMed] [Google Scholar]
- Jatana M, Giri S, Sing AK. Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci. Lett. 2002;330:183–187. doi: 10.1016/s0304-3940(02)00655-9. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Thomas R, Elliot-Smith E, Smith DA, van der Spoel AC, d’Azzo A, Perry VH, Butters TD, Dwek RA, Platt FM. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003;126:974–987. doi: 10.1093/brain/awg089. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Smith DA, Williams IM, Borja MC, Neville DC, Butters TD, Dwek RA, Platt FM. NSAIDs increase survival in the Sandhoff disease mouse: synergy with N-butyldeoxynojirimycin. Ann. Neurol. 2004;56:642–649. doi: 10.1002/ana.20242. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Dwek RA, Butters TD, Platt FM. Storage solutions: treating lysosomal disorders of the brain. Nat. Rev. Neurosci. 2005;6:713–725. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- Kagitani-Shimono K, Mohri I, Fujitani Y, Suzuki K, Ozono K, Urade Y, Taniike M. Anti-inflammatory therapy by ibudilast, a phosphodiesterase inhibitor, in demyelination of twitcher, a genetic demyelination model. J. Neuroinflammation. 2005;2:10. doi: 10.1186/1742-2094-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kean RB, Spitsin SV, Mikheeva T, Scott GS, Hooper DC. The peroxynitrite scavenger uric acid prevents inflammatory cell invasion into the central nervous system in experimental allergic encephalomyelitis through maintenance of blood-central nervous system barrier integrity. J. Immunol. 2000;165:6511–6518. doi: 10.4049/jimmunol.165.11.6511. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Yamanaka T, Jacobs JM, Teixeira F, Suzuki K. The Twitcher mouse: an enzymatically authentic model of human globoid cell leukodystrophy (Krabbe disease) Brain Res. 1980;202:479–483. doi: 10.1016/0006-8993(80)90159-6. [DOI] [PubMed] [Google Scholar]
- Kondo A, Nakano T, Suzuki K. Blood-brain barrier permeability to horseradish peroxidase in twitcher and cuprizone-intoxicated mice. Brain Res. 1987;425:186–190. doi: 10.1016/0006-8993(87)90499-9. [DOI] [PubMed] [Google Scholar]
- Krivit W, Shapiro EG, Peters C, Wagner JE, Cornu G, Kurtzberg J, Wenger DA, Kolodny EH, Vanier MT, Loes DJ, Dusenbery K, Lockman LA. Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. New Engl. J. Med. 1998;338:1119–1126. doi: 10.1056/NEJM199804163381605. [DOI] [PubMed] [Google Scholar]
- Kurkowska-Jastrzebska I, Babiuch M, Joniec I, Przybylkowski A, Czlonkowski A, Czlonkowska A. Indomethacin protects against neurodegeneration caused by MPTP intoxication in mice. Int. Immunopharmacol. 2002;2:1213–1218. doi: 10.1016/s1567-5769(02)00078-4. [DOI] [PubMed] [Google Scholar]
- LeVine SM, Brown DC. IL-6 and TNFα expression in brains of twitcher, quaking and normal mice. J. Neuroimmunol. 1997;73:47–56. doi: 10.1016/s0165-5728(96)00166-x. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Fantz CR, Levy B, Rafi MA, Vogler C, Wenger DA, Sands MS. AAV2/5 vector expressing galactocerebrosidase ameliorates CNS disease in the murine model of globoid-cell leukodystrophy more efficiently than AAV2. Mol. Ther. 2005;12:422–430. doi: 10.1016/j.ymthe.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Luzi P, Rafi MA, Zaka M, Curtis M, Vanier MT, Wenger DA. Generation of a mouse with low galactocerebrosidase activity by gene targeting: A new model of globoid cell leukodystrophy (Krabbe disease) Molec. Genet. Metab. 2001;73:211–223. doi: 10.1006/mgme.2001.3194. [DOI] [PubMed] [Google Scholar]
- Luzi P, Rafi MA, Zaka M, Rao HZ, Curtis M, Vanier MT, Wenger DA. Biochemical and pathological evaluation of long-lived mice with globoid cell leukodystrophy after bone marrow transplantation. Mol. Genet. Metab. 2005;86:150–159. doi: 10.1016/j.ymgme.2005.06.023. [DOI] [PubMed] [Google Scholar]
- Netland EE, Newton JL, Majocha RE, Tate BA. Indomethacin reverses the microglia response to amyloid beta-protein. Neurobiol. Aging. 1998;19:201–204. doi: 10.1016/s0197-4580(98)00047-5. [DOI] [PubMed] [Google Scholar]
- Phares TW, Kean RB, Mikheeva T, Hooper DC. Regional differences in blood-brain barrier permeability changes and inflammation in the apathogenic clearance of virus from the central nervous system. J. Immunol. 2006;176:7666–7675. doi: 10.4049/jimmunol.176.12.7666. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Rao HZ, Passini MA, Curtis M, Vanier MT, Zaka M, Luzi P, Wolfe JH, Wenger DA. AAV-mediated expression of galactocerebrosidase in brain results in attenuated symptoms and extended life span in murine models of globoid cell leukodystrophy. Mol. Ther. 2005;11:734–744. doi: 10.1016/j.ymthe.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Scott GS, Kean RB, Fabis MJ, Mikheeva T, Brimer CM, Phares TW, Spitsin SV, Hooper DC. ICAM-1 upregulation in the spinal cords of PLSJL mice with experimental allergic encephalomyelitis is dependent upon TNF-α production triggered by the loss of blood-brain barrier integrity. J. Neuroimmunol. 2004;155:32–42. doi: 10.1016/j.jneuroim.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ezoe T, Tohyama J, Matsuda J, Vanier MT, Suzuki K. Are animal models useful for understanding the pathophysiology of lysosomal storage disease? Acta Paediatr. Suppl. 2003;443:54–62. doi: 10.1111/j.1651-2227.2003.tb00223.x. [DOI] [PubMed] [Google Scholar]
- Taniike M, Suzuki K. Spacio-temporal progression of demyelination in twitcher mouse: with clinico-pathological correlation. Acta Neuropathol. 1994;88:228–236. doi: 10.1007/BF00293398. [DOI] [PubMed] [Google Scholar]
- Taniike M, Mohri I, Eguchi N, Irikura D, Urade Y, Okada S, Suzuki K. An apoptotic depletion of oligodendrocytes in the twitcher, a murine model of globoid cell leukodystrophy. J. Neropathol. Exp. Neurol. 1999;58:644–653. doi: 10.1097/00005072-199906000-00009. [DOI] [PubMed] [Google Scholar]
- Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA. 2000;97:10954–10959. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA. Krabbe disease: Globoid Cell Leukodystrophy. In: Rosenberg RN, Di Mauro S, Paulson HL, Ptacek L, Nestler EJ, editors. The Molecular and Genetic Basis of Neurologic and Psychiatric Disease. Fourth edition Wolters Kluwer/Lippincott Williams & Wilkins; Philadelphia: 2008. pp. 239–244. [Google Scholar]
- Wenger DA, Suzuki K, Suzuki K. Galactosylceramide lipidosis. Globoid cell leukodystrophy (Krabbe disease) In: Scriver CR, Beaudet AL, Sly DS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. Eight edition McGraw-Hill; New York: 2001. pp. 3669–3694. [Google Scholar]
- Wu YP, McMahon EY, Masuda J, Suzuki K, Matsushima GK, Suzuki K. Expression of immune-related molecules is downregulated in twitcher mice following bone marrow transplantation. J. Neurophatol. Exp. Neurol. 2001;60:1062–1074. doi: 10.1093/jnen/60.11.1062. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona VO, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]