

Abstract
The hepoxilin pathway was discovered over two decades ago. Products in this pathway are derived through the 12S-lipoxygenase/hepoxilin synthase enzyme system and contain intrinsic biological activity. This activity relates to the reorganization of calcium and potassium ions within the cell, and in inflammation and insulin secretion. Although the natural hepoxilins are chemically unstable, chemical analogues (PBTs) have been synthesized with chemical and biological stability. The PBTs antagonize the natural hepoxilins. The PBTs showed bioavailability, excellent tolerance and stability in vivo. In proof of principle studies in vivo in animal models, the PBTs have shown actions as anti-inflammatory agents, anti-thrombotic agents, anti-cancer agents and anti-diabetic agents. These studies demonstrate the effectiveness of the base structure of the hepoxilin (and PBT) molecule and serve as an excellent framework for the design and preparation of second-generation compounds with improved pharmaceutical properties as therapeutics for the above-mentioned diseases.
This article is part of a themed issue on Mediators and Receptors in the Resolution of Inflammation. To view this issue visit http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Keywords: hepoxilins, hepoxilin stable analogues, bioavailability, efficacy, tolerance, inflammation, thrombosis, diabetes, cancer, novel therapeutics
Nomenclature
The natural hepoxilins (Hx) are 20-carbon tri-olefinic molecules derived through metabolism of arachidonic acid. They have an open chain structure with an (S, S) epoxide group located at C11, 12 of the chain. Two Hxs are formed enzymatically from 12(S)-hydroperoxy eicosatetraenoic acid (HPETE), that is, HxA3 and HxB3; HxA3 has cis delta 5 (5Z) and delta 14 (14Z) double bonds, while the double bond at C9 is trans (9E). HxA3 has a hydroxyl group at C8 (racemic) (Pace-Asciak et al., 1995). HxB3 also has cis double bonds at delta 5 and delta 14, but also at delta 8. The racemic hydroxyl group in HxB3 is at the C10 location (see structures, Figure 1A).
Figure 1.
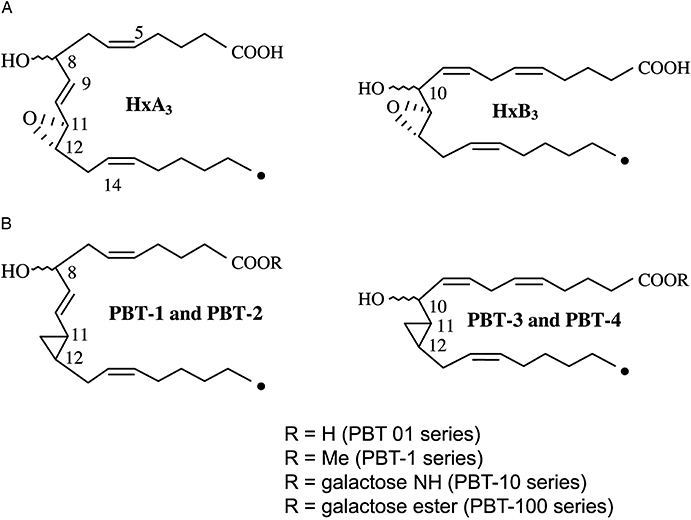
(A) Structures of the natural hepoxilins epimeric at C8 or C10. The wavy line for the OH group indicates both S and R configuration. (B) Structures of the hepoxilin analogues (PBTs). Note that families of analogues are indicated in the inset for the ‘1’ series depending on the definition of R, but similar analogues are named for the other three families (2–4). Hx, natural hepoxilins.
Although only 12S-HPETE (derived from arachidonic acid via 12S-lipoxygenase) acts as substrate for enzymatic Hx formation in many cells/tissues (i.e. enzymatic conversion is stereoselective yielding the S, S epoxide configuration (Pace-Asciak, 1984a)], both 12S- and 12R-HPETE can be converted to Hxs non-enzymatically through the use of haemin or haemoglobin as catalyst (Pace-Asciak, 1984a,b; 1985;) to produce epoxide with S, S and R, R configuration and racemic hydroxyl groups at C8 and C10 (Reynaud et al., 1994).
Because the natural Hxs are chemically and biologically unstable, studies of their actions in vivo are limited. Despite this, published data are available showing, through use of mass spectrometry, that the natural Hxs are released into the rat circulation after bolus intra-arterial administration of arachidonic acid, the eicosanoid lipid precursor, in both the normal rat and the diabetic rat (Pace-Asciak et al., 1988). The formation of Hxs and thromboxane was enhanced in the blood of the diabetic rat. Still, the use of the natural Hxs in vivo is not warranted due to their chemical and biological instability, unless a topical application is investigated and local effects are monitored, for example, skin vascular permeability, intraocular pressure. Hence, analogues were prepared in which the epoxide was replaced by a cyclopropyl group (PBTs). This afforded considerable chemical and biological stability in vivo. In fact, a single intravenous injection of the PBT as methyl ester showed its detection in blood up to 23 h as the parent methyl ester and its free acid metabolite.
The unstable epoxide moiety of the native structure was replaced with a stable cyclopropyl group through total chemical synthesis (Demin and Pace-Asciak, 1993). Four cyclopropyl analogues corresponding to the four Hx molecules (two C8 and two C10 enantiomers each of HxA3 and HxB3) were prepared and termed PBT-1 and PBT-2 (corresponding to the HxA3 structures) and PBT-3 and PBT-4 (corresponding to the HxB3 structures). Several derivatives/analogues were prepared as detailed in Figure 1B.
These compounds proved to be chemically and biologically stable, and were mostly used as the methyl ester derivative. The methyl ester group affords good uptake into the cell where it is hydrolysed to release the free carboxylic acid through abundant esterases present in cells.
The chemical synthesis of the natural Hxs and of the PBTs have been described (Hx: Chabert et al., 1989; Corey and Su, 1990; Demin et al., 1990; 1991; Lumin, et al. 1992; Wu and Wu, 1992; 1993; Vasiljeva et al., 1993; Belosludtsev et al., 1994; Pivnitsky et al., 1994; Vasiljeva and Pivnitsky, 1996); PBTs: Demin and Pace-Asciak, 1993; Demin et al., 1996). The chemical synthesis of a sulphur analogue of HxA3 has been described in which the epoxide ‘O’ has been replaced by ‘S’ (Demin et al., 1996). The sulphur analogues were found as active as the natural Hxs on calcium mobilization in vitro in human neutrophils (Demin et al., 1996). Peptido metabolites of the natural Hxs resulting from the opening of the epoxide group through Glutathione S-transferase and the attachment of the peptide group at C11 have been isolated from the liver (Laneuville et al., 1990) and brain (Pace-Asciak et al., 1990a,b;); the glutathione conjugate was named HxA3-C in keeping with the leukotriene nomenclature (Samuelsson, 1982). Limited studies in vitro have shown these products to be biologically active. Other analogues of the PBTs that were prepared and proved to be biologically active in vitro and in vivo were the galactose esters and galactose amides (see Figure 1B; C.R. Pace-Asciak and D. Reynaud, unpubl. obs.).
While the natural Hxs are unstable chemically and biologically in vivo due to the presence of an allylic epoxide, the PBTs are stable. The cyclopropyl group in the PBTs that replaced the epoxide in the natural compounds affords them their stability. Studies designed to investigate the lifetime of the PBTs in the rat circulation demonstrated that the PBT was still detectable in the circulation 23 h after a single bolus dose of PBT-1 had been administered (400 µg) as methyl ester. Thus, PBT-1, as the methyl ester, and its free acid metabolite (PBT-01) were monitored by mass spectrometry (LC-MSMS) (C.R. Pace-Asciak, unpubl. obs.). These experiments demonstrated the bioavailability of the compound in the body and its appearance in the circulation. The animals tolerated this amount of PBT well, and this is far above the amount needed to cause a biological effect (threshold dose in mice in the cancer model was 30 µg·mouse−1 (see later section on cancer)). These findings add support to the reports in other sections of this review that the PBTs are biologically available in vivo. The lipidic nature of the PBT probably accounts for its long lifetime in the body as the compound would likely enter lipid pools and be released slowly; this slow release would account for its desirable long actions in the body (see effects in cancer).
Biological properties
The natural Hxs – HxA3 and HxB3
The naturally occurring Hxs are unstable, degrading to stable trihydroxy products (TrXA3 and TrXB3) through epoxide ring opening by epoxide hydrolases in the cell cytosol. The TrXAs and the TrXBs have no biological activity in the applications mentioned here within the same dose range of the Hxs or PBTs. As mentioned previously, the unstable epoxide functionality of the HxAs (and to a lesser extent the HxBs) can be opened enzymatically by glutathione S-transferases to produce biologically active peptido conjugates (HxA3-C and presumably also the respective conjugates D, E and F as in the leukotriene series). HxA3-C is biologically active in brain and on platelet cell volume regulation (see below).
Insulin secretion
The first biological action reported for the naturally derived and isolated HxA3 was on isolated rat pancreatic islets of Langerhans; the Hxs were discovered to be present in these cells (Pace-Asciak and Martin, 1984). This study was carried out in collaboration with Dr Julio Martin at our institute. Indeed, the compound stimulated the release of insulin in vitro in a glucose-dependent fashion.
Calcium regulation
In an attempt to investigate whether calcium ions were involved in the actions of Hxs, we investigated whether HxA3 affected the transport of calcium across membranes. We used a foetal-maternal membrane system (the guinea pig yolk sac) to investigate this in the laboratory of Dr Ingeborg Radde at the Hospital for Sick Children. Transport of calcium 45 across the membrane was stimulated to the compartment containing HxA3. These studies were undertaken with synthetic natural compounds provided by Professor E. J. Corey that were obtained through total chemical synthesis (Corey and Su, 1990). In collaboration with Professor Sergio Grinstein, we showed that HxA3 stimulated the rapid release of intracellular calcium in fluorescent dye-loaded human neutrophils (Dho et al., 1990). This rapid increase in intracellular calcium slowly returned towards baseline but stayed above baseline within the 5 min observation period. Interestingly, the slow second component of the curve after the initial calcium spike depended on the presence of calcium in the medium; that is, in the absence of external calcium, the return to baseline was rapid and occurred within 5 min, unlike when the medium contained calcium (Dho et al., 1990). This demonstrated that HxA3 affects two phases of intracellular calcium concentrations, the most pronounced being an initial rapid spike of calcium (independent of the presence of extracellular calcium) derived from the release of calcium from intracellular stores. A second but less dramatic rise in intracellular calcium (dependent on the presence of extracellular calcium) is derived through calcium influx from the external medium. Further interesting observations were derived through the use of GTP analogues, GTP-γ-S and GppNHp. These analogues, loaded into human neutrophils, inhibited the rapid calcium spike evoked by HxA3, suggesting that the HxA3 effect was mediated by a G-protein (Reynaud et al., 1995a). Studies with carbonyl cyanide m-chloro phenyl hydrazone (CCCP), an uncoupler of mitochondrial oxidation, revealed that the pattern of intracellular calcium changes evoked by HxA3 resembled that of the pattern observed in the absence of external calcium, suggesting that the second slow wave of calcium represented uptake of intracellular calcium by the mitochondria, and this was inhibited by CCCP (Mills et al., 1997).
Exposure of human neutrophils to HxA3 blunts the rise in intracellular calcium evoked by a variety of different agonists, for example, leukotriene B4 (LTB4), platelet activating factor and formyl methionyl leucyl phenylalanine (fMLP) (Laneuville et al., 1993). The diversity in structure of these agonists suggests that HxA3 acts at some site common to all of these agonists; presumably, this may be at a post-receptor site or it may act primarily at calcium channels, blunting the release of calcium into the cell cytosol. There is insufficient evidence at this time to point to a precise mode of action.
Is the calcium regulation evoked by HxA3 caused by depletion of calcium stores?
Other actions of other Hxs that do not cause calcium release, but also cause an inhibition of the action of the inflammatory mediators mentioned in the previous section suggest that depletion of calcium stores is not the mechanism responsible for the actions of these Hxs. For example, HxB3 does not cause release of intracellular calcium at the doses used for HxA3, yet it inhibits dose-dependently the release of calcium caused by LTB4 or fMLP. Similar findings were observed with the PBT analogues. These experiments suggest that an intrinsic common pathway is involved in the release of calcium from internal stores that is activated by HxA3 leading to the release of calcium on one hand, and the inhibition of the action of inflammatory mediators on the other hand; however, the other analogues such as HxB3 and the PBTs (as methyl esters) that, although lacking intrinsic actions to release calcium, may still ‘bind’ to the same protein to block the actions of other calcium mediators. This blockade is mediated by G-proteins (see above and section below).
Inflammatory mediators
HxA3 is equipotent to LTB4 as a chemotactic factor to human neutrophils, but has higher potency than that of fMLP (Sutherland et al., 2000). More recently, HxA3 was identified as pathogen-elicited epithelial chemoattractant (PEEC), a low-molecular-weight naturally occurring mediator of mucosal inflammation (Mrsny et al., 2004). PEEC stimulates G-protein-coupled Ca2+ mobilization that is pertussis toxin-sensitive. Mrsny et al. (2004) have shown that PEEC is distinguished from the effect of other known polymorphonuclear (PMN) chemoattractants in that it does not cause degranulation or oxidative burst. PEEC (HxA3) appears to be formed by the apical epithelial cells and is secreted from their apical surface in response to inflammatory events, for example, bacterial pathogens. It then acts by drawing PMNs via the establishment of a gradient across the epithelial tight junction complex. HxA3 is more active than LTB4 in causing basolateral to apical transepithelial migration of human PMNs in vitro. Additional confirmation of the role of HxA3 in these events was obtained by disruption of its synthesis in vivo through use of 12-lipoxygenase blockers that reduced tissue inflammation (Mrsny et al., 2004). These studies were designed to probe into the mechanisms and potential treatment of inflammatory bowel disease, and indeed the positive results obtained demonstrate that the Hx pathway is an essential mediator in causing neutrophil emigration to inflammatory sites, the control of which can be accomplished through the use of the 12-lipoxygenase inhibitors that block formation of endogenous Hx. Interestingly, and as anticipated, confirming the mediation of inflammation by HxA3, in vitro studies have shown efficacy of the PBT analogues (as methyl esters) in blocking neutrophil emigration (B.A. McCormick and C.R. Pace-Asciak, unpubl. obs.), further confirming the Hx antagonistic effects of the PBTs.
Cell volume regulation
Cells regulate their volume through mechanisms innate within the cell (Sarkadi and Parker, 1991). Platelets, for example, respond to hypotonic-induced swelling by elevation of K + conductance, which occurs simultaneously with that of an independent conductive Cl- transport. Thus, the outward clearance of KCl, driven by the K+ gradient, results in an osmotically obliged water efflux, causing a volume loss known as RVD (regulatory volume decrease) (Livne et al., 1987). RVD was discovered to be controlled by HxA3, formed endogenously during hypotonic volume expansion. Thus, when 12-lipoxygenase activity is blocked (thereby formation of endogenous HxA3 is inhibited), and the cell is subjected to volume expansion, the cell volume stays expanded; addition of exogenous HxA3 in the presence of TCPO to prevent enzymatic hydrolysis of HxA3 causes the cell volume to retract (Margalit et al., 1993). Interestingly, the glutathione conjugate of HxA3, HxA3-C, was also very active in regulating platelet cell volume (C.R. Pace-Asciak and A. Margalit unpubl. obs.).
Potassium regulation
Studies in the marine snail Aplysia have shown that the Hxs mimic the effects of histamine to produce a dual depolarizing–hyperpolarizing synaptic action in a class of motor neurones (Piomelli et al., 1987a,b; 1989;). In the mammalian brain, Hxs were shown to possess synaptic actions and to inhibit the release of tritiated norepinephrine from brain slices that were pre-labelled with the neurotransmitter (Pace-Asciak et al., 1990c and see section below). In Aplysia sensory neurones, indirect evidence suggests that the Hxs may help mediate the action of the neuropeptide FMRFamide to open the S-type K+ channels (Belardetti et al., 1989). In this study, S-type channels in inside-out membrane patches from Aplysia sensory neurones failed to respond to the Hx precursor, 12-HPETE. However, co-application of 12-HPETE with haematin, which catalyses Hx formation, produced a robust increase in S-type channel opening. This suggests that 12-HPETE may need to be converted into Hxs to enhance channel opening and that an Hx-generating system exists in the cytosol. Indeed, heme-proteins present in the cytosol are capable of converting 12-HPETE into the Hxs (Pace-Asciak, 1984b; 1985; Belardetti et al., 1989). Independently, Buttner et al. (1989) reported that outside-out patches were more responsive to 12-HPETE than inside-out patches, presumably due to the conversion into the active Hxs by the former preparation. Although the molecular identity of the S-type channels in Aplysia remains unknown, they appear to be closely related to the mammalian K2P channel, TREK1 (see review by Honore, 2007). Interestingly, TREK1 activation has been shown to play an important role in neuroprotection, anaesthesia, pain and depression. Are these actions of TREK1 ones that the native Hxs are associated with? If so, the PBT antagonists described herein could serve important functions as novel therapeutics in the central nervous system.
Effects in the mammalian brain
Both HxA3 and HxA3-C showed biological activity in the brain. In electrophysiological recordings from intracellular and whole-cell hippocampal CA1 neurones, HxA3 and HxA3-C displayed excitatory effects of lowering spike threshold and decreasing spike frequency adaptation and the inhibitory actions of membrane hyperpolarization, enhanced post-spike train after hyperpolarizations and increased inhibitory postsynaptic potentials or currents (Carlen et al., 1989; Pace-Asciak et al., 1990b). This appeared to be structure-specific as a synthetic analogue of HxA3-C, provided by Professor E. J. Corey, having the glutathione moiety at carbon 9 instead of carbon 11, was totally devoid of activity. The minimal threshold activity for HxA3-C was at 3 nmol·L−1.
HxA3 showed interesting possibilities in that it caused an enhancement of neurite regeneration in neurones after axotomy. Hence, HxA3 caused a remarkable potentiation of nerve growth factor-evoked neurite outgrowth in superior cervical ganglion neurones in vitro (Amer et al., 2003). Could this observation be useful in treating optic nerve damage as in glaucoma or in spinal cord damage?
Potentiation of vasoconstriction
The Hxs possessed vascular properties that potentiated the effects of vasoconstrictor hormones on smooth muscle. Thus, with a concentration of 10 nmol·L−1 HxA3 in the bath containing helicoidal strips of rat thoracic aorta, the EC50 for norepinephrine was reduced from 5 to 0.5 nmol·L−1. The Hx glutathione metabolite, 8S HxA3-C, was also effective, but the sensitivity was somewhat decreased (EC50 at 1 nmol·L−1 of norepinephrine). Interestingly, the responses were enantiomer-specific, with the 8S enantiomer of the Hx being active, while the 8R enantiomer was totally inactive at the same bath concentration of 10 nmol·L−1 (Laneuville et al., 1992a). The isolated portal vein was also stimulated by 10 nmol·L−1 of 8S HxA3 to contraction by norepinephrine. Here, the frequency and strength of contraction to norepinephrine were enhanced by the presence of 4.88 nmol·L−1 of the 8S enantiomer of HxA3 in the bath. The actions of the Hxs were dependent on the presence of calcium in the bath. Thus, the EC50 for norepinephrine changed from 13.3 ± 3.3 nmol·L−1 in the presence of calcium to 93.1 ± 6.2 nmol·L−1 in the absence of calcium in the medium. Importantly, the threshold dose of norepinephrine required to cause a contraction in the presence of HxA3 was 0.49 nmol·L−1 in calcium-containing medium and 8.3 nmol·L−1 in a calcium-free medium (Laneuville et al., 1992a). HxA3 and HxA3-C also potentiated the contraction of guinea pig isolated trachea to neurokinin A, although on their own, the Hxs had no effect. The maximal response to neurokinin A was also increased in the presence of the Hxs. The 8R enantiomer of HxA3 was active. But in contrast to the aorta and the portal vein studies described above, the guinea pig trachea responded only to the 8R enantiomer of the glutathione metabolite, HxA3-C (Laneuville et al., 1992b). Could different Hx receptors be involved?
A Hx receptor?
We have generated ample evidence for the presence of a binding protein in human neutrophils that is specific to the natural Hxs (Reynaud et al., 1995a,b; 1996;). The binding protein appears to be on the inside of the cell as the methyl ester of Hx binds but not the free acid form (Reynaud et al., 1995a). We demonstrated a high degree of binding with a single population of binding sites (Bmax 8.86 ± 1.4 pmol·mL−1 per 2 × 106 cells = 2.6 × 106 binding sites per cell) and a Kd of 79.9 nmol·L−1 (Reynaud et al., 1996). This binding was specific to Hx as other related eicosanoids ranging from 12-HETE through to leukotrienes and the prostaglandins were unable to compete for the binding (Reynaud et al., 1996). The binding was specific to the hydroxy-epoxide functionality of the Hx molecule, a central feature in its structure, although related compounds having only an epoxide group appeared to compete for Hx binding. Interestingly, this protein displayed different binding properties between neutrophils from normal and type 1 diabetic human volunteers (Kd 79.9 nmol·L−1 for normals; Kd 38.7 for type 1 diabetes; Kd 68.4 for type 2 diabetes). When analysed on 2D PAGE after photoaffinity covalent binding with a radioiodinated Hx analogue, the protein showed differences between normal and diabetic neutrophils. The protein from normal neutrophils appeared as a single radiolabelled spot while a triplet was seen with the type 1 diabetic protein (C.R. Pace-Asciak, unpubl. obs.). Is this protein involved in the mechanism of action of HxA3 (and the antagonism by PBTs) in causing release of intracellular calcium and inhibition of action of the inflammatory mediators mentioned in the previous sections?
The Hx analogues – PBTs
PBTs are Hx antagonists
The PBTs antagonize the actions of the natural Hxs. They inhibit the mobilization of intracellular calcium evoked by HxA3 and other calcium agonists in human neutrophil suspensions (Pace-Asciak et al., 1999), while on their own, they have marginal effects on calcium mobilization. The PBTs compete for the binding sites on human neutrophils occupied by tritiated HxA3 (see below).
Inhibition of platelet aggregation
Probably, the most remarkable activity of the PBTs lies in their ability to inhibit human platelet aggregation. This was totally unexpected when compared with other eicosanoids. For example, PGE2 and thromboxane A2 contain a five- and a strained four-member ring structure respectively, the former antagonizing aggregation of platelets while the latter being one of the most potent aggregating agents. The PBTs possess a stable three-member ring structure. It is also unexpected that one of the PBTs is far more active in opposing platelet aggregation than the others. Structurally, the distance between the carboxyl end of thromboxane A2 and the strained four-member ring is actually quite similar to the distance between the carboxyl end of PBT and the three-member ring.
PBT-3 was the most active of the series in this in vitro assay (Reynaud et al., 2001; Pace-Asciak et al., 2002), with the natural compounds being virtually inactive, possibly due to the rapid enzymatic hydrolysis of the latter compounds by epoxide hydrolases in the cell (Pace-Asciak et al., 1986a; Pace-Asciak and Lee, 1989). IC50 for PBT-3 = 0.06 µmol·L−1 versus aggregation by the thromboxane agonist, I-BOP, while its IC50 versus aggregation by collagen was 0.8 µmol·L−1 (Pace-Asciak et al., 2002). PBT-3 was much more effective as an antagonist to I-BOP binding in washed platelets. Hence, the IC50 against I-BOP binding was 8.1 nmol·L−1 (Pace-Asciak et al., 2002). Intravenous administration of PBT-3 (3 mg·kg−1 rat body weight) reduced thrombus formation induced by deposition of FeCl3 onto the abdominal aorta in pentobarbital anaesthetized rats; however at 1 mg·kg−1, the PBT was ineffective in this in vivo assay (J.M. Dogne and C.R. Pace-Asciak, unpubl. obs.).
Using an ex vivo model of bleeding time employing the PFA-100, PBT-3 was shown to inhibit primary platelet-related haemostasis in whole human blood. PBT-3 caused a significantly longer closure time only in membranes coated with collagen/epinephrine but not coated with collagen/ADP. In this study, PBT-3 was 250 times less active than the thromboxane receptor antagonist, SQ 29,548 and about 20 times less active than pinane thromboxane A2 (Reynaud et al., 2003). The lesser activity of PBT-3 may be related to its expected greater binding to plasma protein.
The actions of PBT-3 were mediated through binding to the platelet thromboxane receptor as PBT-3 inhibited aggregation evoked by the TP receptor agonists, I-BOP and U-46619 (Pace-Asciak et al., 2002). The possible selectivity of PBT-3 may be related to its selective binding to the TP receptor. This was further confirmed through binding studies of the TP receptor antagonist, 3H-SQ 29,548, in COS-7 cells that had been transfected with the α and the β isoforms of the TP receptor cDNA (Qiao et al., 2003a). These isoforms possess a different tail length, the α form being shorter than the β form. The TPα-isoform-transfected cells were selectively inhibited by PBT-3. It should be noted that PBT-3 is both a TP receptor antagonist and a thromboxane synthase (TS) inhibitor, although its actions on the receptor far exceed those as a TS inhibitor (IC50 for inhibition of I-BOP binding was 8 nmol·L−1 while that for TS inhibition was 400 nmol·L−1) (Qiao et al., 2003a).
The galactose ester of PBT-3, PBT-300 (see Figure 1B), was as active in inhibiting collagen-induced platelet aggregation as PBT-3 (methyl ester); however, other galactose-esterified PBTs (100, 200 and 400) were more potent than the methyl esters. Interestingly, all galactose esters were more potent than the corresponding galactose amides (C.R. Pace-Asciak and D. Reynaud, unpubl. obs.; see Figure 2). The galactose esters are also potent in antagonizing the HxA3-evoked intracellular calcium rise in human neutrophils in vitro. This probably relates to an expected decrease in the rate of hydrolysis of the galactose ester group in comparison with the methyl ester.
Figure 2.
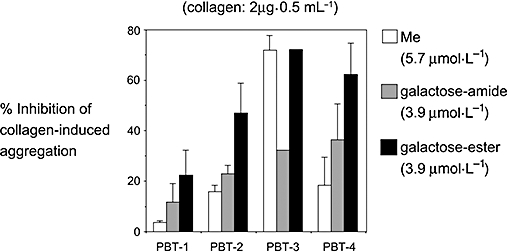
Comparison of the inhibition of collagen-induced human platelet aggregation by three analogues of the PBTs, the methyl ester (Me), the galactose amide and the galactose ester.
Cancer
The PBTs (as methyl esters) cause apoptosis of neoplastic cells in vitro. Thus, PBT-1 through to PBT-4 were approximately equally active in inhibiting the growth of the BCR-ABL-positive human chronic myelogenous leukaemic cell line, K-562, in vitro. Neither the natural Hxs nor thromboxane analogues had much effect at the doses used for the PBTs (Qiao et al., 2003b), so the actions of the PBTs in cancer are not mediated via antagonism of the TP receptor. Further actions of the PBTs in apoptosing neoplastic cells were observed in the breast cancer cell lines, MCF-7 and MDA MB 231, the prostate cancer cell line DU 145, and the cervical cancer cell line HeLa (C.R. Pace-Asciak and N. Qias, unpubl. obs.). Of all cell lines tested, the K562 line was the most responsive with an IC50 of 0.4 µmol·L−1 for PBT-3 as the methyl ester (Qiao et al., 2003b). The effects of PBTs compare favourably in doses with those of Gleevec (STI571) in vitro as well as in their characteristics of growth inhibition. While BCR-ABL opposes apoptosis through its ability to delay the release of mitochondrial cytochrome c (Amarante-Mendes et al., 1998), Gleevec opposes this effect, thereby causing the release of cytochrome c from the mitochondria into the cell cytosol. PBT-3 mimics the apoptotic effect of Gleevec in a time-dependent manner. Both Gleevec and PBT-3 cause the activation of caspase 3 (Qiao et al., 2003b).
An interesting observation showed that PBT-3 was active in causing apoptosis of K562 cells that were made resistant to Gleevec by long-term exposure of the cells to small amounts of Gleevec in the growth medium. These observations indicate that the mechanisms of action of PBT-3 may not be entirely related to those evoked by Gleevec (Qiao et al., 2007).
PBT-3 was advanced to xenograft animal studies in which K562 cells (1 × 10−7 cells) were transplanted into the left flank of 6-week-old female CD-1 nude mice (20 g body weight). Solid tumours were visible 2–3 weeks after subcutaneous injection of the cells. PBT-3 and Gleevec were administered in several groups of mice, saline being used as control in another group of mice. Results showed that PBT-3 at 300 µg per injection (twice daily for 8 days, intravenous and intra-tumour) was as effective as the same dose of Gleevec (Li et al., 2005a,b;), reducing tumour volume relative to the saline group by 84%. The tumours at the end of the study were analysed for apoptosis by DNA laddering as well as DNA fragmentation (TUNEL assay), showing that significant apoptosis had taken place in the PBT-3 and Gleevec groups. Additional studies in vivo indicated that PBT-3 was well tolerated and that inhibition of tumour growth was long-term, up to 50 days after the drug was stopped (after an 8 day period of administration) before tumours started to grow again (Li et al., 2005a). Additional studies demonstrated that if PBT-3 was administered a second time (another 8 day period) before tumours began to grow (i.e. around 40 days), an additional long period of tumour growth suppression was observed extending the start of tumour growth to approximately 100 days (C.R. Pace-Asciak unpubl. obs.). Although in vitro studies had shown that PBT-3 and PBT-4 actively apoptosed K562 cells that were made resistant to Gleevec (Qiao et al., 2007), combination of Gleevec with PBT-3 in vivo caused a further but small delay in growth of solid tumours (C.R. Pace-Asciak and X. Li, unpubl. obs.). The threshold concentration of PBT-3 that caused apoptosis of K562-induced tumours in vivo was 30 µg per animal, that is, 1.2 mg·kg−1 for a 25 g mouse; partial response was observed at 0.40 mg·kg−1, that is, 10 µg per animal twice daily for 8 days.
The PBTs were active in inhibiting growth in nude mice of solid tumours derived from subcutaneous implantation of the breast cancer cell lines, MBA MD 231 and MT-3. Their actions were similar to those of doxorubicin used as a positive control (C.R. Pace-Asciak and X. Li, unpubl. obs.).
Lung inflammation
The PBTs inhibit pulmonary fibrosis evoked by bleomycin (Jankov et al., 2002). Bleomycin is an antibiotic with potent cancer chemotherapeutic actions (Adamson, 1984; Nici et al., 1998); however, it may cause interstitial lung fibrosis in men (Yagoda et al., 1972). This observation has led to the development of an animal model in which a single intratracheal administration of bleomycin sulphate caused pulmonary changes that resembled histopathologically human idiopathic lung fibrosis (Kelley et al., 1980), where the acute phase is characterized by an accumulation of inflammatory cells and an increase in collagen synthesis and deposition (Cooper et al., 1988; Gurujeyalakshmi and Giri, 1995; Giri and Hollinger, 1996). In collaboration with Dr Keith Tanswell at the Hospital for Sick Children, we showed that PBTs (as the methyl esters) administered intraperitoneally inhibited the bleomycin-evoked effects dose-dependently in mice (Jankov et al., 2002). The best compound was PBT-1, with as little as 10 µg·day−1 (400 µg·kg−1) showing such pronounced effects. In the same animals, while bleomycin caused an increase in the synthesis and deposition of collagen, PBT-1 showed a reduction in the bleomycin-evoked changes. The best compound again was PBT-1. All four PBTs (PBT-1 to PBT-4) caused a decrease in the bleomycin-evoked accumulation of macrophages in the lung. Hence, PBT-1 especially, eliminated the incidence of bleomycin-evoked pulmonary effects including alveolar hemorrhage, macrophage infiltration and collagen deposition.
Vascular permeability
HxA3 increased the vascular permeability of rat skin in vivo (Laneuville et al., 1991; Jankov et al., 2002). This was observed after intradermal application of the Hx and measurement of the amount of Evans blue leakage at the site of application. The effect was time- and concentration-dependent (0–60 min, and 10–1000 ng·site−1). The threshold dose was 10 ng·site−1 of HxA3 at 5 min after administration (139% of control). Similar findings were observed with prostaglandin E2 (Laneuville et al., 1991; Jankov et al., 2002). The PBTs (100 ng·site−1) were found to inhibit plasma leakage in the rat skin that was evoked by 12 µg bleomycin sulphate intradermally (Jankov et al., 2002). PBT-1 and PBT-2 showed significant inhibition of the bleomycin effect at 30 min after application (Jankov et al., 2002).
Glucose control
The natural Hxs are formed and released by isolated perifused rat islets of Langerhans in vitro on which they act to release insulin (Pace-Asciak and Martin, 1984; Pace-Asciak et al., 1985; 1986b;). HxA3 is found in the circulation of the rat after intra-arterial administration of arachidonic acid, the fatty acid precursor (Pace-Asciak et al., 1987). In fact, total profiling of the prostaglandins and the Hxs in blood by mass spectrometry after arachidonic acid administration in the rat showed that the diabetic rat (BB) produced more thromboxane and Hxs than the normal rat. Water-soluble galactose amide analogues of PBTs (i.e. PBT-10, -20, -30, -40) (C.R. Pace-Asciak and P.M. Demin, unpubl. obs.) were prepared and tested on rat plasma glucose concentrations in vivo. At 100 µg bolus intra-arterial injection of each of the galactose amide derivatives, only PBT-10 gave a statistically significant reduction in plasma glucose (see Figure 3).
Figure 3.
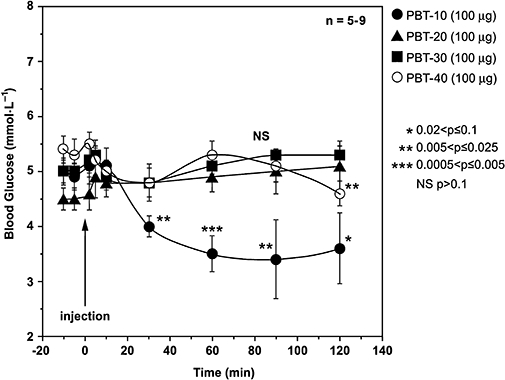
Effect of four hepoxilin galactose amide analogues on plasma glucose concentrations in the normal fed rat. Analogues were injected intra-arterially 2 h after surgery. Animals were anaesthetized with Inactin, and indwelling cannulas were inserted in the carotid artery and jugular veins for blood sampling and drug injection respectively. A 2 h period of stabilization after surgery was allowed prior to starting the experiment. Data are reported for five to nine animals per test. Statistics shown relate to corresponding values at the time of injection of drug. All animal studies had Animal Care Committee approval at our institution.
The threshold concentration for this hypoglycemic response was 50 µg bolus dose and the drop in glucose was dose-responsive up to 200 µg tested. When insulin concentrations were measured in these animals, a time-dependent decrease in plasma insulin was observed that paralleled the decrease in plasma glucose concentrations, indicating that the effect of PBT-10 was primarily on glucose concentration and not on insulin; the latter would have been expected to rise (not fall) to cause a decrease in blood glucose. In control animals, there was no decrease in insulin levels within the time period monitored (−10 min up to 1 h after injection of vehicle). In contrast, when natural HxA3 and HxB3 were administered, a small gradual rise in glucose and insulin was observed (C.R. Pace-Asciak, unpubl. obs.). It appears that the galactose amide derivative (PBT-10) may be active in vivo on its own rather than through its hydrolysis product, PBT-01, as preliminary studies on smooth muscle L6 cells in vitro, carried out in collaboration with Dr Amira Klip at our hospital, showed that the four compounds in the PBT 0 series (i.e. PBT-01, 02, 03, 04, all free acids) caused a dose-dependent uptake of tritiated 2-deoxyglucose in the cells. PBT-03, not PBT-01, was the most active in these experiments at 3 µmol·L−1 and this compared favourably in response to 100 nmol·L−1 insulin (A. Klip and C.R. Pace-Asciak, unpubl. obs.). Interestingly, the methyl esters were mostly inactive in the in vitro study (and the in vivo study), further confirming that the galactose amide derivative was the active species.
PBT-10 lowered plasma glucose when it was administered orally through a gastric tube, indicating that the compound is bioavailable through this route and is effective (C.R. Pace-Asciak and R. Aslam, unpubl. obs.). The potential advantages of PBT-10 as an oral hypoglycemic drug are that its parent compound PBT-01 (see Figure 1B), which is formed after hydrolysis of PBT-10 in vivo, possesses potent anti-inflammatory potential inhibiting bleomycin-induced lung fibrosis in mice and that it belongs to a class of compounds which appear to have neuro-generative actions (Amer, 2000). Hence, PBT-10 may serve multiple functions not only to manage glucose plasma levels independent of insulin, but also to control complications associated with diabetes, that is, to oppose neurodegeneration, thrombosis and inflammation. Animal studies have shown that the compound is well tolerated at the doses employed in this study (800 µg·kg−1). In animal toxicity studies, the lethal dose was not reached up to a PBT dose of 30 mg·kg−1.
Sulphur analogues of the Hxs
Replacing the epoxide functionality of HxA3 with sulphur rendered compounds that are also active in mobilizing intracellular calcium in human neutrophils. Their potency was similar to that of the natural Hxs (Demin et al., 1996). Due to the restriction in the availability of compounds, no other biological tests were carried out on these analogues.
Conclusions
This article reviews much of the published and unpublished studies carried out on the Hxs and related stable analogues, PBTs. The PBTs are antagonists of the natural Hxs, but also provide compounds that revealed many interesting biological properties when applied in vivo not seen or anticipated with the natural compounds; for example, these relate to: the efficacy of the PBTs in vivo, their anti-cancer effects (the natural Hxs have no effects), their anti-inflammatory actions (the natural Hxs are pro-inflammatory), their anti-thrombotic actions (the natural Hxs have no effects on platelet aggregation), their anti-diabetic actions (the natural compounds are insulin secretagogues). We have just skimmed the surface exploring the actions of this promising class of eicosanoids. Although current information points to the presence of a specific receptor for the Hxs that may be linked to G-proteins, little published information is available on the putative Hx binding protein. It is interesting to note that one specific compound in the family, PBT-3, is a potent anti-thrombotic agent that functions as a selective antagonist to the TPα isoform of the thromboxane receptor. Furthermore, stable galactose amide and ester analogues possess unexpected potent biological activities. Importantly, the PBTs appear to have a low toxicity as evidenced by lack of mortality even at the high dose of 30 mg·kg−1. This author feels that the studies to date point to the recognition that the base structure of the Hxs, especially modified and stabilized as the cyclopropyl analogues (PBT), possesses properties that allow good bioavailability and efficacy and possesses the successful framework for expansion into second-generation compounds with improved pharmaceutical properties. The present PBTs as described herein appear suitable for advancement into clinical trials.
Acknowledgments
The author wishes to acknowledge the financial support of the Canadian Institutes of Health, the Ontario Institute of Cancer Research and the Hospital for Sick Children. The postdoctoral involvement of Drs Peter Demin, Denis Reynaud, Na Qiao, Xiang Li, Alon Margalit and the collaboration of Drs Mohamed Abdelhaleem and Linda Mills is gratefully acknowledged for the various studies reported from my laboratory. I wish to thank Professor Steven Siegelbaum for his helpful suggestions to the section on potassium regulation in the Aplysia.
Glossary
Abbreviations:
- ABL
Abelson gene
- BCR
breakpoint cluster region
- CCCP
carbonyl cyanide m-chloro phenyl hydrazone
- CD-1
cluster of differentiation 1, a family of glycoproteins expressed on the surface of various human antigen-presenting cells
- COS-7
a cell line derived from the CV-1 cell line by transformation with a replication origin defective mutant of SV40 virus
- DU 145
a cell line established from the tumour tissue removed from the metastatic central nervous system lesion of a man with human prostate cancer
- fMLP
formyl methionyl leucyl phenylalanine
- FMRFamide
Phe-Met-Arg-Phe-NH2
- GppNHp
guanosine-5′-(βγ-imino) triphosphate
- HeLa
a cell line derived from cervical cancer cells taken from Henrietta Lacks
- K562
the first immortalized myelogenous leukaemia line that is bcr:abl-positive erythroleukaemia line derived from a female chronic myelogenous leukaemia patient in blast crisis
- HX or Hx
natural hepoxilins
- HxA3-C
11-glutathionyl-8,12-dihydroxy-5Z,9E,14Z-trienoic acid
- HPETE
hydroperoxy eicosatetraenoic acid
- I-BOP
a thromboxane A2 receptor agonist, [[1S-[1α,2α (Z),3β(1E,3S*),4α]]-7-[3-[3-hydroxy-4-(4-iodophenoxy)-1-butenyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid]
- K2P
potassium channels with two P domains (pore-forming regions)
- L6
a rat myogenic cell line derived from thigh muscle from newborn rats
- LTB4
leukotriene B4
- MCF-7
Michigan Centre Foundation, a cancer cell line derived from a pleural effusion from a woman with breast cancer
- MDA MB 231
M. D. Anderson Cancer Center, a cancer cell line derived from a pleural effusion from a woman with breast cancer
- MT-3
middle-T-antigen breast cancer cells
- PBT
stable hepoxilin analogues
- PEEC
pathogen-elicited epithelial chemoattractant
- PFA-100
platelet function analyser
- PMN
polymorphonuclear
- RVD
regulatory volume decrease
- SQ 29,548
[[1S-[1α,2α (Z),3α,4α]]-7-[3-[[2-[(phenylamino) carbonyl]hydrazino]methyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid]
- TCPO
3,3,3-trichloropropene-1, 2-oxide
- TP
thromboxane receptor protein
- TREK1
a two-pore domain potassium ion channel
- TrXA3
trihydroxy metabolite of HxA3, 8,11,12-trihydroxy-5Z,9E,14Z-trienoic acid
- TrXB3
trihydroxy metabolite of HxB3, 10,11,12-trihydroxy-5Z,8Z,14Z-trienoic acid
- TS
thromboxane synthase
References
- Adamson I. Drug-induced pulmonary fibrosis. Environ Health Perspect. 1984;55:25–36. doi: 10.1289/ehp.845525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarante-Mendes GP, Kim CN, Liu L, Huang U, Perkins C, Green DR, et al. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome c and activation of caspase-3. Blood. 1998;91:1700–1705. [PubMed] [Google Scholar]
- Amer R. Hepoxilins and neuronal repair: effects on SCG neurons after in vitro injury. MSc thesis, Department of Pharmacology, University of Toronto. [Google Scholar]
- Amer RK, Pace-Asciak CR, Mills LR. A lipoxygenase product, hepoxilin A3, enhances nerve growth factor-dependent neurite regeneration post-axotomy in rat superior cervical ganglion neurons in vitro. Neuroscience. 2003;116:935–946. doi: 10.1016/s0306-4522(02)00764-9. [DOI] [PubMed] [Google Scholar]
- Belardetti F, Campbell WB, Falck JR, Demontis G, Rosolowsky M. Products of heme-catalyzed transformation of the arachidonate derivative 12-HPETE open S-type K+ channels in Aplysia. Neuron. 1989;3:497–505. doi: 10.1016/0896-6273(89)90208-0. [DOI] [PubMed] [Google Scholar]
- Belosludtsev YY, Kollah RO, Falck JR, Capdevila JH. Hepoxilins B3: Synthesis of all four stereoisomers and a glutathione adduct. Tetrahedron Lett. 1994;35:5327–5330. [Google Scholar]
- Buttner N, Siegelbaum S, Volterra A. Direct modulation of Aplysia S-K+ channels by a 12-lipoxygenase metabolite of arachidonic acid. Nature. 1989;347:553–555. doi: 10.1038/342553a0. [DOI] [PubMed] [Google Scholar]
- Carlen PL, Gurevich N, Wu PH, Su W-G, Corey EJ, Pace-Asciak CR. Actions of arachidonic acid and hepoxilin A3 on mammalian CA1 neurons. Brain Res. 1989;497:171–176. doi: 10.1016/0006-8993(89)90984-0. [DOI] [PubMed] [Google Scholar]
- Chabert P, Mioskowski C, Falck JR. Synthesis of (±)-hepoxilin A3 utilizing arsonium ylides. Tetrahedron Lett. 1989;30:2545–2548. [Google Scholar]
- Cooper J, Zitnik R, Matthay R. Mechanisms of drug-induced pulmonary disease. Ann Rev Med. 1988;39:395–404. doi: 10.1146/annurev.me.39.020188.002143. [DOI] [PubMed] [Google Scholar]
- Corey EJ, Su W-G. 8R)- and (8S)-Hepoxilin A3. Assignment of configuration and conversion to biologically active conjugates with glutathione. Tetrahedron Lett. 1990;31:2113–2116. [Google Scholar]
- Demin P, Reynaud D, Pace-Asciak C. Chemical synthesis and actions of 11,12-thiirano-hepoxilin A3. J Lipid Med Cell Signalling. 1996;13:63–72. doi: 10.1016/0929-7855(95)00045-3. [DOI] [PubMed] [Google Scholar]
- Demin PM, Pace-Asciak CR. Synthesis of racemic 11,12-cyclopropyl analogs of hepoxilins A3 and B3. Tetrahedron Lett. 1993;34:4305–4308. [Google Scholar]
- Demin PM, Vasiljeva LL, Belosludtsev YY, Myagkova GI, Pivnitsky KK. Hepoxilins A3 configuration and their synthesis by means of hepoxilins B3 isomerization. Bioorg Khim. 1990;16:571–572. [Google Scholar]
- Demin PM, Vasiljeva LL, Myagkova GI, Pivnistsky KK. Synthetic research of hepoxilins. 2. Hepoxilins A3: synthesis, configuration and composition of the hydrolysis products – trioxilins A3. Bioorg Khim. 1991;17:1133–1140. [Google Scholar]
- Dho S, Grinstein S, Corey EJ, Su WG, Pace-Asciak CR. Hepoxilin A3 induces changes in cytosolic calcium, intracellular pH and membrane potential in human neutrophils. Biochem J. 1990;266:63–68. doi: 10.1042/bj2660063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S, Hollinger M. Effect of nordihydroguaiaretic acid and ibuprofen on bleomycin and hyperoxia-induced changes in lung superoxide dismutase, prostaglandins and lethality. Arch Toxicol. 1996;70:271–276. doi: 10.1007/s002040050273. [DOI] [PubMed] [Google Scholar]
- Gurujeyalakshmi G, Giri S. Molecular mechanisms of anti-fibrotic effect of interferon-gamma in Bleomycin-mouse model of lung fibrosis: downregulation of TGF-beta and procollagen I and III gene expression. Exp Lung Res. 1995;21:791–808. doi: 10.3109/01902149509050842. [DOI] [PubMed] [Google Scholar]
- Honore E. The neuronal background K2P channels: focus on TREK1. Nat Rev Neurosci. 2007;8:251–261. doi: 10.1038/nrn2117. [DOI] [PubMed] [Google Scholar]
- Jankov RP, Luo X, Demin P, Aslam R, Hannam V, Tanswell AK, et al. Hepoxilin analogs inhibit bleomycininduced pulmonary fibrosis in the mouse. J Pharm Exp Ther. 2002;301:435–440. doi: 10.1124/jpet.301.2.435. [DOI] [PubMed] [Google Scholar]
- Kelley J, Newman R, Evans J. Bleomycin-induced pulmonary fibrosis in the rat. J Lab Clin Med. 1980;96:954–964. [PubMed] [Google Scholar]
- Laneuville O, Chang M, Reddy CC, Corey EJ, Pace-Asciak CR. Isozyme specificity in the conversion of hepoxilin A3 (HxA3) into a glutathionyl hepoxilin (HxA3-C) by the Yb2 subunit of rat liver glutathione S-transferase. J Biol Chem. 1990;265:21415–21418. [PubMed] [Google Scholar]
- Laneuville O, Corey EJ, Pace-Asciak CR. Hepoxilin A3 increases vascular permeability in the rat skin. Eicosanoids. 1991;4:95–97. [PubMed] [Google Scholar]
- Laneuville O, Couture R, Pace-Asciak CR. Hepoxilins sensitize blood vessels to noradrenaline – stereospecificity of action. Br J Pharmacol. 1992a;105:297–304. doi: 10.1111/j.1476-5381.1992.tb14249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laneuville O, Couture R, Pace-Asciak CR. Neurokinin A-induced contraction of guinea-pig isolated trachea: potentiation by hepoxilins. Br J Pharmacol. 1992b;107:808–812. doi: 10.1111/j.1476-5381.1992.tb14528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laneuville O, Reynaud D, Grinstein S, Nigam S, Pace-Asciak CR. Hepoxilin A3 inhibits the rise in free intracellular calcium evoked by formyl-methionyl-leucylphenylalanine, platelet-activating factor and leukotriene B4. Biochem J. 1993;295:393–397. doi: 10.1042/bj2950393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Qiao N, Reynaud D, Abdelhaleem M, Pace-Asciak CR. PBT-3, a hepoxilin stable analog, causes long term inhibition of growth of K562 solid tumors in vivo. Biochem Biophys Res Commun. 2005a;338:158–160. doi: 10.1016/j.bbrc.2005.07.180. [DOI] [PubMed] [Google Scholar]
- Li X, Qiao N, Reynaud D, Abdelhaleem M, Pace-Asciak CR. The hepoxilin analog, PBT-3, inhibits growth of K-562 CML solid tumors in vivo in nude mice. IN VIVO. 2005b;19:185–190. [PubMed] [Google Scholar]
- Livne A, Grinstein S, Rothstein A. Volume-regulating behaviour of human platelets. J Cell Physiol. 1987;131:354–363. doi: 10.1002/jcp.1041310307. [DOI] [PubMed] [Google Scholar]
- Lumin S, Falck JR, Capdevila J, Karara A. Palladium mediated allylic Mitsunobu displacement: Stereocontrolled synthesis of hepoxilin A3 and trioxilin A3 methyl esters. Tetrahedron Lett. 1992;33:2091–2094. [Google Scholar]
- Margalit A, Sofer Y, Grossman S, Reynaud D, Pace-Asciak CR, Livne A. Hepoxilin A3 is the endogenous lipid mediator opposing hypotonic swelling of intact human platelets. Proc Natl Acad Sci USA. 1993;90:2589–2592. doi: 10.1073/pnas.90.7.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills L, Reynaud D, Pace-Asciak CR. Hepoxilin-evoked intracellular reor ganization of calcium in human neutrophils: a confocal microscopy study. Exp Cell Res. 1997;230:337–341. doi: 10.1006/excr.1996.3425. [DOI] [PubMed] [Google Scholar]
- Mrsny RJ, Gewirtz AT, Siccardi D, Savidge T, Hurley BP, Madara JL, et al. Identification of hepoxilin A3 in inflammatory events: A required role in neutrophil migration across intestinal epithelia. Proc Natl Acad Sci (USA) 2004;101:7421–7426. doi: 10.1073/pnas.0400832101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nici L, Santos-Moore A, Kuhn C, Calabresi P. 1998). Modulation of bleomycin-induced pulmonary toxicity in the hamster by the antioxidant amifostine. Cancer. 83:2008–2014. [PubMed] [Google Scholar]
- Pace-Asciak CR. Arachidonic acid epoxides. Demonstration through oxygen-18 labeled oxygen gas studies of an intramolecular transfer of the terminal hydroxyl group of 12S-hydroperoxy-eicosa-5, 8, 10, 14-tetraenoic acid to form hydroxy epoxides. J Biol Chem. 1984a;259:8332–8337. [PubMed] [Google Scholar]
- Pace-Asciak CR. Hemoglobin-and hemin-catalyzed transformation of 12L-hydroperoxy-5, 8, 10, 14-eicosatetraenoic acid. Biochim Biophys Acta. 1984b;793:485–488. doi: 10.1016/0005-2760(84)90267-4. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak CR. Hematin-assisted intramolecular oxygen transfer mechanism is involved in the formation of 8-hydroxy-11, 12-epoxyeicosa-5,9,14-trienoic acid (8H-11, 12-EPETE) from 12-HPETE. In: Bailey JM, editor. Prostaglandins, Leukotrienes and Lipoxins. New York: Plenum Press; 1985. pp. 111–116. [Google Scholar]
- Pace-Asciak CR, Martin JM. Hepoxilin, a new family of insulin secretagogues formed by intact rat pancreatic islets. Prostagl Leukotriene Med. 1984;16:173–180. doi: 10.1016/0262-1746(84)90069-6. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak CR, Lee W-S. Purification of hepoxilin epoxide hydrolase from rat liver. J Biol Chem. 1989;264:9310–9313. [PubMed] [Google Scholar]
- Pace-Asciak CR, Martin JM, Corey EJ, Su W-G. Endogenous release of hepoxilin A3 from isolated perifused pancreatic islets of Langerhans. Biochem Biophys Res Commun. 1985;128:942–946. doi: 10.1016/0006-291x(85)90137-8. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak CR, Klein J, Spielberg SP. Epoxide hydratase assay in human platelets using hepoxilin A3 as a lipid substrate. Biochim Biophys Acta. 1986;875:406–409. doi: 10.1016/0005-2760(86)90193-1. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak CR, Martin JM, Corey EJ. Hepoxilins, potential endogenous mediators of insulin release. Prog Lipid Res. 1986;25:625–628. doi: 10.1016/0163-7827(86)90127-x. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak CR, Lee SP, Martin JM. In vivo formation of hepoxilin A3 in the rat. Biochem Biophys Res Commun. 1987;147:881–884. doi: 10.1016/s0006-291x(87)80152-3. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak CR, Martin JM, Lee S-P. Appearance of prostaglandins, thromboxane B2 and hepoxilin A3 in the circulation of the normal and diabetic (BB) rat after arachidonic acid administration – correlation with plasma insulin. Biochem Cell Biol. 1988;66:901–909. doi: 10.1139/o88-102. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak CR, Laneuville O, Gurevich N, Wu P, Carlen PL, Su W-G, et al. Novel hepoxilin glutathione conjugates with biological activity. In: Reddy CC, Hamilton GA, Kadyastha KM, editors. Biological Oxidation Systems 2. New York: Academic Press; 1990. pp. 725–735. [Google Scholar]
- Pace-Asciak CR, Laneuville O, Su W-G, Corey EJ, Gurevich N, Wu P, et al. A glutathione conjugate of hepoxilin A3: formation and action in the rat central nervous system. Proc Natl Acad Sci (USA) 1990;87:3037–3041. doi: 10.1073/pnas.87.8.3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace-Asciak CR, Wong L, Corey EJ. Hepoxilin A3 blocks the release of norepinephrine from rat hippocampal slices. Biochem Biophys Res Commun. 1990;173:949–953. doi: 10.1016/s0006-291x(05)80877-0. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak CR, Reynaud D, Demin PM. Hepoxilins: a review on their enzymatic formation, metabolism and chemical synthesis. Lipids. 1995;30:107–114. doi: 10.1007/BF02538262. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak CR, Reynaud D. The Hepoxilins – A Review. Adv Exp Med Biol. 1999;447 Nigam, S., and Pace-Asciak, C.R., Eds.), Kluwer Academic/Plenum, New York.: 123–132. [PubMed] [Google Scholar]
- Pace-Asciak CR, Reynaud D, Demin P, Aslam R, Sun A. A new family of thromboxane receptor antagonists with secondary thromboxane synthase inhibition. J Pharmacol Exper Ther. 2002;301:618–624. doi: 10.1124/jpet.301.2.618. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Shapiro E, Feinmark SJ, Schwartz JH. Metabolites of arachidonic acid in the nervous system of Aplysia: possible mediators of synaptic modulation. J Neurosci. 1987a;7:3675–3686. doi: 10.1523/JNEUROSCI.07-11-03675.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli DA, Volterra A, Dale N, Siegelbaum SA, Kandel E, Schwartz JH, et al. Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory neurons. Nature. 1987b;328:38–43. doi: 10.1038/328038a0. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Shapiro E, Zipkin R, Schwartz JH, Feinmark SJ. Formation and action of 8-hydroxy-11,12-epoxy-5,9,14-eicosatrienoic acid in Aplysia: a possible second messenger in neurons. Proc Natl Acad Sci USA. 1989;86:1721–1725. doi: 10.1073/pnas.86.5.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivnitsky KK, Vasiljeva LL, Melnikova VI. Synthesis of hepoxilins and trioxilins. 207th ACS National Meeting.
- Qiao N, Reynaud D, Demin P, Halushka PV, Pace-Asciak CR. The thromboxane receptor antagonist PBT-3, a hepoxilin stable analog, selectively antagonizes the TPα isoform in transfected COS-7 cells. J Pharmacol Exper Therap. 2003a;307:1142–1147. doi: 10.1124/jpet.103.056705. [DOI] [PubMed] [Google Scholar]
- Qiao N, Lam J, Reynaud D, Abdelhaleem M, Pace-Asciak CR. The hepoxilin analog PBT-3 induces apoptosis in BCR-ABL-positive K562 leukemia cells. Anticancer Res. 2003b;23:3617–3622. [PubMed] [Google Scholar]
- Qiao N, Reynaud D, Abdelhaleem M, Pace-Asciak C. Hepoxilin analogs, PBT-3 and PBT-4, cause apoptosis of Gleevec-resistant K562 cells in vitro. IN VIVO. 2007;21:267–271. [PubMed] [Google Scholar]
- Reynaud D, Demin P, Pace-Asciak CR. Hepoxilin A3 formation in the rat pineal gland selectively utilises 12(S)-HPETE but not 12(R)-HPETE. J Biol Chem. 1994;269:23976–23980. [PubMed] [Google Scholar]
- Reynaud D, Demin P, Pace-Asciak CR. Coupling of hepoxilin A3-specific binding with calcium-mobilizing actions in human neutrophils. In: Schrör K, Pace-Asciak CR, editors. Mediators in the Cardiovascular System – Regional Ischemia. Agents and Actions Supplements. Basel: Birkhauser Verlag; 1995a. pp. 291–296. [DOI] [PubMed] [Google Scholar]
- Reynaud D, Demin PM, Pace-Asciak CR. Hepoxilin binding in human neutrophils. Biochem Biophys Res Comm. 1995b;207:191–194. doi: 10.1006/bbrc.1995.1171. [DOI] [PubMed] [Google Scholar]
- Reynaud D, Demin P, Demin P, Pace-Asciak CR. Hepoxilin A3-specific binding in human neutrophils. Biochem J. 1996;313:537–541. doi: 10.1042/bj3130537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud D, Sun A, Demin PM, Pace-Asciak CR. Novel platelet antiaggregating substances. Biochem Biophys Res Commun. 2001;284:580–582. doi: 10.1006/bbrc.2001.5012. [DOI] [PubMed] [Google Scholar]
- Samuelsson B. The Leukotrienes: an Introduction. In: Samuelsson B, Paoletti R, editors. Leukotrienes and Other Lipoxygenase Products. New York: Raven Press; 1982. pp. 1–17. [Google Scholar]
- Sarkadi B, Parker J. Activation of ion transport pathways by changes in cell volume. Biochim Biophys Acta. 1991;1071:407–427. doi: 10.1016/0304-4157(91)90005-h. [DOI] [PubMed] [Google Scholar]
- Sutherland M, Schewe T, Nigam S. Biological actions of the free acid of hepoxilin A3 on human neutrophils. Biochem Pharmacol. 2000;59:435–440. doi: 10.1016/s0006-2952(99)00345-7. [DOI] [PubMed] [Google Scholar]
- Vasiljeva LL, Manukina TA, Demin PM, Lapitskaja MA, Pivnitsky KK. Synthesis, properties, and identification of epimeric hepoxilins (-)-(10R)-B3 and (+)-(10S)-B3. Tetrahedron. 1993;49:4099–4106. [Google Scholar]
- Vasiljeva LL, Pivnitsky KK. Synthesis of trioxilins B3 from hepoxilins B3. Mendeleev Commun. 1996;6:249–251. [Google Scholar]
- Wu WL, Wu YL. Synthesis of (10R)-hepoxilin B3 methyl ester and (10R)-trioxilin B3 methyl ester. J Chem Soc Perkin Trans I. 1992;1992:2705–2707. [Google Scholar]
- Wu WL, Wu YL. Formal syntheses of hepoxilin B3, trioxilin B3 and substances against rice blast disease from D-mannitol. J Chem Soc Chem Commun. 1993;1993:821–822. [Google Scholar]
- Yagoda A, Mukherji B, Young C, Etcubanas E, Lamonte C, Smith JR, et al. Bleomycin: an antitumour antibiotic: clinical experience in 274 patients. Ann Intern Med. 1972;77:861–865. doi: 10.7326/0003-4819-77-6-861. [DOI] [PubMed] [Google Scholar]