

Abstract
The cyclin-dependent kinase inhibitor (CDKi) drugs such as R-roscovitine have emerged as potential anti-inflammatory, pharmacological agents that can influence the resolution of inflammation. Usually, once an inciting inflammatory stimulus has been eliminated, resolution proceeds by prompt, safe removal of dominant inflammatory cells. This is accomplished by programmed cell death (apoptosis) of prominent effector, inflammatory cells typified by the neutrophil. Apoptosis of neutrophils ensures that toxic neutrophil granule contents are securely packaged in apoptotic bodies and expedites phagocytosis by professional phagocytes such as macrophages. A panel of CDKi drugs have been shown to promote neutrophil apoptosis in a concentration- and time-dependent manner and the archetypal CDKi drug, R-roscovitine, overrides the anti-apoptotic effects of powerful survival factors [including lipopolysaccharide (LPS) and granulocyte macrophage-colony stimulating factor (GM-CSF)]. Inflammatory cell longevity and survival signalling is integral to the inflammatory process and any putative anti-inflammatory agent must unravel a complex web of redundancy in order to be effective. CDKi drugs have also been demonstrated to have significant effects on other cell types including lymphocytes and fibroblasts indicating that they may have pleiotropic anti-inflammatory, pro-resolution activity. In keeping with this, CDKi drugs like R-roscovitine have been reported to be efficacious in resolving established animal models of neutrophil-dominant and lymphocyte-driven inflammation. However, the mechanism of action behind these powerful effects has not yet been fully elucidated. CDKs play an integral role in the regulation of the cell cycle but are also recognized as participants in processes such as apoptosis and transcriptional regulation. Neutrophils have functional CDKs, are transcriptionally active and demonstrate augmented apoptosis in response to CDKi drugs, while lymphocyte proliferation and secretory function are inhibited. This review will discuss current understanding of the processes of inflammation and resolution but will focus on CDKis and their potential mechanisms of action.
This article is part of a themed issue on Mediators and Receptors in the Resolution of Inflammation. To view this issue visit http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Keywords: cyclin-dependent kinase, resolution of Inflammation, apoptosis, neutrophil, anti-inflammatory drugs, R-roscovitine, transcriptional regulation
Introduction
Cyclin-dependent kinase inhibitor (CDKi) drugs
CDKi drugs such as R-roscovitine have emerged as potential, anti-inflammatory agents that augment neutrophil apoptosis (Rossi et al., 2006) and suppress lymphocyte proliferation and secretory function (Obligado et al., 2008). This former finding seems counter-intuitive because the neutrophil is a terminally differentiated cell and its cell cycle machinery ought to be effectively vestigial. Further research is required before we develop a functional understanding of the mechanism of action of CDKi drugs in inflammation, but already, some tantalizing clues are emerging. This review will provide background material on inflammation research in general but will focus on the CDKs, their inhibitors, the role of CDK inhibition in inflammation and areas for further research. We anticipate that CDK inhibition will provide the basis for the development of novel therapeutic agents that drive resolution of inflammation and counter inflammatory disease.
Inflammation
Inflammation is part of the beneficial anti-microbial, immune defence system that has been honed and conserved by evolution over millions of years (Marchalonis et al., 2002). The system has become increasingly sophisticated because of the breadth of micro-organisms the human body has encountered and because of the mechanisms these organisms have evolved to enable evasion of its front-line defences. Ideally, following prompt detection of a micro-organism by immune mechanisms, an inflammatory reaction should contain and destroy the organism before it multiplies, spreads, becomes established or causes harm. Self-regulation and limitation are the key, final components of the response as the system must actively drive resolution of inflammation to restore tissue homeostasis (Lawrence and Gilroy, 2007).
Granulocytes are the foot soldiers of the inflammatory response and are dispatched in large numbers to overcome many challenges to the host organism. Neutrophils and eosinophils are leucocytes of the granulocyte lineage that are key players in the immune response to bacteria, fungi and parasites. They are attracted by, and are believed to follow a concentration gradient of, chemotactic stimuli released by invading pathogens or tissues under challenge. They migrate from the circulation across post-capillary venule endothelial cells (or capillary endothelial cells in pulmonary inflammation) (Downey et al., 1993) and employ a formidable armamentarium to overcome their adversaries. Granulocytes are named for the numerous granules within their cytoplasm (e.g. neutrophilic granulocytes contain at least four different types of granules). Each of these granules contains a range of toxic products such as proteases, lysozyme and lactoferrin. Eosinophils have an armament (including major basic protein, eosinophil cationic protein and eosinophil peroxidase) more specific to the killing and digestion of parasites but, in asthma, respond to allergens or airway irritants in a concerted reaction which involves IgE, mast cells, lymphocytes, basophils (the third member of the granulocyte lineage) and smooth muscle. Neutrophils can either phagocytose an offending organism and then drown it in toxic chemicals (e.g. granule products and reactive oxygen species (ROS) such as superoxide anion) within the phagolysosome or, particularly when frustrated by an indigestible opponent, disgorge the same array of chemicals in the general direction of the target (exocytosis). A fascinating recent development has been the identification of neutrophil extracellular traps (Brinkmann and Zychlinsky, 2007; Fuchs et al., 2007). It appears that a proportion of activated neutrophils can extrude web-like fibres of granule constituents and chromatin in order to ensnare, disarm and kill invading pathogens. This appears to be a terminal event for the neutrophil occurring coincidentally with cell membrane rupture and death. Similarly, eosinophils can catapult (by an unidentified but ROS-independent mechanism) mitochondrial DNA which is also toxic to various pathogens but does not result in eosinophil death (Yousefi et al., 2008). In this review, we will focus on the effects of CDKi drugs on neutrophil granulocytes and, to a lesser degree, lymphocytes.
Assuming the influx of neutrophils is successful in overcoming a threat, the next stage is to remove the invading organisms, the neutrophils themselves (alive, dying and dead) and other cellular debris from the inflammatory site. Neutrophils are short-lived cells that leave the circulation within hours but can be influenced by survival factors present at inflammatory sites to extend their longevity to days. During neutrophil apoptosis (Figure 1), cell membrane integrity is maintained (ensuring toxic granule contents are secured) and specific phospholipid or glycoprotein signals are displayed on the outer membrane that are recognized by professional phagocytes. Macrophages, the most common professional phagocyte, are triggered by these signals to engulf the apoptotic neutrophil (Savill et al., 1989; Haslett et al., 1990). Following engulfment, macrophages express a pro-resolution phenotype which allows them to switch from secretion of pro-inflammatory stimuli such as TNF-α to tissue repair mediators such as TGF-β and IL-10 (Fadok et al., 1998; Girkontaite et al., 2007). Additionally, lymphocytes of the adaptive immune response which had previously served to focus (via B-cell-mediated antibody opsonization of non-self antigens) and amplify (via T-cell-mediated interferon-γ secretion leading to macrophage activation) the inflammatory reaction now aid in resolution as interferon-γ dampens down the production of pro-inflammatory chemokines by macrophages (Schultz, 1987; Chung, 2001; Ozato et al., 2002). Streptococcal lobar pneumonia can be paradigmatic of a beneficial inflammatory response as despite a massive, neutrophil-dominant inflammatory reaction, resolution of inflammation occurs (and did so in the majority of cases in the pre-antibiotic era) with no pathological damage (Haslett, 1999).
Figure 1.
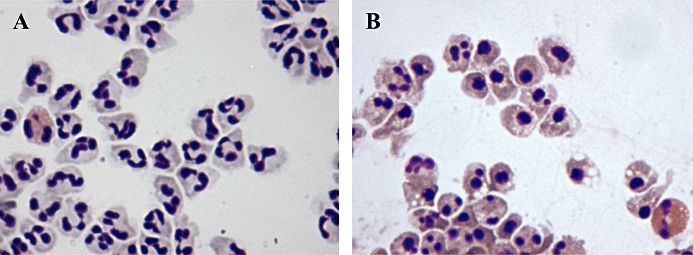
In (A), untreated neutrophils cultured for 8 h (and one eosinophil) are shown at ×200 magnification displaying typical viable neutrophil morphology including polymorphic, multilobed nuclei and pale cytoplasm. In (B), neutrophils (and one eosinophil) treated with the CDK inhibitor R-roscovitine for 8 h are shown at 200× magnification displaying typical apoptotic morphology including condensed, rounded nuclei, darker cytoplasm and blebbing.
The common alternative to granulocyte apoptosis is necrosis where membrane integrity is lost and cell contents are free to spill out into the tissues, initiating all the damaging sequelae associated with chronic inflammatory and autoimmune disease. This may occur where neutrophils are overproduced, over-recruited, have inappropriate longevity or are inadequately cleared. It has been a central tenet of our research that an ideal anti-inflammatory, pro-resolution of inflammation pharmacological agent should drive neutrophil apoptosis without overburdening, or causing detriment to, the ability of professional phagocytes to phagocytose and remove apoptotic neutrophils (Figure 2). There is now abundant evidence that inflammatory cell- (especially neutrophil) programmed cell death followed by non-phlogistic clearance of apoptotic cells by phagocytes such as macrophages plays a key role in ensuring efficient (and in the majority complete) resolution of inflammation (Hallett et al., 2008; Leitch et al., 2008). It is clear that resolution of inflammation may also be driven by strategies that prevent excessive amplification and prolongation of inflammation by the adaptive immune response and allow stop signals and pro-resolution signals to predominate.
Figure 2.
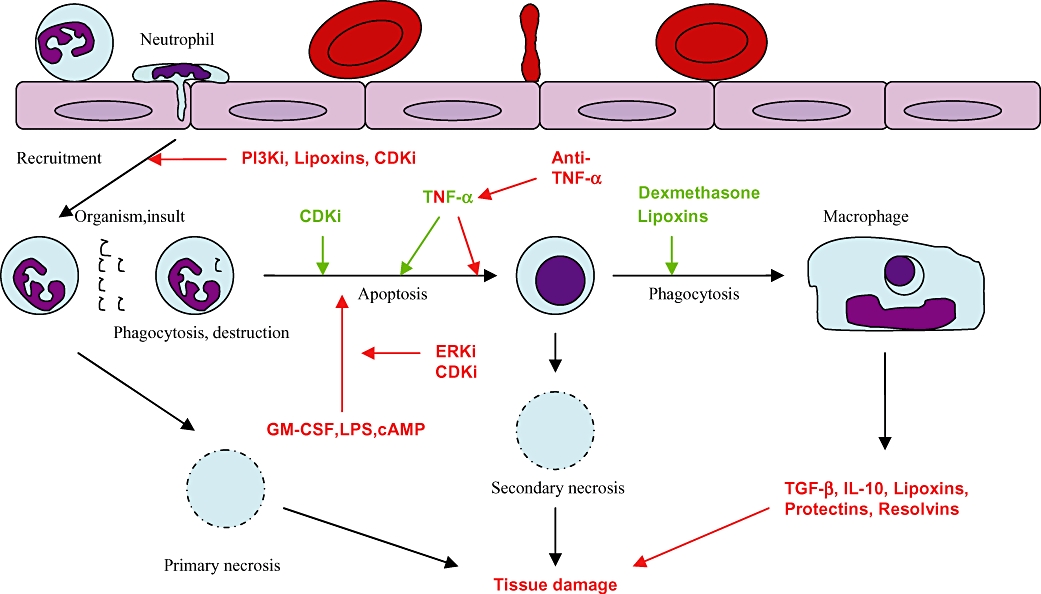
The effects of pro-resolution of inflammation agents on chronic inflammation.
Inflammatory disease
Inflammatory diseases arise for a number of reasons; some of which are not fully understood. If the inflammatory system cannot destroy organisms or foreign particles (such as asbestos fibre and silica), it malfunctions, initiating excessive or prolonged inflammatory reactions which are likely to lead to irreversible fibrosis and scarring. Alternatively, reactions may be sited where none are apparently needed or self-antigens become recognized as foreign. In these settings, chronic inflammatory and autoimmune disease may supervene. The outcome of excessive or persistent (chronic) inflammation is tissue damage caused by the secretion or loss of histotoxic products from overactivated, under-regulated or necrotic inflammatory cells (Rossi et al., 2007). Autoimmune disease is thought to occur by a combination of inappropriate, excessive proliferation and activation of B and T-lymphocytes and the generation of autoantibodies to endogenous antigens. It is possible that endogenous antigens externalized on apoptotic cells that fail to be efficiently cleared contribute to this mechanism (Munoz et al., 2005). The spectrum of inflammatory disease includes all the major organ systems and, increasingly, we are recognizing a role for inflammation in other disease processes such as cancer (Coussens and Werb, 2002), neurological disorders (Gilgun-Sherki et al., 2006), atheroma (Hansson et al., 2006) and menstrual dysfunction (Critchley et al., 2001). There are many similarities between inflammatory diseases but also important differences which may occur even within the same organ system. For example, in lung-based disease, inflammation in asthma is usually eosinophil dominant (although neutrophil-dominant sub-types exist), while in chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis, arguably, neutrophils play a major role, although a plethora of other inflammatory cells have been implicated (Bianchi et al., 2006).
There are always competing hypotheses with regard to putative key cellular determinants of given diseases and it is, perhaps, therefore important (therapeutically, if less helpful scientifically) that novel pro-resolution of inflammation therapies should have pleiotropic actions across the different inflammatory cell populations. Work with CDKi drugs, so far, indicates that they influence not only neutrophils but also lymphocytes, suggesting that they may have a role to play in the management of a number of inflammatory diseases.
Current anti-inflammatory therapies
Many inflammatory diseases are treated with glucocorticosteroid drugs either independently or in combination regimens. Glucocorticoids are very successful at treating eosinophil-dominant inflammatory disease such as asthma, while they have had more modest success with neutrophil-dominant diseases such as COPD. It is now known that glucocorticoids promote eosinophil apoptosis but actually prolong neutrophil survival (Meagher et al., 1996). The latter effect is, presumably, partially compensated by glucocorticoid-mediated, enhanced macrophage phagocytosis of apoptotic neutrophils (Liu et al., 1999; Heasman et al., 2003; Michlewska et al., 2009). The development of non-steroidal anti-inflammatory drugs (NSAIDs) and subsequently cyclo-oxygenase (COX)-2- specific NSAIDs demonstrates further the difficulties associated with combating a well-established, integral host response such as inflammation. NSAIDs work mainly by inhibiting the COX enzymes 1 and 2. COX-1 is constitutively expressed in all tissues, while COX-2 is largely an inducible enzyme subject to various regulatory factors. These enzymes convert arachidonic acid to prostaglandin (PG)-H2 which can be converted by isomerase enzymes to: PGE2, PGF2α, PGD2, prostacyclin (PGI2), and thromboxane (TX)A2. PGE2 is responsible for some of the classic features of inflammation. Traditional NSAIDs inhibited COX-1 and -2 resulting in side effects related to sites where COX-1 has constitutive housekeeping duties (such as in the gut leading to peptic ulcer disease); COX-2-specific inhibitors were an attempt to avoid these side effects. Unfortunately, this specific inhibition appeared to confer a greater risk of vascular thrombotic events (such as heart attack and stroke) thought to be related to enhanced suppression of PGI2, an atheroprotective agent, in comparison to COX-1-derived TXA2, a pro-aggregatory and vasoconstrictor mediator. However, recent work has suggested that it may be possible to exploit the anti-inflammatory and pro-resolution effects of PGD2 metabolites (usually responsible for negative feedback inhibition of the inflammatory response) which inhibit NF-κB and have been shown to resolve inflammation in animal models (Ward et al., 1999; Rossi et al., 2000; Lawrence et al., 2001). The lipid-derived mediators of the lipoxin, resolvin and protectin family may provide a novel therapeutic approach. These compounds have demonstrable pro-resolution properties in vivo including inhibition of neutrophil chemotaxis, recruitment of mononuclear cells and enhanced phagocytosis. In addition, aspirin-triggered lipoxins utilize the same receptor as glucocorticoid-induced annexin 1 suggesting an intriguing interconnectivity between pro-resolution strategies (Lawrence et al., 2002; Perretti and Solito, 2004; Serhan, 2007). A significant barrier to the successful design of novel anti-inflammatory agents has been the complex, multilayered redundancy that characterizes the inflammatory system. Where one cytokine pathway is knocked down, another may compensate and maintain the inflammatory response. Only the removal of certain essential cytokines or pathways will allow resolution. Recent anti-inflammatory approaches have utilized ‘biological therapies’ in the targeting of specific cytokines. Anti-TNF therapy is the standout success of this genre and there is now considerable evidence that TNF-α is the sought-after essential cytokine in inflammatory disease, although other agents are showing promise (Feldmann and Maini, 2002; Williams et al., 2007). It is clear that with such redundancy in the system, there will always be room for new pharmacological agents that have novel targets within the inflammatory system (Hallett et al., 2008).
CDK
CDKs are serine/threonine kinases and part of the diverse protein kinase family. They are essential facilitators of life at the molecular level via their ubiquitous phosphorylation reactions. The majority of CDKs identified rely on binding partners called cyclins for their activation. In all, there are 13 CDKs and 25 identified cyclins so far and although there is a high level of sequence and structural homology between them, knowledge of their function varies. A typical protein kinase consists of a small N-terminal domain formed of β-sheets and a large C-terminal domain (CTD) formed of α-helices; between the terminals, there is an ATP-binding pocket and this pattern is also observed in CDKs. CDKs switch to an active conformation on binding to cyclin proteins which reveals enzymatic elements crucial to the catalytic function of their ATP-binding clefts. CDKs were named for this interaction with cyclin proteins which facilitates passage of cells through the G1, S, G2 and M phases of the cell division cycle (Meijer and Raymond, 2003). Progression of the cell cycle is, of necessity, heavily regulated as cancers arise from its dysfunction. This regulation is more dependent on the concentrations of endogenous CDKi (e.g. the Ink4 and Cip/Kip families) and cyclins than on the relatively stable CDK population. Extra layers of regulation are provided by phosphorylation and dephosphorylation of CDKs. We commented earlier that CDKi drug action on neutrophils appears counter-intuitive but given the diversity of function that CDKs have been demonstrated to possess, it is perhaps not surprising that another role for them has been found. Although initially identified as key components of the cell cycle machinery, they have subsequently been shown to play roles in cell differentiation, cell death (especially apoptosis), transcription and neuronal function. These alternative roles are mediated by different CDKs, an important factor in the drive to develop increasingly specific pharmacological CDKi drugs (Knockaert et al., 2002).
Endogenous CDKis
There are two major families of endogenous CDKis, the Ink4 family (which includes p15, p16, p18 and p19) and the Cip/Kip family (including p21, p27 and p57). Ink4 family proteins are responsible for the inhibition of cyclin D-associated CDK activity and can therefore inhibit CDK4 and CDK6 to cause G1 arrest. They achieve inhibition by competing with D-type cyclins at CDK-binding sites. Of the Ink4 family, the tumour suppressor, p16, has perhaps been studied in most detail and the p16 knockout mouse develops normally but has an increased susceptibility to development of tumours. This is in keeping with the known importance of p16 to the induction of senescence and anoikis (a form of programmed cell death caused by detachment of anchorage-dependent cells from surrounding extracellular matrix) and negative impact on various tumour activities including angiogenesis, cell spreading, vascular endothelial growth factor expression and cell growth. The Cip/Kip family inhibit the kinase activity of cyclin-CDK complexes, in particular, those formed by CDK2 leading once again to G1 arrest. p21 has been implicated in p53-dependent DNA damage-induced cell cycle cessation and also binds proliferating cell nuclear antigen leading to inhibition of DNA synthesis without affecting DNA repair. Interestingly, it appears that the p21 may be induced by both the signal transducers and activators of transcription (STAT) signal transduction pathway and TGF-β and negatively regulates TGF-β-induced apoptosis. Additionally, p21 has been shown to bind pro-caspase-3 in some cell types keeping a check on apoptosis (Vidal and Koff, 2000; Lee and Yang, 2001). This has led researchers to suggest the induction of p21 as a therapeutic strategy to prevent epithelial cell loss due to inflammatory injury. Paradoxically, however, another group has cited p21 as a pro-inflammatory mediator and have noted that the p21 knockout mouse is less susceptible to inflammatory injury caused by cigarette smoke. They attributed these findings to decreased NF-κB activation and decreased ROS release due to down-modulation of p21-activated kinase (Yao et al., 2008). Possible therapeutic applications of endogenous CDKis will be discussed later in the text, but it is clear that the Byzantine complexity of regulation of cyclins, CDKs and CDKi and their differing expression between cells and tissues will mean that conflicting results are almost to be expected.
CDKs 1 and 2
CDKs 1 and 2 have integral roles in the cell cycle, putative roles in transcriptional regulation and a controversial role in apoptosis (Golsteyn, 2005). Traditionally, it has been believed that CDK2 interaction with cyclin E facilitates G1/S transition, although this role has recently been called into question by knockout mouse studies suggesting that the complex is dispensable for transition, although it does have a role in centrosome duplication (Roberts and Sherr, 2003). A second CDK2 complex, CDK2–cyclin A, phosphorylates multiple substrates to inactivate G1 transcription factors and allow DNA replication. CDK1 binds cyclin A at S/G2 transition but binds cyclin B to trigger G2/M transition and, subsequently, has a role in the completion of mitosis by facilitating transition to anaphase (Golsteyn, 2005). CDKs 1 and 2 have been implicated in induction and facilitation of the apoptotic process in some experimental scenarios, while in others, it is clear that their inhibition promotes apoptosis. CDK2 knockout in HeLa cells and CDK1 knockout in an FT210 cell line resulted in a reduction in apoptosis, while levels of CDK1-cyclin complexes were noted to be elevated during apoptosis in HL-60 cells, YAC lymphoma cells and Jurkat cells. This observation had led some investigators to speculate that apoptosis might represent a form of ‘failed mitosis’ (Golsteyn, 2005). Additionally, CDK inhibition with R-roscovitine appeared to prevent apoptosis in human hepatoma cell lines and rat cerebellar neurons. In the latter model, it was suggested that CDK1 phosphorylates BAD (a pro-apoptotic bcl-2 homologue) which facilitates its liberation from sequestration by 14-3-3 proteins. This allows BAD to translocate to the mitochondrial membrane where it mediates apoptosis (Konishi et al., 2002). This work is in complete contrast to experimental experience with CDKi in various cancer cell lines where induction of apoptosis has been demonstrated (MacCallum et al., 2005).
Several CDKs have roles in transcriptional regulation which is perhaps not surprising given the extensive degree of regulation that the transcription process receives from protein kinases. CDK2 has been implicated in cisplatin-related acute kidney injury through involvement in the release of E2F transcriptional machinery by phosphorylation of Retinoblastoma (Rb) protein which promotes transcription of pro-apoptotic genes (Obligado et al., 2008). Additionally, CDK2 inhibition has been implicated in the induction of apoptosis in diffuse large B-cell lymphoma where CDK2 was linked to transcription of Mcl-1 (an important survival protein). Apoptosis induced by CDK2 inhibition was associated with down-regulation of Mcl-1 and reduced phosphorylation of the enzyme responsible for Mcl-1 gene transcription, RNA polymerase II (RNA Pol II) (Faber and Chiles, 2007). CDK1-mediated phosphorylation of the CTD of RNA Pol II has also been reported in vitro (Bregman et al., 2000). Despite a degree of controversy, the involvement of CDKs 1 and 2 in apoptosis and transcription (particularly of an important neutrophil survival protein) remains interesting when trying to define a role for CDK inhibition in the resolution of inflammation.
CDK5
CDK5 is perhaps most renowned as an important player in regulation of neuronal cytoarchitecture and migration, although evidence for its extra-neuronal effects is increasing. So far, there is literature describing involvement in apoptosis, transcription, differentiation and endocytosis. Interestingly, there is no evidence that it has any role to play in the cell cycle and as it can function independently of cyclin proteins, it is essentially misnamed. CDK5 has been implicated in the progression of apoptosis in mouse models of ovarian follicle degeneration, embryonic limb interdigital web apoptosis and digoxin-mediated prostate apoptosis (Rosales and Lee, 2006). CDK5 has also been shown to phosphorylate the important transcription factor STAT3 whose gene products include c-fos and jun-b as well as monocyte non-specific esterase. Of particular relevance to inflammation and apoptosis CDK5 complexed to p35 was expressed in HL60 cells induced to differentiate towards a monocytic phenotype. Specifically, this complex was shown to confer monocyte morphology-defining features such as CD14 and non-specific esterase suggesting a role for CDK5 in myeloid differentiation. Additionally, a key neutrophil function, GTP-dependent secretion, was shown to depend on CDK5-p35 (Rosales et al., 2004). Numerous CDK5-p35 substrates were found within neutrophil granules, while neutrophils treated with a CDKi drug lost GTP-dependent secretory function as well as CD63 and CD66b expression. Evidence of CDK5 involvement in transcriptional regulation, apoptosis and specific effects on inflammatory cells appears to be convincing (Dhavan and Tsai, 2001).
CDKs 4 and 6
CDKs 4 and 6 are required for progression through G1 phase and are activated by cyclin D in the presence of appropriate growth factors. As we have previously noted, neutrophils are terminally differentiated granulocytes. This irreversible cell cycle arrest is mediated by withdrawal of CDK4 and 6 and up-regulation of p27kip1 (an endogenous CDKi). CDK4 and 6, like CDK2, phosphorylate Rb to relieve E2F transcription factors that initiate the switch to S-phase transcription (Klausen et al., 2004). CDK4 and 6 have been implicated in inflammatory events within the rheumatoid joint where they are said to modulate rheumatoid synovial fibroblast production of MMP-3 and MCP-1 via Rb-dependent and -independent mechanisms (Nonomura et al., 2006). Importantly, cyclin D/CDK4 complexes have been shown to activate the STAT transcription pathway independently of JAK in Drosophila and CDK4 has been shown to be potentially important in leukocyte adhesion and migration (discussed later) (Silver and Montell, 2003; Liu et al., 2008). CDK4 and 6 appear to have roles in inflammatory cell differentiation, adhesion and recruitment as well as inflammatory cytokine production and possibly inflammatory signalling (Silver and Montell, 2003; Klausen et al., 2004; Nonomura et al., 2006; Liu et al., 2008). Targeting these CDKs would be facilitated by the current availability of specific pharmacological inhibitors of CDK4 and 6.
CDKs 7 and 9
CDK7 is a CDK-activating kinase responsible for enhancing the kinase activity of CDK1 and 2 by phosphorylation of the activation segment or T-loop. CDK7 associated with cyclin H and MAT1 is responsible for initiation of transcription by the holoenzyme RNA Pol II and mediates this effect by phosphorylation of the CTD. This is a function it shares with CDK9 which, when associated with cyclin T, is responsible for transcriptional elongation by RNA Pol II (Oelgeschlager, 2002). CDK7 and 9 have been implicated in transcription during early infection by CMV and are known to be important in activation of HIV1 transcription (Fisher, 2005; Kapasi and Spector, 2008). Perhaps, most interestingly, CDK7 and 9 with their respective binding partners have been shown to play an integral role in the aberrant survival of multiple myeloma cells. This effect is thought to be mediated by RNAPol II transcription of the survival protein Mcl-1. R-roscovitine, a CDKi which has specificity for CDKs 1,2,5,7 and 9, was able to promote apoptosis in these cells which was associated with down-regulation of Mcl-1 (MacCallum et al., 2005). Additionally CDK9 has also been shown to bind TRAF2, a protein that is of known importance in the activation of NF-κB mediated by TNF-α (MacLachlan et al., 1998; Wang and Fischer, 2008). Regulation of transcription of Mcl-1 (a key neutrophil survival protein) and involvement in pivotal inflammatory signalling via NF-κB are functions of CDKs 7 and 9 that might be applicable to the regulation of neutrophil apoptosis in inflammatory resolution.
CDKs 3,8,10 and 11
These CDKs are considered last because they are perhaps the least well studied to date. CDK3 has been implicated in G0 exit and G1/S phase transition in the cell cycle and binds cyclins C and E2 (van den and Harlow, 1993; Ren and Rollins, 2004). CDK8 has been shown to phosphorylate the CTD of RNA Pol II negatively affecting the activity of CDK7 but also sterically inhibits the interaction between mediator complex and RNA Pol II to repress transcription (Tassan et al., 1995; Elmlund et al., 2006). CDK10 which has a role in transition from G2 to M phase has been shown to bind the Ets 2 transcription factor and has been identified as a gene with potential significance in the aetiology of non-small cell lung cancer (Kasten and Giordano, 2001; Sun et al., 2005). CDK11 associates with cyclin L and cyclin D3 and has potentially dual roles in transcription that do not involve direct phosphorylation of RNA Pol II CTD. CDK11 has been linked to creatine kinase-2 (CK2)-mediated phosphorylation of RNAPol II CTD and also may increase the histone-acetyltransferase activity of HBO1 (Trembley et al., 2003; Zong et al., 2005). Interestingly, CDK11 has been implicated in melanoma cell apoptosis, a contribution that may be mediated by phosphorylation of death substrates and/or direct effects on mitochondrial permeability (Trembley et al., 2003). As more information becomes available, inhibition or activation of these CDKs may well prove important scientific and therapeutic targets.
CDKs in granulocytes
As neutrophils do not undergo cell division and can progress no further in the cell cycle (they are thought to remain in G0 phase), CDKs had not been thought relevant to their biology. There is, therefore, a paucity of information about CDKs, their interactions and potential roles in these cells. CDK1 and CDK2 proteins had been identified in neutrophils, but there was little indication of their function. No change in CDK1 or CDK2 protein expression was found in a variety of neutrophil populations including fresh human neutrophils, aged human neutrophils, survival factor-treated neutrophils at various time points and CDKi drug-treated neutrophils (Rossi et al., 2006). This is perhaps not a surprising finding as levels of CDKs 1 and 2 are relatively constant throughout the cell cycle, but changes in expression of their binding partner cyclins and endogenous CDKis modulate effects on cell cycle progression (Knockaert et al., 2002). It cannot be assumed, therefore, that these CDKs are not important in the induction of apoptosis even though they are not targets for degradation. As previously discussed, CDK1 and CDK2 can bind to various cyclin sub-types, whereas CDK5 binds non-cyclin partners including p35 and p39 regulatory proteins. In addition, CDK5 is expressed in human neutrophils as evidenced by detection of CDK5 mRNA and protein (Rosales et al., 2004). Evidence of CDK1, CDK2 and CDK5 activity in neutrophils is scarce but available. Our group has demonstrated a prompt reduction in CDK1 activity during induction of apoptosis by the activating Fas antibody CH11. The CDK-binding partners and regulatory proteins in neutrophils remain to be identified (Rossi et al., 2006). It is known that despite the terminally differentiated status of neutrophils, they are capable of phenotypic alteration. Indeed, it has been postulated following work with the ‘MacGreen’ mouse that mouse neutrophils sufficiently stimulated with CSF-1 in vitro can trans-differentiate into dendritic-type cells (Sasmono et al., 2007). This has implications for the role of neutrophils in the resolution of inflammation as it may significantly extend their capability to positively contribute. Another phenotypic switch from generation of pro-inflammatory mediators to anti-inflammatory and/or pro-resolving mediators such as lipoxins and resolvins has also been identified (Serhan, 2007). This raises the possibility that a transcriptional effect might be mediated by up- or down-regulation of protein kinases such as the CDKs. More information is required about CDKs, cyclins, endogenous CDKis and their function in inflammatory cells.
CDKs in lymphocytes
Lymphocytes would be expected to possess a full complement of functional cell cycle machinery given their retained capacity to proliferate and differentiate in response to the presence or withdrawal of mitogenic factors. CDK2 is central to T-lymphocyte proliferation and it has previously been shown that in combination with antigenic stimulation, IL-2 can down-regulate the endogenous CDKi, p27 to promote peripheral T-lymphocyte proliferation. By contrast, the immunosuppressant drug rapamycin mediates some of its effects through stabilization of p27, thus preventing IL-2-mediated T-lymphocyte proliferation. It is increasingly clear that CDKs have a pivotal role in the regulation of T-cell immune responsiveness versus anergy (Wells, 2007). p21 knockout mice have been shown to have an autoimmune phenotype including excessive T-cell activation and proliferation with reactance to self-nuclear antigens. This is said to resemble the autoimmune disease lupus, and reconstitution of p21 functionality was shown to halt disease progression (Zoja et al., 2007). It is expected that targeting CDK2 may have dual therapeutic applicability first in promoting immune tolerance in transplant recipients and secondarily by switching off auto-reactive T-cell responses in autoimmune disease. B-cell development, proliferation and differentiation are also dependent on cell cycle control. The archetypal adaptive immune response, antibody generation, requires activation and expansion of antigen-specific B-cells and their differentiation into plasma cells. Plasma cells arrest at G1 phase of the cell cycle exiting as the major arm of the humoral immune response. p18, an endogenous CDKi, has been implicated in both cell cycle exit and differentiation of antibody-secreting plasma cells from plasmacytoid cells (Morse et al., 1997; Chen-Kiang, 2003). Cell cycle competent lymphocytes may be beneficially affected by exposure to CDKi drugs. Experience with animal models and applicability of CDKi drugs will be discussed later in the review.
Pharmacological CDK inhibition
Pioneering work on the cell cycle in starfish oocytes in the 1990s, driven by the realization that anti-mitotic agents would make effective anti-cancer drugs, led to the discovery of CDKi drugs (for a fascinating account of this discovery, see Meijer and Raymond, 2003). The widely available, non-specific kinase inhibitors staurosporine, 6 D-MAP and isopentanyladine were found to potently inhibit CDK1/cyclin B but were too non-specific in action to yield useful information about the functional impact. A laborious screening process subsequently uncovered olomoucine, a purine analogue that showed enhanced specificity for CDKs but some action against MAPKs. This compound was shown to mediate its inhibitory effect by competing with ATP for a binding site on CDK1. Further structural analysis of this ATP-binding site interaction identified the 2,6,9 trisubstituted purine family as likely candidates for effective CDK inhibition. This was the development that brought R-roscovitine and Purvalanol B into the frame and led to the identification of a family of approximately 50 CDKi drugs (Meijer and Raymond, 2003) (Table 1).
Table 1.
IC50 values for selected CDK inhibitor drugs. (Knockaert et al., 2002; Obligado et al., 2008), (Krystof et al., 2002; Bettayeb et al., 2007)
| CDKi |
IC50 values for kinase inhibited by CDKi (µM) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| CDK1/cyclinb | CDK2/cyclinE | CDK4 | CDK5 | CDK7 | CDK9 | GSK | ERK1 | ERK2 | |
| R-roscovitine | 0.45,2.7 | 0.13,0.7 | 14.2,14.7, >100 | 0.16 | 0.49 | 0.74,0.78 | 130 | 34 | 14 |
| Olomoucine | 7 | 7 | >1000 | 3 | 100 | 30 | 50 | ||
| Olomoucine II | 7.6 | 0.1 | 19.8 | 0.45 | 0.06 | >100 | 32 | ||
| Purvalanol B | 0.006 | 0.009 | >10 | 0.006 | >10 | 3.33 | |||
| Flavopiridol | 0.06,0.4 | 0.15 | 0.4 | 0.17 | 0.3 | 0.006 | 0.45 | ||
| Aminopurvalanol | 0.033 | 0.028 | 0.02 | 12 | 2.4 | ||||
| Hymenialdisine | 0.022 | 0.04 | 0.6 | 0.028 | 0.01 | 0.47 | 2 | ||
| Fascaplysin | >100 | >50 | 0.35 | 20 | |||||
| OL567 | 0.23 | ||||||||
| H717 | 0.23 | 0.05 | |||||||
| NU2058 | 5 | 12 | |||||||
| NU6027 | 2.5 | 1.3 | |||||||
| Staurosporine | 0.006 | 0.007 | <10 | 0.02 | 0.02 | ||||
| PD183812 | >40 | 0.17 | 0.008 | ||||||
| Meriolin 1 | 0.78 | 0.09 | 0.51 | 0.026 | 0.63 | ||||
| SU 9516 | 0.04 | 0.2 | |||||||
| Alsterpaullone | 0.035 | 0.2 | >10 | 0.04 | 0.004 | 22 | 4.5 | ||
CDKi drugs are universally (to date) flat, hydrophobic heterocycles of low molecular weight that mediate binding to the ATP pocket via hydrophobic interactions and hydrogen bonds. Surprisingly, despite this uniformity, it is possible to categorize the CDKi drugs on the basis of their relative specificity for CDKs: (i) non-specific (Flavopiridol); (ii) CDKs 1,2,5,7-specific (Olomoucine, R-roscovitine and Purvalanol B); (iii) CDK 4,6-specific (fascaplysin, PD0183812). It is clear that any discussion of CDKi drug specificity for individual CDKs can only be couched in relative terms (Table 1). Additionally, there are some non-CDK targets of these inhibitors identified on random screening which include ERK, GSK3 and pyridoxal kinase (Knockaert et al., 2002). Given the range of activities performed by CDKs on a variety of vital cell processes, a lack of incisive specificity attributable to an inhibitor makes it extremely difficult to assign outcomes to specific actions on defined targets. The basis for this review is that CDKi drugs have proven efficacy in models of inflammation, but the reality is that these effects may not relate to any action on CDKs themselves. An obvious point of interest then is the inhibitory effect of certain CDKi drugs on the ERK signalling pathway. Recent work has shown that specific ERK inhibitors can enhance the resolution of carrageenan-induced pleurisy in rats, an accepted model of acute inflammation (Sawatzky et al., 2006). However, it seems likely that the inhibitory effect of CDKi drugs on the ERK pathway would not account for the anti-inflammatory actions of R-roscovitine. In addition, the biological effect of CDKi and ERK inhibitors on neutrophil apoptosis differs markedly. For example, the ERK inhibitor PD98059 does not induce neutrophil apoptosis per se, whereas CDKi drugs do; R-roscovitine reverses lipopolysaccharide (LPS), granulocyte macrophage-colony stimulating factor (GM-CSF) and db-cAMP-mediated prolongation of neutrophil survival, whereas PD98059 only reverses LPS and GM-CSF survival; finally, PD98059 completely inhibits LPS-induced phospho-ERK expression in neutrophils, whereas R-roscovitine does not (N. Riley, A.E. Leitch, A.G. Rossi, unpubl. data). Regardless of the difficulties associated with non-specificity, the ability of CDKi drugs to selectively induce apoptosis in actively proliferating cells has meant that research into their use as anti-cancer agents has progressed rapidly.
CDKi drugs including R-roscovitine, flavopiridol and SU9516 have anti-cancer actions and R-roscovitine alone is known to reduce the proliferation index of 19 distinct cancer cell lines in vitro (McClue et al., 2002). The mechanism of action of CDKi drugs in cancer cell lines, while not directly applicable to inflammatory cells, is of great interest to those interested in targeting CDKs to resolve inflammation. CDKi drugs were an exciting therapeutic prospect because CDKs interact with the Rb family widely regarded as the master switch of the cell cycle. CDKs are over-abundant in some cancers resulting in excessive phosphorylation and functional inactivation of Rb, allowing unregulated proliferation (Johnson et al., 1994). However, it was noted that R-roscovitine was capable not only of inducing cell cycle arrest, but of active promotion of apoptosis. The induction of apoptosis by R-roscovitine in myeloma cell lines has been reported to be promoted by CDK modulation of RNA Pol II-mediated transcription of Mcl-1. Additionally, down-regulation of XIAP, another Bcl-2 homologous survival protein, was observed in chronic lymphatic leukaemia cells, and apoptosis in both cell lines was found to be caspase dependent (Hahntow et al., 2004; MacCallum et al., 2005). A recent paper has shown inhibition of the well-known inflammatory transcription factor, NF-κB by R-roscovitine in A549, 293, H1299 and ARN8 cancer cell lines (Dey et al., 2008). It is tempting to directly extrapolate from these findings to inflammation studies but given the sometimes directly contrasting effects of pharmacological agents in different cell types (e.g. the anti-apoptotic effects of CDK inhibition in cerebellar neuronal cells), we must await further definitive studies. The availability of this mechanistic information and encouraging safety profiles in animal studies have allowed progression to clinical trials of CDKi drugs in conditions such as B-cell malignancy, non-small cell lung cancer and breast cancer. The side-effect profile has been promising but includes tolerable and limited gastrointestinal disturbance, skin rash, reversible transaminitis and hypokalaemia (Senderowicz, 2003). This transition to human studies makes an elucidation of the mechanism of action of CDKi drugs in inflammation imperative, as trials of these drugs in inflammatory disease are likely to be on the near horizon.
CDK inhibitors in inflammation
The research discussed above led our group to investigate a role for CDKi drugs in inflammation. Our research paradigm has been that inflammation should be driven down resolution pathways by the induction of apoptosis in granulocytes followed by their effective clearance by professional phagocytes such as macrophages (Leitch et al., 2008). We initially examined the in vitro effects of R-roscovitine, NG-75 and hymenialdisine on human neutrophils at different time points and drug concentrations. These CDKi drugs are structurally diverse but their uniform effect was to promote neutrophil apoptosis in a time- and concentration-dependent manner as evidenced by annexin-V binding and morphological assessment. Neutrophils treated with R-roscovitine and the caspase inhibitor zVAD-fmk failed to enter apoptosis, suggesting that R-roscovitine was acting in a caspase-dependent manner. This was evident at 4 h post-incubation when caspase 3 cleavage was already detectable. The most intriguing in vitro result was the ability of the CDKi drugs to overcome diverse survival factors including db-cAMP, GM-CSF and LPS. These survival factors utilize the major inflammatory signalling pathways: PI3K, NF-κB, JAK/STAT and MAPK to augment neutrophil survival. Given that the major hurdle to development of anti-inflammatory agents is the redundancy conferred by these same signalling pathways, this result underlined the potential of CDKi therapy for treatment of inflammatory disease. Inflammation research demands in vivo experimentation as it is impossible to recreate the inflammatory milieu in vitro and hence to predict the efficacious translation of an agent that has been successful in vitro. R-roscovitine was investigated in three mouse models of neutrophil-dominant inflammation including: carrageenan-induced pleurisy, bleomycin lung injury and passively induced arthritis. In the carrageenan pleurisy model, 100 mg·kg−1 of R-roscovitine administered intra-peritoneally reduced an established inflammatory infiltrate to near-basal levels consistent with those found in an untreated mouse pleural cavity. There was a reduction in populations of inflammatory cells (including neutrophils, monocytes and macrophages), oedema formation and pro-inflammatory cytokines. This effect was reversed in vivo by administration of the caspase inhibitor zVAD-fmk. In mice with established bleomycin lung injury, there was a reduction in BAL neutrophil numbers assessed after 3 days, a reduction in histopathological lung inflammation after 7 days and an effect on bleomycin-induced lethality. Finally, in mice with established passively induced arthritis, there was an improvement in clinical scores of arthritis following R-roscovitine administration (Rossi et al., 2006). These in vivo findings suggest encouraging pleiotropic effects of CDKi drug on granulocyte recruitment, survival and removal. Pleiotropic effects of CDKi drug are supported by work from Liu et al. who have shown that CDK4 is important in leukocyte recruitment and adhesion. They studied CDK4−/− knockout mice with bleomycin lung injury and utilized siRNA to CDK4 and CDKi to show that CDK4 inhibition inhibited leukocyte recruitment in the mouse model and leukocyte adhesion in EC matrix models (Liu et al., 2008). In addition, Sekine et al. have now demonstrated positive effects of flavopiridol and a specific CDK4,6 inhibitor on animal models of rheumatoid arthritis (Sekine et al., 2008). Their findings suggest lymphocyte-independent effects of CDKi drugs (including down-regulation of fibroblast proliferation and growth) responsible for improvement in joint histology and clinical arthritis scores in various mouse models. Findings in inflammatory joint and lung disease models are mirrored in kidney disease models where the CDKi drug R-roscovitine has entered phase 1b clinical trials for inflammatory kidney disease. Glomerulonephritides are characterized by inflammation and progressive, scarring destruction of key functional kidney units leading to renal dysfunction and failure. Preclinically, CDKi drugs have been shown to protect renal tubular epithelium from enhanced apoptosis while inhibiting the abnormal proliferation of tubular epithelial and mesangial cells. In vitro and in vivo work has demonstrated that R-roscovitine can restore normal kidney function in animal models of IgA-mediated glomerulonephritis, crescentic glomerulonephritis, lupus nephritis and collapsing glomerulonephropathy (Gherardi et al., 2004; Milovanceva-Popovska et al., 2005; Zoja et al., 2007; Obligado et al., 2008). The work with NZBxNZW mice affected by early or established proliferative lupus nephritis is particularly interesting, as while leucocyte-driven inflammation was shown to be reduced, there was also evidence of a direct effect of CDKi against autoimmune T- and B-lymphocyte responses. It is perhaps less surprising that CDKi should work in this setting, given the proliferative potential/state of differentiation of lymphocytes, but nonetheless, the possibility of pleiotropic action against autoimmune inflammation is an exciting one.
Further evidence for the utility of CDK inhibition has been provided by research into the properties of endogenous CDKis. The physiological CDKi p21 (WAF1, SD1, Cip1), a specific inhibitor of CDK2, 4 and 6, has been shown to negatively regulate macrophage activation by reducing TNF-α and IL-1β production in response to LPS. Additionally, p21−/− mice have an increased susceptibility to LPS-induced shock which is associated with elevated levels of IL-1β (Lloberas and Celada, 2009; Scatizzi et al., 2009). In an inflammatory lung disease model, p21 was over-expressed in the lungs of mice subjected to bleomycin injury by an intra-tracheal adenoviral transfer method (Inoshima et al., 2004). p21 expression in lung epithelial cells led to a reduction in lung inflammation, preservation of epithelial cells and reduced fibrosis. In rheumatoid arthritis patients, p21 gene transfer was shown to down-regulate expression of inflammatory mediators and tissue-degrading proteinases such as: IL-6, -8, type I IL-1 receptor, monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-3alpha, cathepsins B and K, and matrix metalloproteinases-1 and -3 (Nonomura et al., 2003).
In summary, CDK inhibition promotes neutrophil apoptosis in vitro even in the presence of powerful survival factors and promotes resolution of inflammation, in vivo, in various animal models of neutrophil-dominant inflammation. Neutrophil apoptosis has been shown to be central to the resolution of inflammation by caspase inhibition which reversed the beneficial impact of CDKi drugs. In addition, the anti-proliferative and anti-apoptotic effects of CDKi drugs protect epithelia against inflammatory insult. CDKi drugs also prevent lymphocyte proliferation and pro-inflammatory signalling indicating potential effects against chronic and autoimmune, inflammatory disease. An understanding of the mechanism of action by which these powerful anti-inflammatory effects are achieved may allow optimization of CDK inhibition and suggest further targets for pharmacological intervention.
Challenges associated with CDK inhibition as a therapeutic strategy
Apart from the side-effect profile associated with known CDKi drugs and already discussed, there are some potential drawbacks to CDKi therapy. The most obvious of these drawbacks is the degree of redundancy apparent in the cell cycle machinery. The best example of this is the CDK2 knockout mouse which survives (it is subsequently sterile) despite the central cell cycle role attributed to CDK2 (Berthet et al., 2003). The complicated redundancy of the inflammatory system means that single-hit therapies may prove ineffectual, which may be the case with CDK targeting. However, it is proving difficult to achieve more than pan-selectivity for individual CDKs, which may be a blessing in disguise. An expected side effect of the aggressive induction of neutrophil apoptosis would be neutropenia and although this was not reported in early trials, it appears that an early (though recovering) neutropenia is a feature. While this may not be beneficial in cancer patients, in patients with neutrophil-dominant inflammation, it would be hoped that this effect could be dose limited and controlled, especially given some data that suggest that stimulated inflammatory neutrophils are more susceptible to CDKi than unstimulated. It would of course be important to ensure that infective aetiologies for disease had been excluded and antibiotic prophylaxis might be appropriate in some circumstances. Finally, neutrophil apoptosis must not overload macrophage capacity for phagocytosis or the sequelae of secondary necrosis could occur. It may be that combination therapy with phagocytosis-enhancing drugs (e.g., steroids) might prevent this occurrence.
Concluding remarks: Potential mechanisms for anti-inflammatory action of the CDK inhibitors and the future of CDK targeting
The indications from the available CDK/CDKi literature are that CDKs may be important targets of CDKi anti-inflammatory action. They are known to have key roles in inflammatory cell differentiation, in neutrophil function and in influencing established inflammatory signalling pathways. Additionally, they are intimately involved in the regulation of apoptosis and gene transcription in a number of cell lines. There are also strong links between cancer and inflammation as highlighted by a number of recent reviews and CDK inhibition is an efficacious therapeutic approach in a number of cancers (Coussens and Werb, 2002; Hussain and Harris, 2007). Currently, the role of CDKs in granulocyte apoptosis and the resolution of inflammation are unclear, but a number of plausible hypotheses are apparent.
Gene transcription and survival proteins
It is known that R-roscovitine promotes apoptosis in multiple myeloma cell lines via inhibition of CDK- mediated phosphorylation of RNA Pol II which down-regulates Mcl-1 expression. CDKs 1,2,7,8 and 9 are all capable of RNA Pol II CTD phosphorylation. The applicability of this to neutrophil biology is unclear; however, it is established that Mcl-1 is a key survival protein in neutrophils (Edwards et al., 2004; Dzhagalov et al., 2007). Our own experiments have shown a rapid reduction in Mcl-1 protein following R-roscovitine treatment of neutrophils that occurs even in the presence of powerful survival factors (in fact, its reduction is enhanced in the presence of survival factors, unpubl. data). We have not as yet established the mechanism by which this occurs and Mcl-1 is a highly regulated protein, so it is possible or even probable that there is an independent effect at work. It is also possible that R-roscovitine has a more direct effect on Mcl-1 RNA processing or protein expression. A splice variant of Mcl-1 has potentially pro-apoptotic properties and interference with the balance of competing variants of Mcl-1 could result in rapid apoptosis. Mcl-1 protein is subject to ubiquitination with subsequent turnover by the proteasome. Varying levels of phosphorylation have been demonstrated to either postpone or facilitate this process. Different pharmacological agents have been demonstrated to stabilize or destabilize Mcl-1 with down-regulation occurring by both proteasome and caspase-dependent mechanisms (Derouet et al., 2004; Mandelin and Pope, 2007).
CDK4 and leucocyte recruitment
At least part of the pro-resolution of inflammation effect of CDKi may be explained by the impact of CDK4 knockout on leukocyte recruitment and adhesion, although this would not explain the ability of CDKi drugs to promote granulocyte apoptosis, and given that CDK4 is not within the specificity of R-roscovitine, it would not explain the anti-inflammatory effects of this compound (Liu et al., 2008).
CDK balance and apoptosis
An alternative hypothesis is that the complement of CDKs, cyclins and endogenous CDKi is important in the maintenance of neutrophil survival. As the specificity of R-roscovitine extends across CDKs 1,2,5,7 and 9, it is not currently possible to attribute the pro-apoptotic effect to one CDK, but it may be that effects on multiple CDKs are important.
Inflammatory signalling mechanisms
Signalling pathways such as MAPK, NF-κB and PI3K are extremely important in the regulation of the inflammatory response and in neutrophil survival (Suzuki et al., 1999; Ward et al., 1999; Lindemans and Coffer, 2004). At high concentrations, R-roscovitine is known to have a direct inhibitory effect on ERK 1 and 2 (Meijer and Raymond, 2003) and we know from work in our laboratory that specific ERK inhibitors are anti-inflammatory in vitro and in vivo. It is possible that this effect makes an important contribution to the actions of R-roscovitine. NF-κB inhibition was recently identified as playing a role in the anti-cancer actions of R-roscovitine and NF-κB as well as being central to the inflammatory signalling system is a powerful regulator of neutrophil survival (Ward et al., 1999). However, our own experimental results suggest that this effect is not reproduced in neutrophils (unpubl. data). PI3K has importance in neutrophil survival and recruitment and is implicated in the effects of CDK inhibition on xenopus oocyte maturation and the lethal effects of CDK inhibition on the human leukaemia cell lines: U937, HL-60, Jurkat, Raji, and NB4 (Hehl et al., 2001; Yu et al., 2003; Pinho et al., 2005).
An effect on one or multiple inflammatory signalling systems is a potential explanation for observations with regard to CDKi drugs and inflammation. In many respects, it would be ideal if CDKi drugs acted in a pleiotropic manner to counter the inherent redundancy of the inflammatory system (Figure 3).
Figure 3.
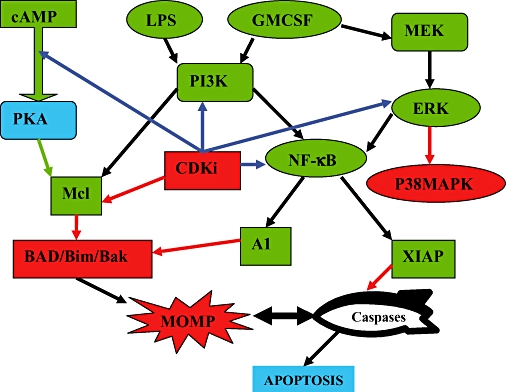
Known and potential CDKi effects on neutrophil survival signalling in inflammation.
It is clear that CDKs hold promise as novel pharmacological targets in inflammation, although further research is required to confirm this hypothesis. The development of new and more specific CDKi drugs continues apace and is increasingly supported by structural elucidation and mechanistic studies. While incisive specificity may not prove beneficial in the treatment of disease, it would certainly prove an important addition to our scientific tool kit. The future of CDK targeting must lie in a human trial of efficacy against neutrophil-dominant or autoimmune inflammatory disease and we expect this to occur in the near future.
Glossary
Abbreviations:
- AA
arachidonic acid
- CDKi
cyclin-dependent kinase inhibitor
- COX
cyclo-oxygenase
- CTD
C-terminal domain
- ERK
extracellular regulated kinase
- GM-CSF
granulocyte macrophage-colony stimulating factor
- IL
interleukin
- JAK
Jun N-terminal Kinase
- LPS
lipopolysaccharide
- MAPK
mitogen activated protein kinase
- Mcl-1
myeloid cell leukaemia-1
- MCP-1
monocyte chemoattractant protein-1
- MMP-3
matrix metalloproteinase-3
- NF-κB
nuclear factor-kappaB
- NSAIDs
non-steroidal anti-inflammatory drugs
- PCNA
proliferating cell nuclear antigen
- PG
prostaglandin
- PI3K
phosphoinositide-3-kinase
- RNA Pol II
RNA polymerase II
- siRNA
small interfering RNA
- STAT
signal transducers and activators of transcription
- TGF-β
transforming growth factor beta
- TNF-α
tumour necrosis factor alpha
- TXA2
thromboxane A2
- VEGF
vascular endothelial growth factor
- zVAD-fmk
Z-Val-Ala-Asp(OMe)-CH2F
Note added in proof
A very recent, important and elegant study by Koedel et al. (2009) has shown that roscovitine induction of neutrophil apoptosis in vivo promoted the resolution of inflammation of mice treated with antibiotics following experimental pneumococcal meningitis.
Conflict of interest
None.
References
- Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–1785. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Bettayeb K, Tirado OM, Marionneau-Lambot S, Ferandin Y, Lozach O, Morris JC, et al. Meriolins, a new class of cell death inducing kinase inhibitors with enhanced selectivity for cyclin-dependent kinases. Cancer Res. 2007;67:8325–8334. doi: 10.1158/0008-5472.CAN-07-1826. [DOI] [PubMed] [Google Scholar]
- Bianchi SM, Dockrell DH, Renshaw SA, Sabroe I, Whyte MK. Granulocyte apoptosis in the pathogenesis and resolution of lung disease. Clin Sci. 2006;110:293–304. doi: 10.1042/CS20050178. [DOI] [PubMed] [Google Scholar]
- Bregman DB, Pestell RG, Kidd VJ. Cell cycle regulation and RNA polymerase II. Front Biosci. 2000;5:D244–D257. doi: 10.2741/bregman. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. 2007;5:577–582. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- Chen-Kiang S. Cell-cycle control of plasma cell differentiation and tumorigenesis. Immunol Rev. 2003;194:39–47. doi: 10.1034/j.1600-065x.2003.00065.x. [DOI] [PubMed] [Google Scholar]
- Chung F. Anti-inflammatory cytokines in asthma and allergy: interleukin-10, interleukin-12, interferon-gamma. Mediators Inflamm. 2001;10:51–59. doi: 10.1080/09629350120054518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley HO, Kelly RW, Brenner RM, Baird DT. The endocrinology of menstruation – a role for the immune system. Clin Endocrinol. 2001;55:701–710. doi: 10.1046/j.1365-2265.2001.01432.x. [DOI] [PubMed] [Google Scholar]
- Derouet M, Thomas L, Cross A, Moots RJ, Edwards SW. Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J Biol Chem. 2004;279:26915–26921. doi: 10.1074/jbc.M313875200. [DOI] [PubMed] [Google Scholar]
- Dey A, Wong ET, Cheok CF, Tergaonkar V, Lane DP. R-Roscovitine simultaneously targets both the p53 and NF-kappaB pathways and causes potentiation of apoptosis: implications in cancer therapy. Cell Death Differ. 2008;15:263–273. doi: 10.1038/sj.cdd.4402257. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Downey GP, Worthen GS, Henson PM, Hyde DM. Neutrophil sequestration and migration in localized pulmonary inflammation. Capillary localization and migration across the interalveolar septum. Am Rev Respir Dis. 1993;147:168–176. doi: 10.1164/ajrccm/147.1.168. [DOI] [PubMed] [Google Scholar]
- Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood. 2007;109:1620–1626. doi: 10.1182/blood-2006-03-013771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SW, Derouet M, Howse M, Moots RJ. Regulation of neutrophil apoptosis by Mcl-1. Biochem Soc Trans. 2004;32:489–492. doi: 10.1042/BST0320489. [DOI] [PubMed] [Google Scholar]
- Elmlund H, Baraznenok V, Lindahl M, Samuelsen CO, Koeck PJ, Holmberg S, et al. The cyclin-dependent kinase 8 module sterically blocks Mediator interactions with RNA polymerase II. Proc Natl Acad Sci USA. 2006;103:15788–15793. doi: 10.1073/pnas.0607483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber AC, Chiles TC. Inhibition of cyclin-dependent kinase-2 induces apoptosis in human diffuse large B-cell lymphomas. Cell Cycle. 2007;6:2982–2989. doi: 10.4161/cc.6.23.4994. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M, Maini RN. Discovery of TNF-alpha as a therapeutic target in rheumatoid arthritis: preclinical and clinical studies. Joint Bone Spine. 2002;69:12–18. doi: 10.1016/s1297-319x(01)00335-9. [DOI] [PubMed] [Google Scholar]
- Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118:5171–5180. doi: 10.1242/jcs.02718. [DOI] [PubMed] [Google Scholar]
- Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherardi D, D'Agati V, Chu TH, Barnett A, Gianella-Borradori A, Gelman IH, et al. Reversal of collapsing glomerulopathy in mice with the cyclin-dependent kinase inhibitor CYC202. J Am Soc Nephrol. 2004;15:1212–1222. doi: 10.1097/01.asn.0000124672.41036.f4. [DOI] [PubMed] [Google Scholar]
- Gilgun-Sherki Y, Melamed E, Offen D. Anti-inflammatory drugs in the treatment of neurodegenerative diseases: current state. Curr Pharm Des. 2006;12:3509–3519. doi: 10.2174/138161206778343091. [DOI] [PubMed] [Google Scholar]
- Girkontaite I, Urbonaviciute V, Maseda D, Neubert K, Herrmann M, Voll RE. Apoptotic cells selectively suppress the Th1 cytokine interferon gamma in stimulated human peripheral blood mononuclear cells and shift the Th1/Th2 balance towards Th2. Autoimmunity. 2007;40:327–330. doi: 10.1080/08916930701356846. [DOI] [PubMed] [Google Scholar]
- Golsteyn RM. Cdk1 and Cdk2 complexes (cyclin dependent kinases) in apoptosis: a role beyond the cell cycle. Cancer Lett. 2005;217:129–138. doi: 10.1016/j.canlet.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Hahntow IN, Schneller F, Oelsner M, Weick K, Ringshausen I, Fend F, et al. Cyclin-dependent kinase inhibitor Roscovitine induces apoptosis in chronic lymphocytic leukemia cells. Leukemia. 2004;18:747–755. doi: 10.1038/sj.leu.2403295. [DOI] [PubMed] [Google Scholar]
- Hallett JM, Leitch AE, Riley NA, Duffin R, Haslett C, Rossi AG. Novel pharmacological strategies for driving inflammatory cell apoptosis and enhancing the resolution of inflammation. Trends Pharmacol Sci. 2008;29:250–257. doi: 10.1016/j.tips.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med. 1999;160:S5–11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- Haslett C, Savill J, Meagher L. Macrophage recognition of senescent granulocytes. Biochem Soc Trans. 1990;18:225–227. doi: 10.1042/bst0180225. [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Giles KM, Ward C, Rossi AG, Haslett C, Dransfield I. Glucocorticoid-mediated regulation of granulocyte apoptosis and macrophage phagocytosis of apoptotic cells: implications for the resolution of inflammation. J Endocrinol. 2003;178:29–36. doi: 10.1677/joe.0.1780029. [DOI] [PubMed] [Google Scholar]
- Hehl S, Stoyanov B, Oehrl W, Schonherr R, Wetzker R, Heinemann SH. Phosphoinositide 3-kinase-gamma induces Xenopus oocyte maturation via lipid kinase activity. Biochem J. 2001;360:691–698. doi: 10.1042/0264-6021:3600691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373–2380. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- Inoshima I, Kuwano K, Hamada N, Yoshimi M, Maeyama T, Hagimoto N, et al. Induction of CDK inhibitor p21 gene as a new therapeutic strategy against pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L727–L733. doi: 10.1152/ajplung.00209.2003. [DOI] [PubMed] [Google Scholar]
- Johnson DG, Ohtani K, Nevins JR. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 1994;8:1514–1525. doi: 10.1101/gad.8.13.1514. [DOI] [PubMed] [Google Scholar]
- Kapasi AJ, Spector DH. Inhibition of the cyclin-dependent kinases at the beginning of human cytomegalovirus infection specifically alters the levels and localization of the RNA polymerase II carboxyl-terminal domain kinases cdk9 and cdk7 at the viral transcriptosome. J Virol. 2008;82:394–407. doi: 10.1128/JVI.01681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten M, Giordano A. Cdk10, a Cdc2-related kinase, associates with the Ets2 transcription factor and modulates its transactivation activity. Oncogene. 2001;20:1832–1838. doi: 10.1038/sj.onc.1204295. [DOI] [PubMed] [Google Scholar]
- Klausen P, Bjerregaard MD, Borregaard N, Cowland JB. End-stage differentiation of neutrophil granulocytes in vivo is accompanied by up-regulation of p27kip1 and down-regulation of CDK2, CDK4, and CDK6. J Leukoc Biol. 2004;75:569–578. doi: 10.1189/jlb.1003474. [DOI] [PubMed] [Google Scholar]
- Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002;23:417–425. doi: 10.1016/s0165-6147(02)02071-0. [DOI] [PubMed] [Google Scholar]
- Koedel U, Frankenberg T, Kirschnck S, Obermaier B, Häcker H, Paul R, et al. Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog. 2009;5:e1000461. doi: 10.1371/journal.ppat.1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Krystof V, Lenobel R, Havlicek L, Kuzma M, Strnad M. Synthesis and biological activity of olomoucine II. Bioorg Med Chem Lett. 2002;12:3283–3286. doi: 10.1016/s0960-894x(02)00693-5. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Gilroy DW. Chronic inflammation: a failure of resolution? Int J Exp Pathol. 2007;88:85–94. doi: 10.1111/j.1365-2613.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med. 2001;7:1291–1297. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol. 2002;2:787–795. doi: 10.1038/nri915. [DOI] [PubMed] [Google Scholar]
- Lee MH, Yang HY. Negative regulators of cyclin-dependent kinases and their roles in cancers. Cell Mol Life Sci. 2001;58:1907–1922. doi: 10.1007/PL00000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch AE, Duffin R, Haslett C, Rossi AG. Relevance of granulocyte apoptosis to resolution of inflammation at the respiratory mucosa. Mucosal Immunol. 2008;1:350–363. doi: 10.1038/mi.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemans CA, Coffer PJ. Regulation of granulocyte apoptosis by phosphatidylinositol 3-kinase. Biochem Soc Trans. 2004;32:480–484. doi: 10.1042/BST0320480. [DOI] [PubMed] [Google Scholar]
- Liu L, Schwartz B, Tsubota Y, Raines E, Kiyokawa H, Yonekawa K, et al. Cyclin-dependent kinase inhibitors block leukocyte adhesion and migration. J Immunol. 2008;180:1808–1817. doi: 10.4049/jimmunol.180.3.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cousin JM, Hughes J, Van DJ, Seckl JR, Haslett C, et al. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol. 1999;162:3639–3646. [PubMed] [Google Scholar]
- Lloberas J, Celada A. p21(waf1/CIP1), a CDK inhibitor and a negative feedback system that controls macrophage activation. Eur J Immunol. 2009;39:691–694. doi: 10.1002/eji.200939262. [DOI] [PubMed] [Google Scholar]
- MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, et al. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65:5399–5407. doi: 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]
- MacLachlan TK, Sang N, De LA, Puri PL, Levrero M, Giordano A. Binding of CDK9 to TRAF2. J Cell Biochem. 1998;71:467–478. [PubMed] [Google Scholar]
- Mandelin AM, Pope RM. Myeloid cell leukemia-1 as a therapeutic target. Expert Opin Ther Targets. 2007;11:363–373. doi: 10.1517/14728222.11.3.363. [DOI] [PubMed] [Google Scholar]
- Marchalonis JJ, Kaveri S, Lacroix-Desmazes S, Kazatchkine MD. Natural recognition repertoire and the evolutionary emergence of the combinatorial immune system. FASEB J. 2002;16:842–848. doi: 10.1096/fj.01-0953hyp. [DOI] [PubMed] [Google Scholar]
- McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM, et al. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine) Int J Cancer. 2002;102:463–468. doi: 10.1002/ijc.10738. [DOI] [PubMed] [Google Scholar]
- Meagher LC, Cousin JM, Seckl JR, Haslett C. Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J Immunol. 1996;156:4422–4428. [PubMed] [Google Scholar]
- Meijer L, Raymond E. Roscovitine and other purines as kinase inhibitors. From starfish oocytes to clinical trials. Acc Chem Res. 2003;36:417–425. doi: 10.1021/ar0201198. [DOI] [PubMed] [Google Scholar]
- Michlewska S, Dransfield I, Megson IL, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-alpha. FASEB J. 2009;23:844–854. doi: 10.1096/fj.08-121228. [DOI] [PubMed] [Google Scholar]
- Milovanceva-Popovska M, Kunter U, Ostendorf T, Petermann A, Rong S, Eitner F, et al. R-roscovitine (CYC202) alleviates renal cell proliferation in nephritis without aggravating podocyte injury. Kidney Int. 2005;67:1362–1370. doi: 10.1111/j.1523-1755.2005.00213.x. [DOI] [PubMed] [Google Scholar]
- Morse L, Chen D, Franklin D, Xiong Y, Chen-Kiang S. Induction of cell cycle arrest and B cell terminal differentiation by CDK inhibitor p18(INK4c) and IL-6. Immunity. 1997;6:47–56. doi: 10.1016/s1074-7613(00)80241-1. [DOI] [PubMed] [Google Scholar]
- Munoz LE, Gaipl US, Franz S, Sheriff A, Voll RE, Kalden JR, et al. SLE – a disease of clearance deficiency? Rheumatology. 2005;44:1101–1107. doi: 10.1093/rheumatology/keh693. [DOI] [PubMed] [Google Scholar]
- Nonomura Y, Kohsaka H, Nagasaka K, Miyasaka N. Gene transfer of a cell cycle modulator exerts anti-inflammatory effects in the treatment of arthritis. J Immunol. 2003;171:4913–4919. doi: 10.4049/jimmunol.171.9.4913. [DOI] [PubMed] [Google Scholar]
- Nonomura Y, Nagasaka K, Hagiyama H, Sekine C, Nanki T, Tamamori-Adachi M, et al. Direct modulation of rheumatoid inflammatory mediator expression in retinoblastoma protein-dependent and -independent pathways by cyclin-dependent kinase 4/6. Arthritis Rheum. 2006;54:2074–2083. doi: 10.1002/art.21927. [DOI] [PubMed] [Google Scholar]
- Obligado SH, Ibraghimov-Beskrovnaya O, Zuk A, Meijer L, Nelson PJ. CDK/GSK-3 inhibitors as therapeutic agents for parenchymal renal diseases. Kidney Int. 2008;3(6):684–690. doi: 10.1038/sj.ki.5002731. [DOI] [PubMed] [Google Scholar]
- Oelgeschlager T. Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. J Cell Physiol. 2002;190:160–169. doi: 10.1002/jcp.10058. [DOI] [PubMed] [Google Scholar]
- Ozato K, Tsujimura H, Tamura T. Toll-like receptor signaling and regulation of cytokine gene expression in the immune system. Biotechniques. 2002;33:S66–S75. [PubMed] [Google Scholar]
- Perretti M, Solito E. Annexin 1 and neutrophil apoptosis. Biochem Soc Trans. 2004;32:507–510. doi: 10.1042/BST0320507. [DOI] [PubMed] [Google Scholar]
- Pinho V, Souza DG, Barsante MM, Hamer FP, De Freitas MS, Rossi AG, et al. Phosphoinositide-3 kinases critically regulate the recruitment and survival of eosinophils in vivo: importance for the resolution of allergic inflammation. J Leukoc Biol. 2005;77:800–810. doi: 10.1189/jlb.0704386. [DOI] [PubMed] [Google Scholar]
- Ren S, Rollins BJ. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell. 2004;117:239–251. doi: 10.1016/s0092-8674(04)00300-9. [DOI] [PubMed] [Google Scholar]
- Roberts JM, Sherr CJ. Bared essentials of CDK2 and cyclin E. Nat Genet. 2003;35:9–10. doi: 10.1038/ng1234. [DOI] [PubMed] [Google Scholar]
- Rosales JL, Lee KY. Extraneuronal roles of cyclin-dependent kinase 5. Bioessays. 2006;28:1023–1034. doi: 10.1002/bies.20473. [DOI] [PubMed] [Google Scholar]
- Rosales JL, Ernst JD, Hallows J, Lee KY. GTP-dependent secretion from neutrophils is regulated by Cdk5. J Biol Chem. 2004;279:53932–53936. doi: 10.1074/jbc.M408467200. [DOI] [PubMed] [Google Scholar]
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- Rossi AG, Hallett JM, Sawatzky DA, Teixeira MM, Haslett C. Modulation of granulocyte apoptosis can influence the resolution of inflammation. Biochem Soc Trans. 2007;35:288–291. doi: 10.1042/BST0350288. [DOI] [PubMed] [Google Scholar]
- Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–1064. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- Sasmono RT, Ehrnsperger A, Cronau SL, Ravasi T, Kandane R, Hickey MJ, et al. Mouse neutrophilic granulocytes express mRNA encoding the macrophage colony-stimulating factor receptor (CSF-1R) as well as many other macrophage-specific transcripts and can transdifferentiate into macrophages in vitro in response to CSF-1. J Leukoc Biol. 2007;82:111–123. doi: 10.1189/jlb.1206713. [DOI] [PubMed] [Google Scholar]
- Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawatzky DA, Willoughby DA, Colville-Nash PR, Rossi AG. The involvement of the apoptosis-modulating proteins ERK 1/2, Bcl-xL and Bax in the resolution of acute inflammation in vivo. Am J Pathol. 2006;168:33–41. doi: 10.2353/ajpath.2006.050058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scatizzi JC, Mavers M, Hutcheson J, Young B, Shi B, Pope RM, et al. The CDK domain of p21 is a suppressor of IL-1beta-mediated inflammation in activated macrophages. Eur J Immunol. 2009;39:820–825. doi: 10.1002/eji.200838683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz RM. Interleukin 1 and interferon-gamma: cytokines that provide reciprocal regulation of macrophage and T cell function. Toxicol Pathol. 1987;15:333–337. doi: 10.1177/019262338701500311. [DOI] [PubMed] [Google Scholar]
- Sekine C, Sugihara T, Miyake S, Hirai H, Yoshida M, Miyasaka N, et al. Successful treatment of animal models of rheumatoid arthritis with small-molecule cyclin-dependent kinase inhibitors. J Immunol. 2008;180:1954–1961. doi: 10.4049/jimmunol.180.3.1954. [DOI] [PubMed] [Google Scholar]
- Senderowicz AM. Novel small molecule cyclin-dependent kinases modulators in human clinical trials. Cancer Biol Ther. 2003;2:S84–S95. [PubMed] [Google Scholar]
- Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- Silver DL, Montell DJ. A new trick for Cyclin-Cdk: activation of STAT. Dev Cell. 2003;4:148–149. doi: 10.1016/s1534-5807(03)00028-5. [DOI] [PubMed] [Google Scholar]
- Sun M, Li Z, Zhang Y, Zheng Q, Sun CC. Homology modeling and docking study of cyclin-dependent kinase (CDK) 10. Bioorg Med Chem Lett. 2005;15:2851–2856. doi: 10.1016/j.bmcl.2005.03.088. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Hino M, Hato F, Tatsumi N, Kitagawa S. Cytokine-specific activation of distinct mitogen-activated protein kinase subtype cascades in human neutrophils stimulated by granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, and tumor necrosis factor-alpha. Blood. 1999;93:341–349. [PubMed] [Google Scholar]
- Tassan JP, Jaquenoud M, Leopold P, Schultz SJ, Nigg EA. Identification of human cyclin-dependent kinase 8, a putative protein kinase partner for cyclin C. Proc Natl Acad Sci USA. 1995;92:8871–8875. doi: 10.1073/pnas.92.19.8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembley JH, Hu D, Slaughter CA, Lahti JM, Kidd VJ. Casein kinase 2 interacts with cyclin-dependent kinase 11 (CDK11) in vivo and phosphorylates both the RNA polymerase II carboxyl-terminal domain and CDK11 in vitro. J Biol Chem. 2003;278:2265–2270. doi: 10.1074/jbc.M207518200. [DOI] [PubMed] [Google Scholar]
- van den HS, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- Vidal A, Koff A. Cell-cycle inhibitors: three families united by a common cause. Gene. 2000;247:1–15. doi: 10.1016/s0378-1119(00)00092-5. [DOI] [PubMed] [Google Scholar]
- Wang S, Fischer PM. Cyclin-dependent kinase 9: a key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol Sci. 2008;29:302–313. doi: 10.1016/j.tips.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN, et al. NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem. 1999;274:4309–4318. doi: 10.1074/jbc.274.7.4309. [DOI] [PubMed] [Google Scholar]
- Wells AD. Cyclin-dependent kinases: molecular switches controlling anergy and potential therapeutic targets for tolerance. Semin Immunol. 2007;19:173–179. doi: 10.1016/j.smim.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RO, Paleolog E, Feldmann M. Cytokine inhibitors in rheumatoid arthritis and other autoimmune diseases. Curr Opin Pharmacol. 2007;7:412–417. doi: 10.1016/j.coph.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Yao H, Yang SR, Edirisinghe I, Rajendrasozhan S, Caito S, Adenuga D, et al. Disruption of p21 attenuates lung inflammation induced by cigarette smoke, LPS, and fMLP in mice. Am J Respir Cell Mol Biol. 2008;39:7–18. doi: 10.1165/rcmb.2007-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–953. doi: 10.1038/nm.1855. [DOI] [PubMed] [Google Scholar]
- Yu C, Rahmani M, Dai Y, Conrad D, Krystal G, Dent P, et al. The lethal effects of pharmacological cyclin-dependent kinase inhibitors in human leukemia cells proceed through a phosphatidylinositol 3-kinase/Akt-dependent process. Cancer Res. 2003;63:1822–1833. [PubMed] [Google Scholar]
- Zoja C, Casiraghi F, Conti S, Corna D, Rottoli D, Cavinato RA, et al. Cyclin-dependent kinase inhibition limits glomerulonephritis and extends lifespan of mice with systemic lupus. Arthritis Rheum. 2007;56:1629–1637. doi: 10.1002/art.22593. [DOI] [PubMed] [Google Scholar]
- Zong H, Li Z, Liu L, Hong Y, Yun X, Jiang J, et al. Cyclin-dependent kinase 11(p58) interacts with HBO1 and enhances its histone acetyltransferase activity. FEBS Lett. 2005;579:3579–3588. doi: 10.1016/j.febslet.2005.05.039. [DOI] [PubMed] [Google Scholar]