

Abstract
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors that belong to the nuclear hormone receptor superfamily. PPARγ regulates several metabolic pathways by binding to sequence-specific PPAR response elements in the promoter region of genes involved in lipid biosynthesis and glucose metabolism. However, more recently PPARγ, PPARα and PPARβ/δ agonists have been demonstrated to exhibit anti-inflammatory and immunomodulatory properties thus opening up new avenues for research. The actions of PPARγ and PPARα activation are thought to be due to their ability to down regulate pro-inflammatory gene expression and inflammatory cell functions, and as such makes them an attractive target for novel drug intervention. Interestingly, PPARβ/δ has been shown to be involved in wound healing, angiogenesis, lipid metabolism and thrombosis. In this review we will focus on the data describing the beneficial effects of these ligands in the airway and in the pulmonary vasculature and in vivo in animal models of allergic and occupational asthma, chronic obstructive pulmonary disease and pulmonary fibrosis. A clinical trial is underway to examine the effect of rosiglitazone in asthma patients and the outcome of this trial is awaited with much anticipation. In conclusion, PPARs are novel targets for lung disease and continued work with these ligands may result in a potential new treatment for chronic inflammatory lung diseases.
This article is part of a themed issue on Mediators and Receptors in the Resolution of Inflammation. To view this issue visit http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Keywords: peroxisome proliferator-activated receptor; asthma; COPD; inflammation; fibrosis; 15-deoxy-Δ12,14-PGJ2 (15-PGJ2)
Introduction
Inflammatory diseases of the lung such as asthma and chronic obstructive pulmonary disease (COPD) represent a major worldwide health problem. And while there are potent anti-inflammatory drugs available to treat asthma, such as the glucocorticoids, these drugs suffer from unwanted side effects and exhibit limited efficacy in the treatment of COPD. Consequently, the search for novel drug targets leading to new therapies for these diseases is ongoing (Belvisi et al., 2004). The suggestion that peroxisome proliferator-activated receptors (PPARs) may possess potent immunomodulatory and anti-inflammatory activity (Serhan, 1996; Serhan & Devchand, 2001) has led to increased interest in these receptors and to the study of their involvement in a variety of disease states including type 2 diabetes, atherosclerosis, inflammatory bowel disease, arthritis, myocarditis, cancer and endotoxic shock (Spears et al., 2006).
PPARs
Peroxisome proliferator-activated receptors are a family of ligand-activated transcription factors belonging to the nuclear hormone receptor family and related to retinoid, glucocorticoid, and thyroid hormone receptors (Evans, 1988). The three recognized subtypes, PPARα (also known as NR1C3), γ (NR1C1) and δ (β or NR1C2), are widely expressed and have a wide range of effects on metabolism, cellular proliferation and immune responses (Berger et al., 2005; Kota et al., 2005).
All members of this superfamily have a similar structural organization: an amino-terminal region that allows ligand-independent activation, a DNA-binding domain and a ligand-dependent activation domain. Three alternative promoters have been identified for PPARγ so far, giving rise to at least four different transcripts and two different protein isoforms, γ1 and γ2, which differ in their amino-terminal, with γ2 carrying 30 additional amino acids. PPARγ2 is expressed exclusively in adipose tissue whereas PPARγ1 is more widely expressed, although it is most abundant in adipocytes (Moras and Gronemeyer, 1998).
Peroxisome proliferator-activated receptors were first identified for their role in lipid and glucose regulation and until recently their actions were thought to be limited to specific tissue types. PPARα is highly expressed in tissues exhibiting high carbolic rates of fatty acids such as the liver, heart, kidney and intestinal mucosa. PPARγ is also expressed in lung epithelium, submucosa and airway smooth muscle. PPARβ/δ is ubiquitously expressed and was initially shown to play a role in regulating energy homeostasis, thermogenesis, keratinocyte proliferation and differentiation (Braissant et al., 1996).
Recently, two of the PPARs, PPARγ and PPARα, have been identified as important immunomodulators and to have potential as novel anti-inflammatory targets for diseases of the airways (Belvisi et al., 2004; 2006; Becker et al., 2006). PPARβ/δ was not thought to possess these properties (Trifillieff et al., 2003) but recent evidence suggests it may have a role to play in regulating the transition from inflammation to wound healing and may enhance the anti-fibrotic actions of PPARγ agonists (Lakatos et al., 2007). Furthermore, the PPARβ/δ ligand GW0742 was recently shown to inhibit fibroblast proliferation consistent with a proposed anti-fibrotic role for this receptor (Ali et al., 2006b). Most recently, work from our group has revealed acute inhibitory effects of PPARβ/δ agonists in platelets (Ali et al., 2006a) and in blood vessels (Reed et al., 2007). These acute effects were seen within seconds or minutes of PPARβ/δ agonists being added to tissues and as such must be mediated independently of gene induction. The potential mechanism by which PPARβ/δ agonists affect inflammation dependently or independently of gene induction are illustrated on Figure 1 and discussed below.
Figure 1.
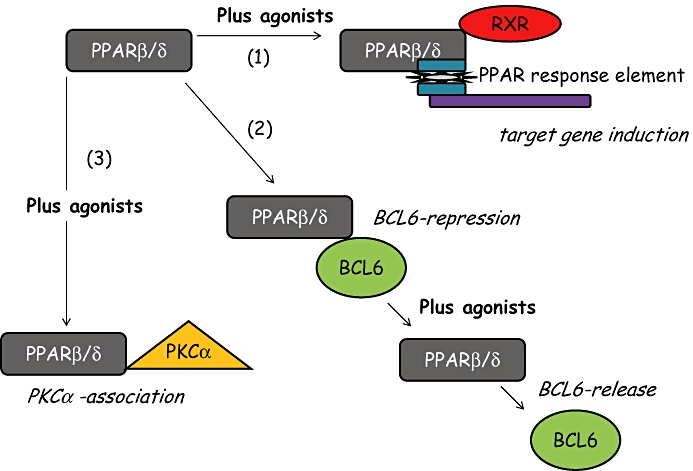
Pathways by which PPARβ/δ can influence inflammatory pathways. (1) After binding to its ligand PPARβ/δ can affect classic PPAR genomic responses by binding to RXR and the PPAR response element leading to the induction or repression of target genes. (2) In its inactivated state PPARβ/δ can bind and repress the transcription factor BCL6. BCL6 is displaced and released in the presence of PPARβ/δ ligands. BCL6 is then free to influence gene induction. (3) Following activation of PPARβ/δ with a specific ligand it can bind and repress PKCα, without the involvement of RXR. These properties of PPARβ/δ happens within seconds and may contribute to the acute actions of ligands in platelets and vessels. PPAR, peroxisome proliferator-activated receptor.
PPARs: mechanism of action
The anti-inflammatory actions of PPARγ and PPARα observed in cell-based assays and animal models are thought to be mainly due to their ability to regulate inflammatory gene expression. PPARs, including PPARβ/δ, regulate gene expression after binding as a heterodimer with the retinoid X receptors (RXRs), a member of the nuclear hormone receptor superfamily activated by binding of 9-cis-retinoic acid. The RXR family comprises three different gene isoforms: RXRα, RXRβ and RXRγ. RXR is widely expressed in several tissues and cells including adipose tissue, liver, kidneys, small intestine, cardiac myocytes and monocytes/macrophages (Dubuquoy et al., 2002).
The PPAR/RXR heterodimer binds to sequence-specific PPAR response elements in the promoter region of target genes and acts as a transcriptional regulator. To prevent PPAR/RXR binding to DNA, high-affinity complexes are formed between the inactive PPARγ/RXR heterodimers and co-repressor molecules, such as nuclear receptor co-repressor or silencing mediator for retinoic receptors. On ligand binding and activation, these co-repressors are displaced and the heterodimer is free to bind to the response element in the promoter region of the relevant target genes, resulting in either activation or suppression of a specific gene. Recruitment of co-activator proteins along with chromatin remodelling proteins is also required for transcriptional interaction of PPAR with motifs in the PPAR response elements (Desvergne and Wahli, 1999). Therefore, to summarize, the actions of PPARγ and PPARα are thought to be mainly due to their ability to down regulate pro-inflammatory gene expression either by: (i) sequestration of shared co-activators such that competition for co-activators would reduce the ability of inflammatory transcription factors to access their target DNA; (ii) ligand-dependent transrepression which does not involve sequence-specific DNA binding and involves the physical interaction of PPARγ ligands with other transcription factors (e.g. NF-κB, STAT, NFAT) preventing their association with DNA sequences; (iii) SUMOylation of PPARγ and subsequent PPARγ binding to the NCoR-containing corepressor complex interferes with the removal of repressor complexes and hence suppresses inflammatory gene transcription (Ghisletti et al., 2007; Straus and Glass, 2007).
The other PPAR isoforms also induce ligand-dependent transrepression but the mechanisms involved have not been fully elucidated. However, direct interaction of PPARα with the p65 subunit of NF-κB suggests that transrepression of pro-inflammatory genes may occur via this mechanism (Delerive et al., 1999). Furthermore, for PPARβ/δ another mechanism of transrepression has been identified that involves the binding of the transcriptional repressor BCL-6 by unliganded PPARβ/δ. Binding of ligand to PPARβ/δ then releases BCL-6 which then is free to repress inflammatory genes (Lee et al., 2003). It should also be noted that PPARβ ligands may have anti-inflammatory actions via gene induction (in addition to BCL6), for example by induction of anti-inflammatory cytokines and anti-oxidant enzymes. Interestingly, one of the early proposals regarding the mechanism of action of PPARα ligands was that they were anti-inflammatory by affecting lipid mediator metabolism through regulating oxidative degradation of fatty acids (e.g. Leukotiene B4) (Devchand et al., 1996).
Finally, the mechanisms that mediate the acute inhibitory effects of PPARβ/δ agonists, which are clearly mediated independently of any involvement of the nucleus, remain somewhat of a mystery. Nevertheless, unpublished observations from our group suggest that these may be mediated by the binding and repression of PKCα (Ali et al., unpublished observations). While this pathway remains speculative in the case of PPARβ/δ, others have shown that PPARγ receptors bind and repress PKCα in human nucleated cells (Paumelle et al., 2006; von Knethen et al., 2007). The full impact of this and other acute, none gene mediated effects of PPARβ/δ remain the subject of investigation.
Ligands for PPAR receptors
PPARγ
The interest in PPARs led to the identification of PPARγ as a target for intervention and thus more is known about this receptor and more tools are available to study it. PPARγ is bound and activated by a variety of lipophilic ligands, including long-chain polyunsaturated fatty acids and several eicosanoids. The essential fatty acids arachidonic acid, gamolenic acid, docosahexanoic acid and eicosapentaenoic acid, as well as modified oxidized lipids 9- and 13-hydroxyoctadecadienoic acid and 12- and 15-hydroxyeicosatetraenoic acid, bind to and activate PPARγ (Willson et al., 2000). 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2) has been recognized as an endogenous ligand for PPARγ, although it is now known to activate all the PPAR receptors, and is thought to be responsible for many of its anti-inflammatory actions (Sher and Pillinger, 2005).
The cyclopentenone prostaglandin 15d-PGJ2 was first discovered in 1983 (Fitzpatrick and Wynalda, 1983) but received relatively little attention until two independent groups reported that it was capable of activating PPARγ (Forman et al., 1995; Kliewer et al., 1995). Accumulating evidence now suggests that 15d-PGJ2 is the endogenous PPARγ ligand (Sher and Pillinger, 2005). However, what goes against prostanoids (e.g. 15d-PGJ2, prosacyclin) being endogenous PPAR agonists is the fact that NSAIDs, which would globally reduce all the prostanoids, do not have dramatic metabolic side effects, as one might expect if effecting endogenous PPAR pathways. Furthermore, in vivo it has been hard to demonstrate endogenous 15-dPGJ2 activity as it is very difficult to get accurate measurements making it difficult to confirm its role as an endogenous ligand.
15d-PGJ2 and other PPARγ ligands have been shown to possess anti-inflammatory activity in a wide range of inflammatory disease models and recently 15d-PGJ2 has been shown to significantly limit lung injury in an animal model of pulmonary fibrosis, a disease characterized by inflammatory cell infiltration (Genovese et al., 2005a). A reduction in neutrophil influx, oedema, histological parameters and mortality were observed thus supporting the anti-inflammatory potential of PPARγ ligands.
15d-PGJ2 has been widely used as a pharmacological tool for defining the role of PPARγ and it is important to note that it can induce a variety of PPARγ-independent responses, and indeed a recent study demonstrated that 15d-PGJ2 exerts its anti-inflammatory effect in rat chondrocytes by a PPARγ-independent mechanism, which could be attributed to a partial inhibition of inhibitor κBα degradation (Boyault et al., 2004). Other PPARγ-independent effects include inhibition of IκB kinase and inhibition of NF-κB DNA binding (Straus and Glass, 2007). 15d-PGJ2 has also been shown to induce responses in cells devoid of the receptor.
In addition to natural ligands, a wide range of synthetic PPARγ agonists have been developed. The most widely used belong to the thiazolidinedione or glitazone class of anti-diabetic drugs used in the treatment of type 2 diabetes (Yki-Jarvinen, 2004). These include rosiglitazone, pioglitazone, ciglitazone and troglitazone. The first to be developed, troglitazone, has since been withdrawn from the market following the emergence of a serious hepatotoxicity in some patients. The two PPARγ agonists currently available for the treatment of type 2 diabetes in the United States are rosiglitazone and pioglitazone.
Thiazolidinediones exert their insulin-sensitizing and hypoglycaemic effects through stimulation of PPARγ (Berger et al., 1996). The involvement of PPARγ in the pharmacological effects of thiazolidinediones has been supported by studies showing that their binding affinity to PPARγ closely parallels their in vivo hypoglycaemic potency (Willson et al., 2000). In the last few years, there have been numerous reports indicating that the therapeutic benefits of PPARγ agonists may go far beyond their use in diabetes with increasing evidence of anti-inflammatory activities in a range of disease models from Alzheimer's to pancreatitis and evidence is now emerging of the potential benefits of PPARγ ligands in models of inflammatory airways disease. See Table 1 for a comprehensive list (adapted from Michalik et al., 2006).
Table 1.
Ligands for PPARγ receptors
| Endogenous agonists | 15d-PGJ2, 15-hydroxyeicosatetraenoic acid (15-HETE) and 13-hydroxyoctadecadienoic acid (13-HODE) |
| Synthetic agonists | SB-219994 (8.68), LY-510929 (8), AD-5061 (7.7), TZD18 (7.24), L-764406 (7.15), ragaglitazar (7.03), GW0072 (6.96), nTzDpa (6.5), troglitazone (6.27), LY-465608 (6.26), pioglitazone (6.23), fatty acids (6), SB-219993 (5.5), 5-ASA (1.82) [pIC50]. GW1929 (8.84), L-796449 (8.7), GW7845 (8.43), CDDO (8), L-783483 (7.85), L-165461 (7.8), AD5075 (7.66), FMOC-L-leucine (∼6), CS-045 (5.8) [pKi], farglitazar (7.47), indomethacin (7.38), rosiglitazone (7.37), GW2331 (6.52), KRP-297/MK-0767 (6.49), PAT5A (6.35), MCC555 (∼6.3), Iinoleic acid (5.3), BADGE (4) [pKd], GW409544 (9.55), GW9578 (6) BVT0.13 (7.52), TAK-559 (7.5), reglitazar (7.08), GW9578 (6), ciglitazone (4.64), KRP-297/MK-0767 (7) [pEC50]; DRF2519, LG10074, ibuprofen, diclofenac |
| Antagonists | GW9662 (8.48), PD068235 (6.1), BADGE (5), SR-202 (3.85) [pIC50]; CDDO-Me (8), LG100641 (6.36) [pKi]; diclofenac |
PPAR, peroxisome proliferator-activated receptor.
PPARa
A variety of endogenous ligands also activate PPARα including endogenous fatty acids, like the 8-hydroxyeicosatetraenoic acid (8S-HETE) and the arachadonic acid derivative leukotriene B4 (LTB4). Synthetic molecules have also been developed including Wy-14,643 and GW2331 and the fibrates that are used clinically to treat dyslipidaemia (e.g. fenofibrate, ciprofibrate). See Table 2 (adapted from Michalik et al., 2006). In addition to the selective ligands there are also dual PPARα/γ ligands including ragaglitazar, GW-409544 and KRP-297.
Table 2.
Ligands for PPARα receptors
| Endogenous agonists | 8-HETE, LTB4 |
| Synthetic agonists | GW409544 (8.7), LY-518674 (7.6), LY-510929 (7.55), TZD18 (7.55), LTB4 (7), oleylethanolamide (6.92), LY-465608 (6.8), pirinixic acid (6.22), fatty acids (6), ragaglitazar (6), AD-5061 (5.55), fenofibric acid (4.46) [pIC50]; GW7647 (8.22), GW9578 (7.3), TAK-559 (7.17), KRP-297/MK-0767 (6.8), eicosatetraenoic acid (6.7), farglitazar (6.35), reglitazar (5.72), DRF 2519 (∼5), pristanic acid (4.4), bezafibrate (4.3), clofibrate (4.25) [pEC50]; KRP-297/MK-0767 (7.64), 8S-HETE (7), GW2331 (6.8); pterostilbene, tetradecylglycidic acid, ortylthiopropionic acid |
| Antagonists | MK886 (4.6) [pIC50] |
PPAR, peroxisome proliferator-activated receptor.
PPARβ/δ
Perhaps what makes PPARβ/δ particularly intriguing and relevant as a therapeutic target is the fact that prostacyclin is an endogenous ligand. Prostacyclin is a cardio-protective hormone which inhibits thrombosis, vasospasm and lipid accumulation which mediates its effects by actions on cell surface IP receptors as well as on cytosolic PPARβ/δ (Mitchell et al., 2008). In addition, many therapeutic prostacyclin mimetics, including trepostinil sodium, activate PPARβ/δ (Ali et al., 2006a,b;) However, it should be mentioned that other studies, using four different cell types and different experimental strategies, do not support the prevailing opinion that PGI2 plays a significant role in the regulation of PPARβ/δ (Fauti et al., 2006).
Nevertheless, the action of prostacyclin and related mimetics on PPARβ/δ highlights a potential therapeutic opportunity for the treatment of pulmonary hypertension. Pulmonary hypertension is rare, but difficult to treat and associated with a high level of morbidity and mortality. Prostacyclin therapy has been the gold standard treatment for pulmonary hypertension. However, it is associated with severe side effects and risks as well as being expensive. Several small molecule ligands of PPARβ/δ have been synthesized by various Pharma companies including GW 501516, L165041, GW0742, L-783, 483. These are potent activators of PPARβ/δ, but do activate the other PPARs at micromolar concentrations. See Table 3 for a comprehensive list which is adapted from (adapted from Michalik et al., 2006). These agonists have been developed principally for the treatment of hyperlipidaemia; however, if preclinical data are substantiated in clinical trials, they could also be useful treatments for pulmonary hypertension. It should be noted that one study recently demonstrated that a pro-inflammatory effect of the PPARγ ligands (i.e. thiazolidinediones) in a human monocytic cell line was due to low affinity binding of these ligands to the PPARβ/δ receptor (Hall and McDonnell, 2007). These potential side effects and any others remain speculative at the moment as large scale clinical trials in man have yet to be completed and published.
Table 3.
Ligands for PPARβ receptors
| Endogenous agonists | Prostacyclin |
| Synthetic agonists | GW0742X (7.52), GW2433 (6.57), GW9578 (5.9) [pEC50]; GW0742 (9), fatty acids (5.2) [pIC50]; GW501516 (8.96), retinoic acid (7.77) [pKd]; L-796449 (8.7), L-165461 (8.52), L-165041 (8.22) [pKi] |
| Antagonists |
PPAR, peroxisome proliferator-activated receptor.
In vitro activity of PPAR ligands
Inflammatory cells
Peroxisome proliferator-activated receptor γ activation can lead to differentiation of monocytes to macrophages (Tontonoz et al., 1998). Furthermore, the expression of PPARγ in macrophages is up-regulated by interleukin (IL)-4 and IL-4 also enhances the activation of PPARγ via the production of endogenous PPARγ ligands such as 13-hydroxyoctadecadienoic acid and 12- and 15-hydroxyeicosatetraenoic acid (Huang et al., 1999). PPAR ligands have effects on cytokine production. 15d-prostaglandin J2 (15d-PGJ2) and 13-hydroxyoctadecadienoic acid inhibited lipopolysaccharide-induced IL-10 and IL-12 production by macrophages (Azuma et al., 2001). PPARγ agonists also inhibit IL-1β, IL-6 and tumour necrosis factor (TNF)-α, in stimulated human peripheral blood monocytes (Jiang et al., 1998). PPARγ activation suppresses cycloxygenase-2 expression by preventing activation and translocation of NF-κB (Inoue et al., 2000; Maggi et al., 2000). PPARγ is markedly up-regulated in activated peritoneal macrophages and PPARγ ligands inhibit the expression of inducible nitric oxide synthase, gelatinase B and scavenger receptor A genes, in part by antagonizing the activities of the transcription factors AP-1, STAT and NF-κB (Chinetti et al., 1998; Ricote et al., 1998a,b;). Recent studies also show that PPARγ plays an anti-inflammatory role in macrophages by inhibiting cytokine production, increasing CD36 expression and enhancing the phagocytosis of apoptotic neutrophils, an essential process for the resolution of inflammation (Asada et al., 2004). Interestingly, exposure to rosiglitasone led to decreased TNFα, but not cigarette smoke induced cytokine release from a monocyte-macrophage cell line (Caito et al., 2008). PPARα is expressed in various inflammatory cells including human and murine monocytes and macrophages (Cuzzocrea, 2006). PPARα ligands induce apoptosis of activated human macrophages, decrease the lipopolysaccaride (LPS)-induced release of matrix metalloproteinases (MMPs) from human monocytic lines and decrease NOS activity in murine macrophage cell lines (Cuzzocrea, 2006).
Immune cells such as T and B lymphocytes have also been shown to express PPAR α and γ (Yang et al., 2000; Jones et al. 2002). In T cells, PPARγ activation inhibits IL-2 production via a mechanism believed to involve transrepression of NFAT (Yang et al., 2000). PPARγ activation has also been reported to down-regulate CCR2, the receptor for monocyte chemoattractant protein-1, in circulating rat monocytes (Ishibashi et al., 2002). Interestingly, suppression of IFNγ and IL-17, thought to be a key mediator in inflammatory diseases, expression has been observed in cultured splenocytes by the PPARα agonist, fenofibrate (Lee et al., 2007). As mentioned above PPARγ also has a role in apoptosis in several cell types.
Eosinophils may play a pivotal role in the development of allergic diseases such as asthma. IL-5 and eotaxin are critical cytokines/chemokines for eosinophil activation. The PPARγ agonist, troglitazone reduced both IL-5-stimulated eosinophil survival and eotaxin-directed eosinophil chemotaxis suggesting a role for PPARγ agonists in the treatment of allergic diseases such as asthma (Ueki et al., 2004).
Dendritic cells are powerful antigen-presenting cells with a unique capacity to stimulate naïve T cells and the migration of dendritic cells from the epithelia to the lymphoid organs represents a tightly regulated series of events involved in the induction of the immune response. A recent study has shown that PPARγ activation reduces the spontaneous migration of antigen bearing lung dendritic cells (Angeli et al., 2003) suggesting a potential role for PPARγ agonists in the treatment of allergic asthma.
Recently, data have been generated suggesting that platelets have a role in the inflammatory process by releasing mediators such as eicosanoids in addition to their involvement in thrombus formation. Although platelets do not have a nucleus they have been found to possess PPARγ and PPAR β/δ receptors (Akbiyik et al., 2004; Ali et al., 2006a) and PPARs may, separate to DNA binding, directly interact with proteins such as the transcription factor NF-κB. PPARγ ligands have been shown to attenuate CD40L surface expression and sCD40L release from thrombin-activated platelets, thromboxane release and also prevented ATP release and ADP-induced aggregation (O'Brien et al., 2007). PPARβ/δ and PPARα agonists have also been demonstrated to inhibit platelet aggregation (Ali et al., 2006a). In the same study the authors showed that prostacyclin also activates PPARβ/δ selectively and that both agonists can synergize with nitric oxide to inhibit platelet activation to various stimuli (e.g. ADP, collagen, thrombin).
Structural cells
Our group was the first to show that human airway smooth muscle cells express PPARγ and PPARα and that treatment with endogenous and synthetic PPARγ ligands could inhibit serum-induced growth of these cells and promote apoptosis (Patel et al., 2003). This study also revealed that PPARγ activation inhibited the release of the cytokines G-CSF and GM-CSF. The effect on cell growth and G-CSF was greater than that produced by a glucocorticoid, the current drug class of choice for the treatment of inflammatory airways disease. This suggests that PPARγ agonists may provide a novel alternative approach to the treatment of inflammatory diseases of the airways and one that has advantages over the therapies currently used.
PPARγ receptors are present in epithelial cells (e.g. lung alveolar type 2 cells) (Michael et al., 1997) and agonists suppress the production of IL-8 in airway epithelial cells (Wang et al., 2001) thus suggesting the possibility of reducing leukocyte recruitment and airway inflammation. MMPs are known to be involved in airway wall remodelling and are thought to play a role in the development of chronic inflammatory diseases of the airways. In a study using human bronchial epithelial cells Hetzel et al. (2003) found that PPARγ was expressed and was functionally active in these cells. Activation of PPARγ by rosiglitazone or pioglitazone significantly reduced TNFα and PMA-induced MMP-9 gelatinolytic activity, but did not alter the expression of tissue inhibitor of MMPs type 1, the endogenous inhibitor of MMP-9. They also demonstrated a decrease in MMP-9 mRNA expression following treatment with PPARγ which resulted from the inhibition of NF-κB activation in these cells (Hetzel et al., 2003). Limiting the expression of matrix degrading MMP-9 by PPARγ activation might have therapeutic potential in the treatment of chronic inflammatory diseases of the respiratory system.
Myofibroblasts are one of the key effector cells in pulmonary fibrosis and are the primary source of extracellular matrix production. Burgess et al. (2005) have shown that both 15d-PGJ2, ciglitazone and rosiglitazone inhibit transforming growth factor (TGF)-β driven myofibroblast differentiation. PPARγ agonists also potently attenuated TGF-β driven type 1 collagen protein production (Burgess et al., 2005). Thus, PPARγ agonists may provide potential therapy for fibrotic diseases of the lung.
Peroxisome proliferator-activated receptor α is expressed in human aortic smooth muscle (HASMCs) and endothelial cells. PPARα ligands partially inhibit LPS and TNFα-induced VCAM-1 expression in these cell types. In HASMCs they also inhibit IL-1β-induced production of IL-6 and COX-2 expression (Cuzzocrea, 2006).
In vivo studies
Studies in knockout animals have also highlighted a role for PPARs in animal models. A specific role for PPARγ in lung maturation was recently revealed by utilizing mice where PPARγ deletion was initiated specifically in conducting airway epithelium (Simon et al., 2006). This produced persistent enlargement of the airspaces in adult mice, which together with other phenotypic changes in the lungs point to a role for PPARγ in postnatal lung maturation. Targeted animals also had greater smoke-induced emphysema and macrophage number when compared with age-matched, wild-type littermate controls suggesting that epithelial PPARγ is necessary for proper lung maturation and response to injury. PPARγ expression is also decreased in pulmonary hypertension and affects endothelial cell growth. In these studies the authors concluded that fluid shear stress decreases the expression of PPARγ in endothelial cells and induces a loss of PPARγ expression and leads to the development of an abnormal, proliferating, apoptosis-resistant endothelial cell phenotype (Ameshima et al., 2003).
Mice that have the PPARα gene knocked out (PPARα–/– homozygotes) have an increase in the disease phenotype in a model of allergic asthma (Woerly et al., 2003; Delayre-Orthez et al., 2004). Interestingly, a difference between control and PPARα deficient mice was observed in disease severity in the absence of PPAR ligands suggestive of an anti-inflammatory role for unliganded receptors or endogenous ligands bound to the receptor.
Little is known about PPARδ in the lung although it is now known to play a role in wound healing and specifically in the transition from inflammation to healing. A recent study has also suggested that it may be a target for eicosanoids where its activation resulted in the inhibition of lung fibroblast proliferation (Ali et al., 2006b). It is evident then that not only PPARγ but also PPARα and possibly PPARδ should be studied when searching for novel targets and therapies for combating inflammatory diseases of the airways.
PPAR agonists in lung disease models
The PPARs have been identified in various cells in the lung and in lung tissue, they have also been found to be present in cells associated with inflammation in the lung. However, although there are a large number of endogenous ligands many of them bind with low affinity which questions the biological relevance of any effects seen especially in vivo. The suggestion that activation of PPARs may have anti-inflammatory and immunomodulatory effects led to the development of agonists for each of the PPAR isoforms (Table 1). These were then examined in cells known to be involved in inflammation in the airways and then in various animal models of airway disease (Belvisi et al., 2006; Belvisi and Hele, 2008). Some of these data, however, require careful interpretation given that certain ligands; for example, 15-deoxyΔ12,14-prostaglandin J2 can act via both PPAR-dependent (e.g. inhibition of NF-κB mediated transcription) and independent (inhibition of IKβ kinase and NFκB DNA binding) mechanisms (Straus and Glass, 2007).
PPARγ
As described, PPARγ ligands inhibit the release of pro-inflammatory cytokines from activated macrophages, airway epithelial cells and eosinophils and play an important role in regulating cellular differentiation (Belvisi et al., 2006). In addition, PPAR-γ agonists function as regulators of epithelial cell inflammation by reducing cigarette smoke-induced mucin-production in cells in the airway epithelium (Lee et al., 2006b). Various animal models of airway disease such as asthma, COPD, acute lung injury and pulmonary fibrosis have been used to study the anti-inflammatory effects of PPARγ ligands (Belvisi et al., 2006; Spears et al., 2006). In an animal model of airway inflammation the PPARγ agonist, rosiglitazone, inhibited lipopolysaccharide (LPS)-induced neutrophilia and reduced chemoattractants and survival factors (Birrell et al., 2004). Similar results have been achieved by other groups working with LPS-induced models of lung pathology in both mice and rats (Inoue et al., 2003; Liu et al., 2005). This work suggests that PPARγ agonists may have potential in the treatment of acute lung injury and possibly COPD.
Recently, several groups have published work demonstrating that PPARγ agonists have beneficial effects in models of asthma (Narala et al., 2007). Using a murine model of allergic asthma they have demonstrated beneficial effects of PPARγ agonists on allergic airway inflammation and airway hyperresponsiveness. PPARγ expression is increased in airway epithelial cells after allergen exposure in sensitized mice and a nebulized PPARγ agonist, ciglitazone significantly suppressed mucus secretion, and collagen deposition (Honda et al., 2004). Another study with ciglitazone showed significantly reduced lung inflammation and mucus production and T cells from the ciglitazone treated mice produced less interferon-γ, IL-4, and IL-2 upon allergen challenge in vitro (Mueller et al., 2003). A further study in a murine asthma model, using the PPARγ agonist rosiglitazone, demonstrated a reduction in airway hyperresponsiveness (Ward et al., 2006). PPARγ agonists have also been shown to inhibit allergen-induced eosinophilic inflammation in murine lungs through an effect on dendritic cell function where the results suggested that PPARγ activation prevented the induction of Th2-dependent eosinophilic airway inflammation (Hammad et al., 2004). Trifillieff et al. (2003) showed that intranasally administered agonists of PPARα (GW 9578) and PPARγ (GI 262570), but not PPARδ (GW 501516), inhibited allergen-induced bronchoalveolar lavage eosinophil and lymphocyte influx in ovalbumin sensitized and challenged mice. A study where PPAR agonists were administered by aerosol resulted in a reduction in antigen-induced airway hyperresponsiveness, lung inflammation, eosinophilia, cytokine production, GATA-3 expression and serum levels of antigen-specific IgE (Woerly et al., 2003). In a murine model of toluene diisocyanate-induced occupational asthma the administration of PPARγ agonists or adenovirus carrying PPARγ2 cDNA decreased the pathophysiological symptoms of asthma and reduced the levels of Th2 cytokines, adhesion molecules, chemokines and TGF-β1 (Lee et al., 2006a). Employing another lung injury model, bleomycin-induced lung injury, Genovese et al. (2005a) have shown that a PPARγ agonist reduced fibrosis, cellular influx, inflammation and mortality and this observation has recently been confirmed by another study (Milam et al., 2008). These data make a strong case for PPARγ activation as a potential treatment for inflammatory diseases of the airways such as asthma and COPD.
PPARα
Peroxisome proliferator-activated receptor α has been implicated in the control of airway inflammation but as yet little is known about its role. Recent work has shown that the PPARα agonist, fenofibrate dose-dependently reduced inflammation triggered by LPS in mouse lung, as demonstrated by decreased airway neutrophil and macrophage infiltration and reduced release of chemoattractants and metalloproteinases (Delayre-Orthez et al., 2005; Becker et al., 2006). Another interesting and recent study has attempted to characterize the role of PPARα in glucocorticoid-mediated anti-inflammatory activity in the lung. They tested the efficacy of dexamethasone, in an experimental model of lung inflammation, carrageenan-induced pleurisy, comparing mice lacking PPARα with wild-type mice (Cuzzocrea et al., 2007). They also tested for possible synergism with the combined treatment of dexamethasone and a PPARα agonist, clofibrate. Their results showed that dexamethasone-mediated anti-inflammatory activity is weakened in PPARα knockout mice as compared with wild-type controls, and that it is increased in wild-type mice when combined with PPARα agonist treatment. These results suggest that PPARα may contribute to the anti-inflammatory activity of glucocorticoids.
The role of PPARα in lung fibrosis has been investigated in mice using the bleomycin model of lung injury and fibrosis (Genovese et al., 2005b). PPARα knockout mice treated with bleomycin developed more severe inflammation and fibrosis than wild-type mice and exhibited increased expression of TNFα and IL-1β, increased apoptosis of interstitial cells, and decreased survival. Treatment of the wild-type mice with a PPARα agonist, WY-14643, enhanced survival and reduced the severity of fibrosis, as well as reducing the detection of TNF-α and apoptosis by immunohistochemistry. These data show that endogenous PPARα ligands can reduce the fibrotic response to bleomycin in wild-type mice, and that treatment with PPARα ligands may have potential in the treatment of fibrotic diseases of the lung.
PPARδ
Peroxisome proliferator-activated receptor δ appears to play a critical role in regulating the transition from inflammation to wound healing and PPARδ agonists inhibit lung fibroblast proliferation and enhance the anti-fibrotic properties of PPARγ agonists and as such they may prove useful as an adjunct to PPARγ therapy (Lakatos et al., 2007). As mentioned above, one recent study suggested that PPARδ may be a target of prostacyclin mimetics used in the treatment of pulmonary hypertension. Treprostinil sodium activated a PPARδ reporter gene and inhibited proliferation of lung fibroblasts in vitro. This effect was not seen in lung fibroblasts from PPARδ knockout mice, suggesting that the effect was dependent on PPARδ and not on the prostacyclin receptor (Ali et al., 2006b). In addition, it has been reported that PPAR-δ protein content is reduced in the skeletal muscle of COPD patients suggesting that a disturbed expression of these regulatory factors may well underlie the disturbed skeletal muscle functioning in COPD (Remels et al., 2007). Interestingly, a recent paper suggests that activation of PPARβ/δ attenuates the degree of inflammation in a model of LPS-induced pulmonary inflammation (Haskova et al., 2008). This paper adds additional support to the assertion that activation of PPARβ/δ would be of value in the setting of pulmonary inflammation.
Conclusions
Inflammatory diseases of the lung (e.g. asthma, COPD, pulmonary fibrosis) need safe and effective drugs to treat them. Corticosteroids while effective in treating asthma have limited efficacy in the treatment of COPD and pulmonary fibrosis and suffer from unwanted side effects. Therefore, there is a demand for safe and effective anti-inflammatory approaches for these indications. Inflammation in the lung involves several transduction pathways and inhibition of these pathways is a logical anti-inflammatory approach. Therefore, a new therapy that can exert control over many pathways in a similar but different manner to corticosteroids is required for the treatment of these airway diseases.
Peroxisome proliferator-activated receptors are ligand-activated nuclear hormone receptors, PPARγ, PPARα and PPARδ, belong to the nuclear receptor superfamily and there is now sufficient evidence that activation of these receptors induces anti-inflammatory and immunomodulatory effects in the lung as well as in other tissues. It would appear that activation of each of the PPARs results in an anti-inflammatory action, thus providing three potentially novel targets for drug intervention. The PPAR initially generating the most interest was PPARγ because thiazolidinediones, such as rosiglitazone, were established in the management of diabetes in the clinic and the belief was that these compounds could be used to treat inflammatory diseases. Unfortunately, a recent problem which may hamper this strategy has been the suggestion of an association of rosiglitazone treatment with an increased risk of cardiovascular events in patients being treated for type 2 diabetes. It remains to be seen whether this is a class effect of the thiazolidinediones or whether it is due to PPARγ activation. In contrast, in patients with type 2 diabetes mellitus and a high cardiovascular risk the PROactive study demonstrated that pioglitazone significantly reduced the predefined secondary combined endpoint of total mortality, nonfatal myocardial infarction and stroke (Dormandy et al., 2005) suggesting that these are not class effects. Interestingly, a clinical trial is currently underway examining the effect of rosiglitazone on lung function in comparison with low-dose inhaled corticosteroids in steroid naive smokers with asthma (Spears et al., 2006). The results of this trial will be keenly awaited, not only to determine the hoped for efficacy but also to see if this approach is free of unwanted side effects. However, this fact coupled with the recent positive animal model results obtained with PPARα and PPARδ agonists make this a field worthy of continued exploration in the hope of discovering novel and effective treatments for these chronic diseases of the airways.
Glossary
Abbreviations:
- COPD
chronic obstructive pulmonary disease
- LPS
lipopolysaccaride
- PPAR
peroxisome proliferator-activated receptor
- TGFβ
transforming growth factor β
- TNFα
tumour necrosis factor α
References
- Akbiyik F, Ray DM, Gettings KF, Blumberg N, Francis CW, Phipps RP. Human bone marrow megakaryocytes and platelets express PPARγ, and PPARγ agonists blunt platelet release of CD40 ligand and thromboxanes. Blood. 2004;104:1361–1368. doi: 10.1182/blood-2004-03-0926. [DOI] [PubMed] [Google Scholar]
- Ali FY, Davidson SJ, Moraes LA, Traves SL, Paul-Clark M, Bishop-Bailey D, et al. Role of nuclear receptor signaling in platelets: antithrombotic effects of PPARbeta. FASEB J. 2006a;20:326–328. doi: 10.1096/fj.05-4395fje. [DOI] [PubMed] [Google Scholar]
- Ali FY, Egan K, FitzGerald GA, Desvergne B, Wahli W, Bishop-Bailey D, et al. Role of prostacyclin versus peroxisome proliferator-activated receptor β receptors in prostacyclin sensing by lung fibroblasts. Am J Respir Cell Mol Biol. 2006b;34:242–246. doi: 10.1165/rcmb.2005-0289OC. [DOI] [PubMed] [Google Scholar]
- Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92:1162–1169. doi: 10.1161/01.RES.0000073585.50092.14. [DOI] [PubMed] [Google Scholar]
- Angeli V, Hammad H, Staels B, Capron M, Lambrecht BN, Trottein F. Peroxisome proliferator-activated receptor gamma inhibits the migration of dendritic cells: consequences for the immune response. J Immunol. 2003;170:5295–5301. doi: 10.4049/jimmunol.170.10.5295. [DOI] [PubMed] [Google Scholar]
- Asada K, Sasaki S, Suda T, Chida K, Nakamura H. Anti-inflammatory roles of peroxisome proliferator-activated receptor gamma in human alveolar macrophages. Am J Respir Crit Care Med. 2004;169:195–200. doi: 10.1164/rccm.200207-740OC. [DOI] [PubMed] [Google Scholar]
- Azuma Y, Shinohara M, Wang PL, Ohura K. 15-Deoxy-delta (12, 14)-prostaglandin J(2) inhibits IL-10 and IL-12 production by macrophages. Biochem Biophys Res Com. 2001;283:344–346. doi: 10.1006/bbrc.2001.4783. [DOI] [PubMed] [Google Scholar]
- Becker J, Delayre-Orthez C, Frossard N, Pons F. Regulation of inflammation by PPARs: a future approach to treat lung inflammatory diseases? Fundam Clin Pharmacol. 2006;20:429–447. doi: 10.1111/j.1472-8206.2006.00425.x. [DOI] [PubMed] [Google Scholar]
- Belvisi MG, Hele DJ. Peroxisome proliferator-activated receptors as novel targets in lung disease. Chest. 2008;134:152–157. doi: 10.1378/chest.08-0019. [DOI] [PubMed] [Google Scholar]
- Belvisi MG, Hele DJ, Birrell MA. New anti-inflammatory therapies and targets for asthma and chronic obstructive pulmonary disease. Expert Opin Ther Targets. 2004;8:265–285. doi: 10.1517/14728222.8.4.265. [DOI] [PubMed] [Google Scholar]
- Belvisi MG, Hele DJ, Birrell MA. Peroxisome proliferator-activated receptor gamma agonists as therapy for chronic airway inflammation. Eur J Pharmacol. 2006;533:101–109. doi: 10.1016/j.ejphar.2005.12.048. [DOI] [PubMed] [Google Scholar]
- Berger J, Bailey P, Biswas C, Cullinan CA, Doebber TW, Hayes NS, et al. Thiazolidinesdiones produce a conformational change in peroxisome proliferator-activated receptor gamma: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology. 1996;137:4189–4195. doi: 10.1210/endo.137.10.8828476. [DOI] [PubMed] [Google Scholar]
- Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26:244–251. doi: 10.1016/j.tips.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Birrell MA, Patel HJ, McCluskie K, Wong S, Leonard T, Yacoub MH, et al. PPAR-γ agonists as therapy for diseases involving airway neutrophilia. Eur Respir J. 2004;24:18–23. doi: 10.1183/09031936.04.00098303. [DOI] [PubMed] [Google Scholar]
- Boyault S, Bianchi A, Moulin D, Morin S, Francois M, Netter P, et al. 15-Deoxy-delta(12,14)-prostaglandin J (2) inhibits IL-1 beta- induced IKK enzymatic activity and IkappaBalpha degradation in rat chondorcytes through a PPAR gamma-independent pathway. FEBS Letts. 2004;572:33–40. doi: 10.1016/j.febslet.2004.06.090. [DOI] [PubMed] [Google Scholar]
- Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M, et al. PPAR gamma agonists inhibit TGF-induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of fibrosis. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1146–L1153. doi: 10.1152/ajplung.00383.2004. [DOI] [PubMed] [Google Scholar]
- Caito S, Yang S-R, Kode A, Edirisinghe I, Rajendrasozhan S, Phipps RP, et al. Rosiglitasone and 15-Deoxy-Δ12,14-prostaglandin J2, PPARg agonists, differentially resgulate cigarette smoke-mediated pro-inflammatory cytokine release in monocyte/macrophages. Antioxidant and Redox Signalling. 2008;10:253–260. doi: 10.1089/ars.2007.1889. [DOI] [PubMed] [Google Scholar]
- Chinetti G, Griglio S, Antonucci M, Torra IP, Delerive P, Majd Z, et al. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J Biol Chem. 1998;273:25573–25580. doi: 10.1074/jbc.273.40.25573. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S. Peroxisome proliferator-activated receptors and acute lung injury. Curr Opinion in Pharmacology. 2006;6:263–270. doi: 10.1016/j.coph.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Bruscoli S, Mazzon E, Crisafulli C, Donato V, Di Paola R, et al. PPAR-{alpha} contributes to the anti-inflammatory activity of glucocorticoids. Mol Pharmacol. 2007;73:323–337. doi: 10.1124/mol.107.041475. [DOI] [PubMed] [Google Scholar]
- Delayre-Orthez C, Becker J, Auwerx J, Frossard N, Pons F. PPARalpha decreases airway inflammation associated with asthma in the mouse. Fund Clin Pharmacol. 2004;18:220. [Google Scholar]
- Delayre-Orthez C, Becker J, Guenon I, Lagente V, Auwerx J, Frossard N. PPARα downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir Res. 2005;6:91. doi: 10.1186/1465-9921-6-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. PROactive investigators. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- Dubuquoy L, Dharancy S, Nutten S, Pettersson S, Auwerx J, Desreumaux P. Role of peroxisome proliferator-activated receptor gamma and retinoid X receptor heterodimer in hepatogastroenterological diseases. Lancet. 2002;360:1410–1408. doi: 10.1016/S0140-6736(02)11395-X. [DOI] [PubMed] [Google Scholar]
- Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauti T, Müller-Brüsselbach S, Kreutzer M, Rieck M, Meissner W, Rapp U, et al. Induction of PPARbeta and prostacyclin (PGI2) synthesis by Raf signalling: failure of PGI2 to activate PPARbeta. FEBS J. 2006;273:170–179. doi: 10.1111/j.1742-4658.2005.05055.x. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick FA, Wynalda MA. Albumin-catalysed metabolism of prostaglandin D2. Identification of products formed in vitro. J Biol Chem. 1983;258:11713–11718. [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is the ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- Genovese T, Cuzzocrea S, Di Paola R, Mazzon E, Mastruzzo C, Catalano P, et al. Effect of rosiglitazone and 15-deoxy-Δ12,14-prostaglandin J2 on bleomycin-induced lung injury. Eur Respir J. 2005a;25:225–234. doi: 10.1183/09031936.05.00049704. [DOI] [PubMed] [Google Scholar]
- Genovese T, Mazzon E, Di Paola R, Muia C, Crisafulli C, Caputi AP, et al. Role of endogenous and exogenous ligands for the peroxisome proliferator activated receptor α in the development of bleomycin-induced lung injury. Shock. 2005b;24:547–555. doi: 10.1097/01.shk.0000190825.28783.a4. [DOI] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, et al. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JM, McDonnell DP. The molecular mechanisms underlying the pro-inflammatory actions of thiazolidinediones in human macrophages. Mol Endocrinol. 2007;21:1756–1768. doi: 10.1210/me.2007-0060. [DOI] [PubMed] [Google Scholar]
- Hammad H, de Heer HJ, Soullie T, Angeli V, Trottein F, Hoogsteden HC, et al. Activation of peroxisome proliferator-activated receptor-γ in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. Am J Pathol. 2004;164:263–271. doi: 10.1016/s0002-9440(10)63116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskova Z, Hoang B, Luo G, Morgan LA, Billin AN, Barone FC, et al. Modulation of LPS-induced pulmonary neutrophil infiltration and cytokine production by the selective PPARbeta/delta ligand GW0742. Inflamm Res. 2008;57:314–321. doi: 10.1007/s00011-007-7157-4. [DOI] [PubMed] [Google Scholar]
- Hetzel M, Walcher D, Grub M, Bach H, Hombach V, Marx N. Inhibition of MMP-9 expression by PPAR gamma activators in human bronchial epithelial cells. Thorax. 2003;58:778–783. doi: 10.1136/thorax.58.9.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Marquillies P, Capron M, Dombrowicz D. Peroxisome proliferator-activated receptor γ is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J Allergy Clin Immunol. 2004;113:882–888. doi: 10.1016/j.jaci.2004.02.036. [DOI] [PubMed] [Google Scholar]
- Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, et al. Interleukin-4 dependent production of PPAR gamma ligands by macrophages by 12/15 lipoxygenase. Nature. 1999;400:378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- Inoue H, Tanabe T, Umesono K. Feedback control of cyclooxygenase-2 expression through PPAR gamma. J Biol Chem. 2000;275:28028–28032. doi: 10.1074/jbc.M001387200. [DOI] [PubMed] [Google Scholar]
- Inoue K-I, Takano H, Yanagisawa R, Morita M, Ichinose T, Sadakane K, et al. Effect of 15-deoxy-Δ12,14-prostaglandin J2 on acute lung injury induced by lipopolysaccharide in mice. Eur J Pharmacol. 2003;481:261–269. doi: 10.1016/j.ejphar.2003.09.020. [DOI] [PubMed] [Google Scholar]
- Ishibashi M, Egashira K, Hiasa K, Inoue S, Ni W, Zhao Q, et al. Antiinflammatory and antiarteriosclerotic effects of pioglitazone. Hypertension. 2002;40:687–693. doi: 10.1161/01.hyp.0000036396.64769.c2. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Jones DC, Ding X, Daynes RA. Nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) is expressed in resting murine lymphocytes. The PPAR alpha in T and B lymphocytes is both transactivation and transrepression competent. J Biol Chem. 2002;277:6836–6845. doi: 10.1074/jbc.M106908200. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- von Knethen A, Soller M, Tzieply N, Weigert A, Johann AM, Jennewein C, et al. PPARgamma1 attenuates cytosol to membrane translocation of PKCalpha to desensitize monocytes/macrophages. The J Cell Biol. 2007;176:681–694. doi: 10.1083/jcb.200605038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota BP, Huang TH, Roufogalis BD. An overview on biological mechanisms of PPARs. Pharmacol Res. 2005;51:85–94. doi: 10.1016/j.phrs.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Lakatos HF, Thatcher TH, Kottmann RM, Garcia TM, Phipps RP, Sime PJ. The role of PPARs in lung fibrosis. PPAR Res. 2007;2007:713–723. doi: 10.1155/2007/71323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, et al. Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science. 2003;302:453–457. doi: 10.1126/science.1087344. [DOI] [PubMed] [Google Scholar]
- Lee KS, Park SJ, Kim SR, Min KH, Jin SM, Lee HK, et al. Modulation of airway remodeling and airway inflammation by peroxisome proliferator-activated receptor γ in a murine model of toluene diisocyanate-induced asthma. J Immunol. 2006a;177:5248–5257. doi: 10.4049/jimmunol.177.8.5248. [DOI] [PubMed] [Google Scholar]
- Lee SY, Kang EJ, Hur GY, Jung KH, Jung HC, Lee SY, et al. Peroxisome proliferator-activated receptor-γ inhibits cigarette smoke solution-induced mucin production in human airway epithelial (NCI-H292) cells Am J Physiol. Lung Cell Mol Physiol. 2006b;291:L84–L90. doi: 10.1152/ajplung.00388.2005. [DOI] [PubMed] [Google Scholar]
- Lee JW, Bajwa PJ, Carson MJ, Jeske DR, Cong Y, Elson CO, et al. Fenofibrate represses interleukin-17 and interferon-gamma expression and improves colitis in interleukin-10-deficient mice. Gastroenterology. 2007;133:108–123. doi: 10.1053/j.gastro.2007.03.113. [DOI] [PubMed] [Google Scholar]
- Liu D, Zeng BX, Zhang SH, Wang YL, Zeng L, Geng ZL, et al. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor γ, reduces pulmonary inflammatory response in a rat model of endotoxemia. Inflamm Res. 2005;54:464–470. doi: 10.1007/s00011-005-1379-0. [DOI] [PubMed] [Google Scholar]
- Michael LF, Lazar MA, Mendelson CR. Peroxisome proliferator-activated receptor gamma1 expression is induced during cyclic adenosine monophosphate-stimulated differentiation of alveolar type II pneumonocytes. Endocrinology. 1997;138:3695–3703. doi: 10.1210/endo.138.9.5373. [DOI] [PubMed] [Google Scholar]
- Michalik L, Auwerx J, Berger J, Chatterjee KP, Glass CK, Gonzalez FJ, et al. International Union of Pharmacology. LXI. Peroxisome Proliferator-Activated Receptors. Pharmacol Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- Maggi LB, Jr, Sadeghi H, Weigand C, Scarim AL, Heitmeier MR, Corbett JA. Anti-inflammatory actions of 15-deoxy-delta 12, 14-prostaglandin J2 and troglitazone: evidence for heat shock-dependent and independent inhibition of cytokine-induced inducible nitric oxide synthase expression. Diabetes. 2000;49:346–355. doi: 10.2337/diabetes.49.3.346. [DOI] [PubMed] [Google Scholar]
- Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM, et al. PPAR-{gamma} agonists inhibit pro-fibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L891–L901. doi: 10.1152/ajplung.00333.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Ali F, Bailey L, Moreno L, Harrington LS. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp Physiol. 2008;93:141–147. doi: 10.1113/expphysiol.2007.038588. [DOI] [PubMed] [Google Scholar]
- Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Curr Opin Cell Biol. 1998;10:384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- Mueller C, Weaver V, Vanden Heuvel JP, August A, Cantoma MT. Peroxisome proliferator-activated receptor gamma ligands attenuate immunological symptoms of experimental allergic asthma. Arch Biochem Biophys. 2003;418:186–196. doi: 10.1016/j.abb.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Narala VR, Ranga R, Smith MR, Berlin AA, Standiford TJ, Lukacs NW, et al. Pioglitazone is as effective as dexamethasone in a cockroach allergen-induced murine model of asthma. Respir Res. 2007;8:90. doi: 10.1186/1465-9921-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JJ, Ray DM, Spinelli SL, Blumberg N, Taubman MB, Francis CW, et al. The platelet as a therapeutic target for treating vascular diseases and the role of eicosanoid and synthetic PPARgamma ligands. Prostaglandins Other Lipid Mediat. 2007;82:68–76. doi: 10.1016/j.prostaglandins.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Paumelle R, Blanquart C, Briand O, Barbier O, Duhem C, Woerly G, et al. Acute antiinflammatory properties of statins involve peroxisome proliferator-activated receptor-alpha via inhibition of the protein kinase C signaling pathway. Circ Res. 2006;98:361–369. doi: 10.1161/01.RES.0000202706.70992.95. [DOI] [PubMed] [Google Scholar]
- Patel HJ, Belvisi MG, Bishop-Bailey D, Yacoub MH, Mitchell JA. Activation of peroxisome proliferator-activated receptors in human airway smooth muscle cells has a superior anti-inflammatory profile to corticosteroids: relevance for chronic obstructive pulmonary disease therapy. J Immunol. 2003;170:2663–2669. doi: 10.4049/jimmunol.170.5.2663. [DOI] [PubMed] [Google Scholar]
- Reed A, Harrington L, Moreno L, Warner T, Wort SJ, Mitchell JA. Acute vasodilator effects of PPAR-beta agonists in mouse pulmonary artery and aorta. Available at: http://www.pa2online.org/abstract/search.jsp.
- Remels AH, Schrauwen P, Broekhuizen R, Willems J, Kersten S, Gosker HR, et al. Peroxisome proliferator-activated receptor expression is reduced in skeletal muscle in COPD. Eur Respir J. 2007;30:245–252. doi: 10.1183/09031936.00144106. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor gamma is a negative regulator of macrophage activation. Nature. 1998a;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Ricote M, Huang J, Fagas L, Li A, Welch J, Najib J, et al. Expression of the peroxisome proliferator activated receptor gamma (PPAR gamma) in human atherosclerosis and regulation in macrophages by colony stimulating factorsand oxidised low density lipoprotein. Proc Natl Acad Sci USA. 1998b;95:7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN. Inflammation. Signalling the fat controller. Nature. 1996;384:23–24. doi: 10.1038/384023a0. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Devchand PR. Novel anti-inflammatory targets for asthma. A role for PPARγ? Am J Respir Cell Mol Biol. 2001;24:658–661. doi: 10.1165/ajrcmb.24.6.f210. [DOI] [PubMed] [Google Scholar]
- Sher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol. 2005;114:100–109. doi: 10.1016/j.clim.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Simon DM, Arikan MC, Srisuma S, Bhattacharya S, Andalcio T, Shapiro SD, et al. Epithelial cell PPARγ is an endogenous regulator of normal lung maturation and maintenance. The Proceedings of American Thoracic Society. 2006;3:510–511. doi: 10.1513/pats.200603-034MS. [DOI] [PubMed] [Google Scholar]
- Spears M, McSharry C, Thomson NC. Peroxisome proliferator-activated receptor-gamma agonists as potential anti-inflammatory agents in asthma and chronic obstructive pulmonary disease. Clin Exp Allergy. 2006;36:1494–1504. doi: 10.1111/j.1365-2222.2006.02604.x. [DOI] [PubMed] [Google Scholar]
- Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunology. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPAR gamma promotes monocyte/macrophage differentiation and uptake of oxidised LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- Trifillieff A, Bench A, Hanley M, Bayley D, Campbell E, Whittaker P. PPAR-α and –γ but not –δ agonists inhibit airway inflammation in a murine model of asthma: in vitro evidence for an NF-κB-independent model. Br J Pharmacol. 2003;139:163–171. doi: 10.1038/sj.bjp.0705232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki S, Matsuwaki Y, Kayaba H, Oyamada H, Kanda A, Usami A, et al. Peroxisome proliferator-activated receptor gamma regulates eosinophil functions: a new therapeutic target for allergic airway inflammation. Int Arch Allergy Immunol. 2004;134:30–36. doi: 10.1159/000077790. [DOI] [PubMed] [Google Scholar]
- Wang AC, Dai X, Luu B, Conrad DJ. Peroxisome proliferator-activated receptor gamma regulates airway epithelial cell activation. Am J Respir Cell Mol Biol. 2001;24:688–693. doi: 10.1165/ajrcmb.24.6.4376. [DOI] [PubMed] [Google Scholar]
- Ward JE, Fernandes DJ, Taylor CC, Bonacci JV, Stewart AG. The PPARγ ligand, rosiglitazone, reduces airways hyperresponsiveness in a murine model of allergen-induced inflammation. Pulm Pharmacol Ther. 2006;19:39–46. doi: 10.1016/j.pupt.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- Woerly G, Honda K, Loyens M, Papin JP, Auwerx J, Staels B, et al. Peroxisome proliferator-activated receptors α and γ down-regulate allergic inflammation and eosinophil activation. J Exp Med. 2003;198:411–421. doi: 10.1084/jem.20021384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XA, Wang LH, Chen T, Hodge DR, Resau JH, DaSilva L, et al. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPAR gamma) agonists. PPAR-gamma co-association with transcription factor NFAT. J Biol Chem. 2000;275:4541–4544. doi: 10.1074/jbc.275.7.4541. [DOI] [PubMed] [Google Scholar]
- Yki-Jarvinen H. Thiazolidinesdiones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]