

Abstract
Chronic inflammatory lung diseases such as cystic fibrosis and emphysema are characterized by higher-than-normal levels of pulmonary proteases. While these enzymes play important roles such as bacterial killing, their dysregulated expression or activity can adversely impact on the inflammatory process. The existence of efficient endogenous control mechanisms that can dampen or halt this overexuberant protease activity in vivo is essential for the effective resolution of inflammatory lung disease. The function of pulmonary antiproteases is to fulfil this role. Interestingly, in addition to their antiprotease activity, protease inhibitors in the lung also often possess other intrinsic properties that contribute to microbial killing or termination of the inflammatory process. This review will outline important features of chronic inflammation that are regulated by pulmonary proteases and will describe the various mechanisms by which antiproteases attempt to counterbalance exaggerated protease-mediated inflammatory events. These proteases, antiproteases and their modifiers represent interesting targets for therapeutic intervention.
This article is part of a themed issue on Mediators and Receptors in the Resolution of Inflammation. To view this issue visit http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Keywords: protease, antiprotease, lung, cystic fibrosis, chronic obstructive pulmonary disease (COPD), neutrophil elastase, alpha-1 antitrypsin, secretory leucoprotease inhibitor, elafin
Proteinases (herein referred to colloquially as proteases) have a key role in the lung in health and disease. In the healthy lung, proteases fulfil basic homeostatic roles and regulate processes such as regeneration and repair. Chronic inflammatory lung diseases are associated with higher-than-normal levels of proteases. Functionally, this can positively impact on both infection and inflammation. However, unless a perfect balance can be struck between the protective and harmful effects of pulmonary proteases by an appropriate antiprotease protective screen, damage can occur. Thus, effective resolution of inflammation in the lung is associated not only with protease activity but also with appropriate antiproteolytic control mechanisms. Here we will focus on the mechanisms by which pulmonary proteases regulate innate immunity and the role of specific antiproteases in fine-tuning these responses.
The principal classes of protease present in the human lung are the serine, cysteinyl, aspartyl and metalloproteases. These can function either intracellularly or extracellularly to regulate processes as diverse as tissue remodelling, mucin expression, neutrophil chemotaxis and bacterial killing. Members of these protease classes orchestrate a diverse range of changes with respect to infection and inflammation in the lung, with the serine protease neutrophil elastase (NE) occupying an important position at the apex of a specific protease hierarchy. NE has a number of important intrinsic proteolytic properties. However, it can also directly control the inducible expression and biological properties of other pulmonary proteases. For example, NE regulates expression of cathepsin B and MMP-2 in alveolar macrophages (Geraghty et al., 2007b) and also activates proMMP-2, MMP7 and MMP-9 (Imai et al., 1995; Ferry et al., 1997; Shamamian et al., 2001). Thus, in addition to being a protease, NE also behaves as a proinflammatory mediator. In certain circumstances, NE also controls elegant signalling mechanisms regulating innate immunity (Nakamura et al., 1992; Walsh et al., 2001; Kohri et al., 2002; Devaney et al., 2003; Shao and Nadel, 2005a,b; Bergin et al., 2008); its pluripotency distinguishes it as a unique factor controlling many aspects of infection and inflammation in the lung.
NE
NE, as its name suggests, is a neutrophil-derived elastolytic protease. It is expressed as a 267 amino acid pre-proenzyme that is packaged in a processed and activated form in neutrophil primary (azurophilic) granules. Substrates of NE fall into many categories and include elastin and other extracellular matrix proteins, plasma proteins, cell surface receptors, cytokines, protease inhibitors and proteases (Table 1). Other serine proteases stored in the primary granules of neutrophils are proteinase 3 and cathepsin G. Similar to NE, these enzymes are released by activated and disintegrating neutrophils and are detectable at higher-than-normal levels in the airways during chronic inflammation (Goldstein and Doring, 1986; Witko-Sarsat et al., 1999).
Table 1.
Classes and protein targets degraded by neutrophil elastase
| Substrate | Targets |
|---|---|
| Immunoglobulin | IgA, IgG, IgM |
| Plasma protein | C3; C5; plasminogen; fibrinogen; factors V, VII, XII and XIII; platelet IIb/IIIa receptor |
| Matrix protein | Elasin, collagens I–IV, fibronectin, thrombomodulin, proteoglycan |
| Cytokine | IL-1, IL-2, IL-6, TNFα |
| Protease inhibitor | TIMP, elafin, SLPI |
| Protease | MMP-2, MMP-9, cathepsin B, TACE, meprin-α |
| Others | Cadherins, complement receptors, surfactant, ICAM1, gp120 |
TIMP, tissue inhibitor of MMP; SLPI, secretory leucoprotease inhibitor; TACE, TNFα converting enzyme.
While neutrophils play an important role in many inflammatory lung diseases, cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD) are considered to be the classical chronic neutrophil-dominated diseases of the airways. The abundance of neutrophils in the CF and COPD lung generates a milieu rich in NE (Doring, 1994; Griese et al., 2008). Neutrophil accumulation is believed to be due in part to their inability to effectively clear pathogens and thus accumulate and undergo secondary necrosis. This leads to the liberation of NE and other intracellular components (Griese et al., 2008). Foreign organic molecules that have been phagocytosed by neutrophils are degraded by NE intracellularly. NE also contributes not only to the intracellular killing of Gram-negative bacteria by neutrophils but also, once released extracellularly, can play a role in bacterial killing by comprising a key component of neutrophil extracellular traps (NETs). NETs are involved in host defence (Brinkmann et al., 2004). They bind Gram-positive and Gram-negative bacteria and allow neutrophils to deliver high concentrations of serine proteases that degrade virulence factors and kill bacteria. Recently, bacterial virulence factors that counteract NETs have been identified. The mechanisms identified include the expression of DNAses that degrade the NET backbone, expression of capsule that can reduce bacterial trapping and modulation of cell surface charge (Buchanan et al., 2006; Wartha et al., 2007).
In addition to direct killing of microbes, NE has important roles in innate immunity and inflammation in the lung particularly in the processes of neutrophil recruitment and mucin gene expression (Shao and Nadel, 2005b; Bergin et al., 2008). Interestingly, both of these processes are controlled via similar mechanisms. NE also regulates the expression of other classes of proteases.
Hierarchy of protease expression
Bronchoalveolar lavage fluid sampled from individuals with chronic inflammatory lung disease almost invariably contains significant quantities of proteases. The primary families to be released into the extracellular space following cell activation are the serine, MMP and cysteinyl cathepsin groups. There is evidence demonstrating that NE, and possibly other serine proteases, can transcriptionally regulate expression of other classes of proteases. In human macrophages, for example, transcription of both MMP-2 and cathepsin B has been shown to be increased in response to NE in a nuclear factor-κB (NFκB)-dependent manner (Geraghty et al., 2007b). This is one mechanism contributing to the positioning of serine proteases at the apex of one hierarchy of protease regulation. Toll-like receptor 4 (TLR4) has also been implicated in NE-induced expression of MMP-2 and cathepsin B; however, its precise role in NE-induced changes in gene expression is less clear in macrophages than in airway epithelial cells (see below). In addition to its ability to induce the transcription of specific proteases, NE and other serine proteases can also activate MMPs. For example, NE, proteinase 3 and cathepsin G can activate the latent 72 kDa MMP-2 zymogen via membrane type I MMP (Imai et al., 1995; Ferry et al., 1997; Shamamian et al., 2001). NE can also activate proMMP-7, MMP-9 and procathepsin B and members of a disintegrin and metalloprotease (ADAM) and meprin families (Dalet-Fumeron et al., 1993; Imai et al., 1995; Ferry et al., 1997; Kohri et al., 2002; Bergin et al., 2008).
Regulation of mucin production and neutrophil recruitment
The epidermal growth factor receptor (EGFR, alternatively known as Erb1 or HER1) is a receptor tyrosine kinase that can regulate expression of mucin and interleukin-8 (IL-8) gene expression. In the human airway epithelium, EGFR forms homodimers or heterodimers with Erb2 or Erb3 in response to activation by a range of diverse stimuli, including NE (Kohri et al., 2002; Holbro and Hynes, 2004; Bergin et al., 2008). EGFR is directly activated by binding of the EGFR ligand epidermal growth factor (EGF), transforming growth factor-α (TGFα), heparin-binding (HB) EGF, amphiregulin, betacellulin or epiregulin. These ligands are expressed as membrane-tethered proligands on airway epithelial cells, eosinophils, neutrophils, mast cells and macrophages and are released as bioactive molecules in a metalloprotease-dependent manner. While TNFα converting enzyme (TACE/ADAM17) was originally thought to be uniquely responsible for EGFR ligand generation, it is now clear that there is redundancy between TACE, other MMPs, ADAMs and the metzincin meprin-α in this regard (Choudry and Kenny, 1991; Merlos-Suarez et al., 2001; Schlondorff et al., 2001). To date, several cis-acting enzymes including ADAM10, ADAM12, ADAM15, ADAM17, MMP2, MMP9 and meprin-α, among others, have been implicated in EGFR ligand generation (Ohtsu et al., 2006).
In the airways, NE has been shown to activate EGFR via generation of TGFα. This event involves activation of TACE or meprin-α by NE and leads to intracellular signalling cascades that culminate in the enhanced expression of the mucin genes, MUC2 and MUC5AC, or the neutrophil chemokine IL-8. Hypersecretion of mucus is a common pathophysiological feature of CF and other inflammatory lung diseases. In CF, asthma and chronic bronchitis mucus obstruction of the airways contributes significantly to mortality and morbidity in these conditions. Consisting mostly of water and ions, mucus also comprises approximately 5% protein. It plays an important role in host defence by binding bacteria and ensuring their removal via the mucociliary escalator to the upper airways and oesophagus for expectoration or ingestion. To date, nine MUC genes have been described in the lung – MUC1, 2, 4, 5AC, 5B, 7, 8, 13 and 19 (Rose et al., 2001; Chen et al., 2004). MUC2, 5AC and 5B are secreted, gel-forming mucins. MUC5AC represents the most prominent mucin in normal airway secretions and its expression is increased in the nasal epithelium of individuals with CF and allergic rhinitis (Voynow et al., 1998). In airway inflammation, mucin gene expression can be activated by IL-9 via the human calcium-activated chloride channel, hCLCA1 (Hauber et al., 2004) but also by NE via TACE and EGFR (Fischer and Voynow, 2002; Kohri et al., 2002). Other stimuli that regulate MUC5AC or MUC2 expression via the TACE-EGFR pathway include cigarette smoke, lipopolysaccharide (LPS) (Dohrman et al., 1998; Shao and Nadel, 2005a) and Gram-positive lipoteichoic acid (LTA) (Lemjabbar and Basbaum, 2002).
Mechanisms regulating NE-induced IL-8 production
IL-8 is a neutrophil chemokine and consequently represents a key factor present in the lungs during neutrophil-dominated inflammatory disease. Elevated levels of IL-8 are typically present in the bronchoalveolar lavage fluid of patients with COPD, emphysema and CF. Early studies identified a link between high concentrations of IL-8 and NE in the airways in CF (McElvaney et al., 1992; Nakamura et al., 1992). The mechanism by which this correlation exists has been studied in detail, leading to the elucidation of the molecular mechanism by which NE can regulate the transcriptional induction of the IL-8 gene in airway epithelial cells. Preliminary investigations to determine how this may be occurring identified three important features of the regulatory cascade (Walsh et al., 2001). Firstly, in order for NE to induce IL-8 expression from airway epithelial cells, it must retain its biological activity; inactivation of its protease activity with specific serine protease inhibitors abrogated the effect. Secondly, as the effect could be blocked using actinomycin D, NE was affecting the transcription of IL-8. Thirdly, and most surprisingly, NE was shown to induce IL-8 via the transcription factor NFκB in a manner that was dependent on MyD88, IRAK and TRAF6 – known transducers involved in TLR and interleukin-1 type 1 receptor (IL-1R1) signalling. Subsequent studies revealed that a dominant-negative version of MyD88 (ΔMyD88) could inhibit the expression of multiple NFκB-dependent cytokines in response to NE. Furthermore, the adaptor protein Mal, known to be involved in TLR2/TLR4 signalling, had similar inhibitory capacity as ΔMyD88 when expressed as an inactive transgene (Carroll et al., 2005; Greene et al., 2005). Taken together, these data implicated TLR2 and/or TLR4 in NE-induced IL-8 expression. Geraghty and colleagues (Geraghty et al., 2007b) observed a similar phenomenon in macrophages in their studies on NE induction of MMP2 and cathepsin B. Others (Koff et al., 2008) also recently provided evidence for a role for TLRs in EGFR-mediated signalling, while Bergin et al. demonstrated a direct association between EGFR and TLR4 in response to stimulation with NE (Bergin et al., 2008). The mechanism by which NE regulates IL-8 expression in human bronchial epithelial cells is depicted in Figure 1.
Figure 1.
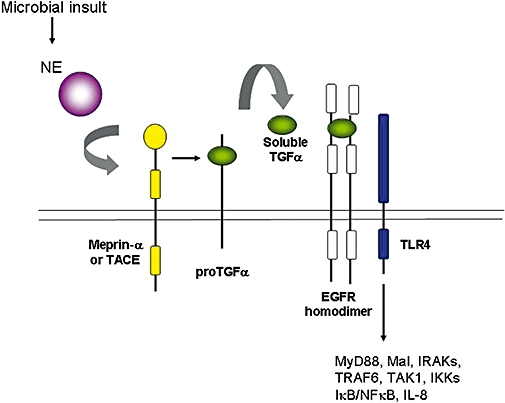
Mechanism of neutrophil elastase (NE) induction of IL-8 in airway epithelial cells. Following its release from the azurophilic granules in response to a microbial insult, NE activates meprin-α or TNFα converting enzyme (TACE), which in turn cleave proTGFα to generate soluble TGFα as a ligand for the epidermal growth factor receptor (EGFR). EGFR co-localizes with TLR4 and a signal transduction cascade is initiated via MyD88 or Mal, IRAKs, TRAF6, transforming growth factor-beta-activated kinase 1 (TAK1) and the IκB kinases (IKKs), leading to a degradation of inhibitor of NFκB (IκB) proteins, activation of nuclear factor-κB (NFκB) and increased IL-8 gene transcription.
Generation of bioactive molecules by pulmonary proteases
In addition to their roles in EGFR trans-activation MMPs have also recently been implicated as important factors regulating the expression of the novel proinflammatory chemotactic peptide proline-glycine-proline (PGP) in both CF and COPD. PGP is a breakdown product of the extracellular matrix protein collagen and shares sequence and structural homology with alpha chemokines (Weathington et al., 2006). Indeed there is mounting evidence that fragments of a number of extracellular matrix proteins, including those derived from collagen and elastin, are important in regulating the recruitment of inflammatory cells to the lung. PGP (and N-acetylated PGP) has been shown to act as a neutrophil chemoattractant via Cys-X-Cys (CXC) receptors 1 and 2 on neutrophils. PGP is generated from collagen via the combined activities of MMP-8, MMP-9 and the serine protease prolyl endopeptidase (Gaggar et al., 2008; Lin et al., 2008). In addition to higher-than-normal levels of NE in the CF and COPD lung, there is evidence that MMP-8, MMP-9 and prolyl endopeptidase are also elevated and that together, these enzymes can degrade collagen in vivo to generate the PGP tripeptide. CF sputum has been shown to contain detectable levels of both PGP and N-Ac-PGP. In addition to being chemotactic for human neutrophils, PGP has been linked to neutrophil superoxide production, alveolar enlargement and right ventricular hypertrophy, which contribute to the pulmonary inflammatory manifestations of CF. Recent elegant studies have demonstrated that the corresponding tripeptide arginine-threonine-arginine (RTR) can bind to PGP sequences, neutralize their effect and inhibit neutrophil infiltration in a model of COPD (van Houwelingen et al., 2008). Furthermore, RTR completely inhibits PGP-induced lung emphysema assessed by changes in alveolar enlargement and right ventricular hypertrophy. Thus, PGP antagonism via RTR is also likely to have therapeutic potential for CF. This represents another example of how protease interactions with structural proteins have an important effect on regulating innate immunity.
Pulmonary proteases and antimicrobial peptides
Important antimicrobial polypeptides of innate pulmonary host defence include lactoferrin, secretory leucoprotease inhibitor (SLPI), lysozyme, the defensins and the cathelicidin family (Zanetti, 2005). The human cathelicidin precursor protein, designated 18 kDa cationic antimicrobial protein (hCAP18) is expressed as a pro-protein composed of a conserved N-terminal pro-domain and a C-terminal antimicrobial peptide domain. In human neutrophils, hCAP18 is processed to an active form by proteinase 3 to generate the cationic α-helical peptide LL-37 (Zanetti et al., 1995; Sørensen et al., 2001). Further proteolytic degradation of LL-37 can lead to the generation of peptide fragments with altered antimicrobial activity and reduced immunomodulatory potential. In addition to degradation of LL-37, a selection of pulmonary proteases has also been shown to be responsible for the degradation and inactivation of other antimicrobial effector molecules, including human β-defensins 2 and 3, SLPI and lactoferrin (Taggart et al., 2001; 2003; Rogan et al., 2004). In particular, the cysteinyl cathepsins B, L and S are implicated in cleavage and inactivation of innate immunity proteins in vivo during inflammatory lung disease.
Human β-defensins 1, 2 and 3 (HBD-1, -2 and -3) are antimicrobial peptides produced by epithelial cells lining the respiratory tract. They are active against Gram-positive and Gram-negative bacteria. In CF, the antimicrobial activity of defensins is compromised therefore predisposing to bacterial colonization of the lung by Pseudomonas aeruginosa and other species. While inactivation of HBDs by the high salt levels present in the CF lung represents one potential mechanism for decreased antimicrobial protection (Goldman et al., 1997), there also exists a protease-mediated mechanism that contributes significantly to this phenomenon. HBD-2 and HBD-3 have been shown to be susceptible to degradation and inactivation by the cysteine proteases cathepsins B, L and S, with all three cathepsins present and active in CF bronchoalveolar lavage fluid (Taggart et al., 2003). In addition to degrading HBDs, these enzymes also cleave and inactivate human lactoferrin in CF, which plays an important role in the inhibition of biofilm formation (Rogan et al., 2004). Furthermore, all three cathepsins have also been shown to cleave and inactivate SLPI in the context of pulmonary emphysema (Taggart et al., 2001). The cleavage of SLPI by cathepsins B, L or S occurs between residues Thr(67) and Tyr(68). This cleavage results in loss of the active site of SLPI and the inactivation of its anti-NE capacity. Cathepsin L has also been shown to cleave and inactivate the serine antiprotease alpha-1 antitrypsin (A1AT) (Johnson et al., 1986). MMP-7 fulfils a similar role in the CF lung where its expression is markedly up-regulated (Sires et al., 1994). Together these findings provide ample evidence for the involvement of cathepsins in damaging the antimicrobial and antiprotease protective screens in the lung.
Bacterial proteases and inflammatory lung disease
Elastolytic activity within the lungs in CF is largely accounted for by the significantly elevated levels of NE, with one report suggesting that up to 90% of the activity in CF sputum is attributable to NE. According to Rees et al., proteinase 3 accounts for a further 7% of this activity, while the remaining 3% derives from macrophage-derived metalloelastases but also elastolytic proteases expressed by Ps. aeruginosa (Rees et al., 1997). Although Ps. aeruginosa represents the classical pathogen associated with colonization of CF airways, other opportunistic Gram-negative and Gram-positive bacteria such as Haemophilus influenzae and Staphylococcus aureus, respectively, are also important (Ramsey, 1996). However, more is known regarding the function and activity of the Pseudomonas-derived metalloproteases in CF. Both Pseudomonas elastase and alkaline protease are present in the CF airway surface liquid (Suter, 1994). Pseudomonas elastase has a number of important biological properties. It promotes secretion of mucus (Adler et al., 1983), degrades surfactant proteins A and D (Mariencheck et al., 2003), cleaves and inactivates A1AT (Morihara et al., 1984), SLPI (Johnson et al., 1982), elafin (Guyot et al., 2008b), lysozyme (Jacquot et al., 1985) and LL-37 (Schmidtchen et al., 2002), and it also impairs the function of cilia (Amitani et al., 1991). Both Pseudomonas elastase and alkaline protease can inactivate lactoferrin (Britigan et al., 1993). These properties represent an ever-growing list and indicate that proteases expressed by bacterial pathogens colonizing the airways should not be overlooked as important factors regulating the inflammatory process.
Pulmonary antiproteases
In order to counterbalance overexuberant and often harmful effects of pulmonary proteases, a battery of antiproteases exists in the lungs. A1AT, SLPI and elafin are three serine antiproteases present, in descending abundance, in the lungs. Monocyte/neutrophil elastase inhibitor (MNEI) is another pulmonary serine protease inhibitor with activity against NE, cathepsin G and proteinase 3 (Cooley et al., 2001). The cysteinyl cathepsins are inhibited by the cystatins, while the tissue inhibitors of metalloproteases (TIMPS) regulate the activities of MMPs and ADAMs.
A1AT
A1AT is an acute phase 52 kDa 418 amino acid glycoprotein that is primarily synthesized and secreted by hepatocytes (Rogers et al., 1983), although it is also actively transcribed and secreted in smaller amounts by cells including neutrophils, mononuclear phagocytes and enterocytes (Molmenti et al., 1993). A1AT is also produced locally in the lung by bronchial epithelial cells (Mason et al., 1991; Venembre et al., 1994; Cichy et al., 1997; Hu and Perlmutter, 2002; Mulgrew et al., 2004). It is present in all tissues of the body and its primary role is to inhibit NE (Travis et al., 1985). A1AT can also inhibit a range of other proteases including trypsin, chymotrypsin, cathepsin G, plasmin, thrombin, tissue kallikrein, factor Xa, plasminogen and proteinase 3.
Although A1AT is principally a serine protease inhibitor, its other properties include the ability to inhibit TNFα and MMP in alveolar macrophages in response to thrombin and cigarette smoke extract (Churg et al., 2003), to impair LPS-induced monocyte activation and to block apoptosis (Daemen et al., 2000; Ikebe et al., 2000; Ikari et al., 2001). A1AT has also been reported to play an immunoregulatory role. It can inhibit neutrophil superoxide production, induce the release of macrophage-derived IL-1 receptor agonist and increase hepatocyte growth factor production in human lung fibroblasts. A1AT can bind to the secreted enteropathogenic Escherichia coli proteins EspB and EspD, thereby reducing their haemolysis of red blood cells (Knappstein et al., 2004). Thus, A1AT may not only afford protection against proteolytic injury but may also have the potential to neutralize microbial activities and to exert effects on the regulation of innate immunity. There is growing evidence that A1AT may also possess the ability to impair LPS-induced inflammatory responses both in vitro and in vivo (Nita et al., 2005).
With respect to apoptosis, A1AT has been shown to have a direct pro-survival effect in a model of apoptosis-dependent emphysema (Petrache et al., 2006b). The same group (Petrache et al., 2006b) demonstrated that A1AT can inhibit apoptosis in alveolar epithelial cells following transduction of an A1AT-expressing adeno-associated virus in a mouse model of apoptosis-dependent emphysema. The mechanism by which A1AT mediates this effect is via direct inhibition of caspase-3 binding to its substrate (Petrache et al., 2006a). Others have reported similar anti-apoptotic effects of A1AT in porcine pulmonary endothelial cells (Aldonyte et al., 2008).
A1AT is susceptible to both cleavage and oxidative inactivation in vivo. Cathepsin L and Pseudomonas elastase are known to cleave A1AT (Morihara et al., 1984; Johnson et al., 1986). A1AT contains nine methionines, two of which are readily oxidizable, Met (351) and Met (358). Met (358) is a key residue located in the active site of A1AT (Johnson and Travis, 1978). When oxidation occurs, A1AT's anti-NE capacity is abolished and its association rate constant for NE is reduced 2000-fold. Cigarette smoke and inflammatory cells in the lower respiratory tract can oxidize Met(358). Studies by Taggart et al. have demonstrated that Met(351) is also susceptible to oxidation, and site-directed mutants of A1AT with alanines substituted for these key methionines are resistant to oxidative inactivation (Taggart et al., 2000).
Augmentation therapy with A1AT is the current treatment for the pulmonary manifestations of A1AT deficiency, a genetic form of emphysema. This approach has the potential not only to redress the protease/antiprotease imbalance and to dampen the inflammatory response on the airway surface but also could potentially inhibit apoptosis associated with the development of emphysema by inactivating caspase-3.
Hartl et al. recently described how CXCR1 fragments released from the surface of neutrophils in vivo in individuals with CF or COPD can act as bioactive molecules signalling via TLR2 in airway epithelial cells (Hartl et al., 2007). In vivo inhibition of proteases by inhalation of A1AT restored CXCR1 expression and improved bacterial killing in individuals with CF. These findings support a novel role for A1AT as a therapeutic for CF and possibly COPD.
SLPI
SLPI is a 11.7 kDa cationic, non-glycosylated serine proteinase inhibitor that is present in fluids lining mucosal surfaces (McElvaney and Crystal, 1997). It inhibits a variety of proteinases, including NE, cathepsin G, trypsin, chymotrypsin, chymase and tryptase (Doumas et al., 2005). The molecule is composed of two highly homologous cysteine-rich domains, and it is the C-terminal domain that contains the elastase inhibitory activity. SLPI is constitutively expressed at many mucosal surfaces and is produced by a number of cell types, including neutrophils, macrophages and epithelial cells lining the respiratory and alimentary tracts. The physiological concentration of SLPI in lung epithelial lining fluid (ELF) can be as high as 670 nM/ELF (McNeely et al., 1995; Taggart et al., 2001). In the lung, SLPI is expressed by clara cells and goblet cells of the surface epithelium and the serous cells of the submucosal glands (Hiemstra, 2002).
In addition to its antiprotease activity, SLPI is well recognized as an antimicrobial factor. Its antimicrobial activity is encoded by the N-terminal domain of the protein (Hiemstra et al., 1996). It has been postulated that due to its high cationicity, SLPI can disrupt microbial cell membranes and that this is the mechanism by which it can inhibit such pathogens as S. aureus, Staphylococcus epidermidis, Ps. aeruginosa and Candida albicans (Wiedow et al., 1998; Williams et al., 2006). SLPI also displays antiviral activity and can inhibit human immunodeficiency virus (HIV) replication in monocytes and can interfere with HIV infection of macrophages via binding to annexin II (McNeely et al., 1997; Ma et al., 2004).
An important property of SLPI is its immunomodulatory activity. SLPI can regulate a variety of important inflammatory processes including decreasing the production of prostaglandin H synthase-2, prostaglandin E2, and MMP-1 and -9 by monocytes (Zhang et al., 1997), inhibiting interferon-γ-induced cathepsin S expression (Geraghty et al., 2007a) and antagonizing the pro-inflammatory activity of bacterial LPS (Jin et al., 1997; Ding et al., 1999). While it has been reported that SLPI can interfere with the interaction between CD14 and LPS (Ding et al., 1999), other reports provide evidence for an intracellular role for SLPI (McNeely et al., 1997; Taggart et al., 2005). It has been shown that SLPI can be internalized by monocytic cells and can be distributed throughout the cytoplasm and nucleus. In addition to its ability to impair the LPS response, SLPI can also inhibit LTA-induced NFκB activation in monocytic cells (Taggart et al., 2002; Greene et al., 2004). Overall, the inhibition has been shown to occur via two mechanisms: firstly, by preventing the proteolytic degradation of IRAK-1, IκBβ and IκBα, and secondly, as a direct result of binding of SLPI to NFκB consensus sequences and of competing with p65 for occupancy of the promoters of NFκB-regulated genes (Taggart et al., 2002; 2005;).
SLPI has been administered by aerosolization to CF patients to suppress respiratory epithelial NE levels and to reduce bronchoalveolar lavage fluid IL-8 levels (McElvaney et al., 1992). A major drawback to its therapeutic potential, however, is its susceptibility to degradation by pulmonary proteases (Taggart et al., 2001).
Elafin
The peptide elafin, also known as skin-derived antileukoprotease (SKALP) or elastase-specific inhibitor (ESI), is a cationic 6 kDa non-glycosylated serine antiproteinase. Elafin belongs to the chelonianin family, a distinct group of canonical inhibitors also including SLPI (Zani et al., 2004). Its compact structure is characteristic of whey acidic proteins (WAPs) and is maintained by four conserved disulphide bonds. Elafin shares 40% sequence identity with SLPI. Tryptase releases elafin from a larger pre-protein molecule called trappin-2 or pre-elafin (Guyot et al., 2005). Trappin-2 possesses an N-terminal WAP domain and a cementoin domain containing repeating GQDPVK motifs that act as a transglutaminase substrate, facilitating the cross-linkage of trappin-2 to extracellular matrix proteins.
Elafin is a secreted protein principally expressed by epithelial surfaces such as skin (Alkemade et al., 1994; Nonomura et al., 1994; Pfundt et al., 1996) or lung epithelium (Sallenave et al., 1994; van Wetering et al., 2000) but also by inflammatory cells including alveolar macrophages (Mihaila and Tremblay, 2001) and neutrophils (Sallenave et al., 1997). It is found in plasma (Alkemade et al., 1995), urine (Streit et al., 1995) and bronchial secretions (Sallenave et al., 1992; Nara et al., 1994) and constitutes up to 20% of the total antielastase activity retrieved from bronchoalveolar lavage fluid in healthy individuals.
First identified by Hochstrasser, elafin was described as an acid-stable inhibitor present in human bronchial mucus that differed from SLPI in that it exerted inhibitory activity towards porcine pancreatic and human granulocytic elastase, but not against trypsin, chymotrypsin or granulocytic cathepsin G (Hochstrasser et al., 1981). Later, the anti-protease spectrum of elafin was found to include activity against proteinase 3 (Wiedow et al., 1991). Based on these properties, elafin was thought to protect tissue from degradation by these enzymes.
Several studies have demonstrated that expression of elafin is inducible and its expression is significantly up-regulated by TNFα or IL-1β in the airway epithelial cell lines NCI-H322 and A549 (Sallenave et al., 1994). Its expression is also induced in response to other proinflammatory stimuli such as LPS and NE (Reid et al., 1999; Simpson et al., 2001). In addition to its antiprotease properties, elafin also possesses both anti-inflammatory and antibacterial activities. Elafin/trappin-2 can inhibit growth of both Ps. aeruginosa (Gram-negative) and S. aureus (Gram-positive) (Simpson et al., 1999) with reported significant killing of both organisms by doses of 2.5–25 µM elafin, concentrations that are potentially achievable in the airway ELF. With respect to its anti-inflammatory activity, the elafin precursor trappin-2 has been shown to dose dependently reduce LPS-induced neutrophil influx into alveoli, to inhibit LPS-induced MMP-9 production and to prevent the generation of CXCL1 and CXCL2 (chemokine ligands 1 and 2) (Simpson et al., 1999). Trappin-2 can also attenuate IL-8 secretion by endothelial cells and/or macrophages in response to TNF, LPS or oxidized low-density lipoprotein via inhibition of NFκB (Henriksen et al., 2004). Recently, Butler et al. reported that elafin also inhibits LPS-induced monocyte chemotatic protein-1 production in monocytes by inhibiting both AP-1 and NFκB activation (Butler et al., 2006).
Notwithstanding elafin's favourable qualities as an antiprotease, antibacterial and anti-inflammatory molecule, in a milieu containing high levels of NE, elafin is known to undergo cleavage at Val(5)-Lys(6) and Val(9)-Ser(10). Although this does not impair elafin's anti-NE capacity, it does diminish its ability to be immobilized by transglutamination and also to bind LPS (Guyot et al., 2008b). This has important implications for the immunmodulatory properties of elafin in vivo at sites characterized by a high-NE burden such as the CF lung.
Cystatins and TIMPs
The activity of cysteinyl cathepsins is regulated by endogenous protein inhibitors called cystatins. Three subfamilies exist based on sequence homology and structure: types 1, 2 and 3 (Rawlings et al., 2004). These are located predominantly intracellularly, extracellularly and intravascularly respectively. The naturally occurring inhibitors of MMPs are the TIMPs. These are small proteins ranging from 21 to 28 kDa in size, which inhibit MMPs in a 1:1 stochiometry. TIMPs are also able to inhibit the metalloproteinase activity of several members of the ADAM family (Huovila et al., 2005). Readers are directed elsewhere for comprehensive reviews of cysatins and TIMPs (Nagase et al., 2006; Turk et al., 2008).
Therapeutics targeting pulmonary proteases
Considerable evidence for the importance of proteases in chronic inflammatory lung disease comes from knockout mouse studies. Both NE and MMP-12 (macrophage metalloelastase) knockout mice are more resistant to cigarette smoke-induced emphysema (Hautamaki et al., 1997; Shapiro et al., 2003). Animal studies have also played a large part in developing our understanding of the therapeutic potential of antiprotease therapies. Cantin et al. performed a number of studies investigating the therapeutic potential of plasma-purified A1AT (Prolastin) and MNEI in rat agar bead models of chronic Ps. aeruginosa infection (Cantin and Woods, 1999; Woods et al., 2005). For example, significantly decreased elastase activity, lung neutrophil counts, bacterial colony counts and a marked decrease in lung inflammation were evident in the A1AT-treated animals compared to controls (Cantin and Woods, 1999). Thiol-specific conjugation of A1AT with polyethylene glycol at Cys232 markedly improved its in vivo pharmacokinetic profile (Cantin et al., 2002). Similar studies may prove useful in animal models of CF. Strangely, CFTR knockout mice show little signs of lung disease (reviewed by (Guilbault et al., 2007); however, mice with airway-specific overexpression of epithelial Na(+) channels (ENaC) show pulmonary characteristics very similar to CF, most likely due to their accelerated Na(+) transport, and represent a more appropriate model for testing antiprotease therapies for CF (Mall et al., 2004).
The potential use of irreversible synthetic inhibitors of NE such as peptide chloromethyl ketones or reversible peptide aldehydes, tripeptide ketones, modified NE-specific β-lactams or peptide boronic acids has been largely superseded by the development of EPI-HNE-4, a rapid acting and potent NE inhibitor (Delacourt et al., 2002) that can potentially be nebulized to CF patients (Grimbert et al., 2003); however, clear clinical efficacy remains to be demonstrated.
Most evidence to date exists for the use of A1AT as an antiprotease-targeted therapeutic for NE-dominated airway diseases. In addition to augmentation studies for A1AT deficiency (Hubbard et al., 1989), a number of human studies have shown that A1AT aerosol therapy has many beneficial effects on airway inflammation in patients with CF (McElvaney et al., 1991; Cantin et al., 2006; Griese et al., 2007). Delivery of SLPI to the lung has yielded less success (McElvaney et al., 1993). Unlike A1AT, SLPI does not accumulate on the epithelial surface due to its degradation by cysteinyl cathepsins (Taggart et al., 2001), and consequently, relatively higher doses are required to inhibit NE. Elafin and its precursor, trappin-2, have both antiproteolytic and anti-inflammatory potential; however, like SLPI, they too are susceptible to degradation by proteases in the CF lung (Guyot et al., 2008a,b;).
Concluding remarks
There is a fine balance between the physiological and deleterious effects of pulmonary proteases. When this balance is disturbed, lung damage results as in the case of CF or COPD where there is dysregulated release of proteases or insufficient inhibition by antiproteases in A1AT deficiency. Therapeutics that target specific pulmonary proteases hold much promise for the treatment of chronic inflammatory lung disease not only with respect to protecting the lungs from protease-mediated tissue damage but also by controlling overexuberant inflammatory responses.
Acknowledgments
Research in Department of Medicine, Royal College of Surgeons in Ireland is generously funded by the Health Research Board, the Higher Education Authority, the Alpha One Foundation, the Department of Health and Children, The Crumlin Research Centre, the CF Association of Ireland, the Medical Charities Research Group and the Royal College of Surgeons in Ireland.
Glossary
Abbreviations:
- A1AT
alpha-1 antitrypsin
- ADAM
a disintegrin and metalloprotease
- CF
cystic fibrosis
- COPD
chronic obstructive pulmonary disease
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- ELF
epithelial lining fluid
- HBD
human β-defensin
- hCAP18
human cathelicidin precursor-18
- IL-1R1
interleukin-1 type 1 receptor
- IRAK
IL-1R-associated kinase
- LPS
lipopolysaccharide
- LTA
lipoteichoic acid
- Mal
MyD88 adaptor-like protein
- MMP
matrix metalloprotease
- MNEI
monocyte/neutrophil elastase inhibitor
- MyD88
myeloid differentiation factor 88
- NE
neutrophil elastase
- NET
neutrophil extracellular trap
- PGP
proline-gylcine-proline
- RTR
arginine-threonine-arginine
- SLPI
secretory leucoprotease inhibitor
- TACE/ADAM17
TNFα converting enzyme
- TGFα
tissue growth factor-α
- TIMP
tissue inhibitor of MMP
- TLR
toll-like receptor
- TRAF6
tumour necrosis factor receptor-associated factor 6
- WAP
whey acidic protein
References
- Adler KB, Winn WC, Jr, Alberghini TV, Craighead JE. Stimulatory effect of Pseudomonas aeruginosa on mucin secretion by the respiratory epithelium. JAMA. 1983;249:1615–1617. [PubMed] [Google Scholar]
- Aldonyte R, Hutchinson ET, Jin B, Brantly M, Block E, Patel J, et al. Endothelial alpha-1-antitrypsin attenuates cigarette smoke induced apoptosis in vitro. COPD. 2008;5:153–162. doi: 10.1080/15412550802092936. [DOI] [PubMed] [Google Scholar]
- Alkemade HA, de Jongh GJ, Arnold WP, van de Kerkhof PC, Schalkwijk J. Levels of skin-derived antileukoproteinase (SKALP)/elafin in serum correlate with disease activity during treatment of severe psoriasis with cyclosporin A. J Invest Dermatol. 1995;104:189–193. doi: 10.1111/1523-1747.ep12612749. [DOI] [PubMed] [Google Scholar]
- Alkemade JA, Molhuizen HO, Ponec M, Kempenaar JA, Zeeuwen PL, de Jongh GJ, et al. SKALP/elafin is an inducible proteinase inhibitor in human epidermal keratinocytes. J Cell Sci. 1994;107:2335–2342. doi: 10.1242/jcs.107.8.2335. [DOI] [PubMed] [Google Scholar]
- Amitani R, Wilson R, Rutman A, Read R, Ward C, Burnett D, et al. Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epithelium. Am J Respir Cell Mol Biol. 1991;4:26–32. doi: 10.1165/ajrcmb/4.1.26. [DOI] [PubMed] [Google Scholar]
- Bergin DA, Greene CM, Sterchi EE, Kenna C, Geraghty P, Belaaouaj A, et al. Activation of the epidermal growth factor receptor (EGFR) by a novel metalloprotease pathway. J Biol Chem. 2008;283:31736–31744. doi: 10.1074/jbc.M803732200. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Britigan BE, Hayek MB, Doebbeling BN, Fick RB., Jr Transferrin and lactoferrin undergo proteolytic cleavage in the Pseudomonas aeruginosa-infected lungs of patients with cystic fibrosis. Infect Immun. 1993;61:5049–5055. doi: 10.1128/iai.61.12.5049-5055.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jt Buchanan, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. 2006;16:396–400. doi: 10.1016/j.cub.2005.12.039. [DOI] [PubMed] [Google Scholar]
- Butler MW, Robertson I, Greene CM, O'Neill SJ, Taggart CC, McElvaney NG. Elafin prevents lipopolysaccharide-induced AP-1 and NF-kappaB activation via an effect on the ubiquitin-proteasome pathway. J Biol Chem. 2006;281:34730–34735. doi: 10.1074/jbc.M604844200. [DOI] [PubMed] [Google Scholar]
- Cantin AM, Woods DE. Aerosolized prolastin suppresses bacterial proliferation in a model of chronic Pseudomonas aeruginosa lung infection. Am J Respir Crit Care Med. 1999;160:1130–1135. doi: 10.1164/ajrccm.160.4.9807166. [DOI] [PubMed] [Google Scholar]
- Cantin AM, Woods DE, Cloutier D, Dufour EK, Leduc R. Polyethylene glycol conjugation at Cys232 prolongs the half-life of alpha1 proteinase inhibitor. Am J Respir Cell Mol Biol. 2002;27:659–665. doi: 10.1165/rcmb.4866. [DOI] [PubMed] [Google Scholar]
- Cantin AM, Berthiaume Y, Cloutier D, Martel M. Prolastin aerosol therapy and sputum taurine in cystic fibrosis. Clin Invest Med. 2006;29:201–207. [PubMed] [Google Scholar]
- Carroll TP, Greene CM, Taggart CC, Bowie AG, O'Neill SJ, McElvaney NG. Viral inhibition of IL-1- and neutrophil elastase-induced inflammatory responses in bronchial epithelial cells. J Immunol. 2005;175:7594–7601. doi: 10.4049/jimmunol.175.11.7594. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhao YH, Kalaslavadi TB, Hamati E, Nehrke K, Le AD, et al. Genome-wide search and identification of a novel gel-forming mucin MUC19/Muc19 in glandular tissues. Am J Respir Cell Mol Biol. 2004;30:155–165. doi: 10.1165/rcmb.2003-0103OC. [DOI] [PubMed] [Google Scholar]
- Choudry Y, Kenny AJ. Hydrolysis of transforming growth factor-alpha by cell-surface peptidases in vitro. Biochem J. 1991;280:57–60. doi: 10.1042/bj2800057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churg A, Wang RD, Xie C, Wright JL. Alpha-1-antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2003;168:199–207. doi: 10.1164/rccm.200302-203OC. [DOI] [PubMed] [Google Scholar]
- Cichy J, Potempa J, Travis J. Biosynthesis of alpha1-proteinase inhibitor by human lung-derived epithelial cells. J Biol Chem. 1997;272:8250–8255. doi: 10.1074/jbc.272.13.8250. [DOI] [PubMed] [Google Scholar]
- Cooley J, Takayama TK, Shapiro SD, Schechter NM, Remold-O'Donnell E. The serpin MNEI inhibits elastase-like and chymotrypsin-like serine proteases through efficient reactions at two active sites. Biochemistry. 2001;40:15762–15770. doi: 10.1021/bi0113925. [DOI] [PubMed] [Google Scholar]
- Daemen MA, Heemskerk VH, van't Veer C, Denecker G, Wolfs TG, Vandenabeele P, et al. Functional protection by acute phase proteins alpha(1)-acid glycoprotein and alpha(1)-antitrypsin against ischemia/reperfusion injury by preventing apoptosis and inflammation. Circulation. 2000;102:1420–1426. doi: 10.1161/01.cir.102.12.1420. [DOI] [PubMed] [Google Scholar]
- Dalet-Fumeron V, Guinec N, Pagano M. In vitro activation of pro-cathepsin B by three serine proteinases: leucocyte elastase, cathepsin G, and the urokinase-type plasminogen activator. FEBS Lett. 1993;332:251–254. doi: 10.1016/0014-5793(93)80643-9. [DOI] [PubMed] [Google Scholar]
- Delacourt C, Herigault S, Delclaux C, Poncin A, Levame M, Harf A, et al. Protection against acute lung injury by intravenous or intratracheal pretreatment with EPI-HNE-4, a new potent neutrophil elastase inhibitor. Am J Respir Cell Mol Biol. 2002;26:290–297. doi: 10.1165/ajrcmb.26.3.4611. [DOI] [PubMed] [Google Scholar]
- Devaney JM, Greene CM, Taggart CC, Carroll TP, O'Neill SJ, McElvaney NG. Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4. FEBS Lett. 2003;544:129–132. doi: 10.1016/s0014-5793(03)00482-4. [DOI] [PubMed] [Google Scholar]
- Ding A, Thieblemont N, Zhu J, Jin F, Zhang J, Wright S. Secretory leukocyte protease inhibitor interferes with uptake of lipopolysaccharide by macrophages. Infect Immun. 1999;67:4485–4489. doi: 10.1128/iai.67.9.4485-4489.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohrman A, Miyata S, Gallup M, Li D, Chapelin C, Coste A, et al. Mucin gene (MUC 2 and MUC 5AC) upregulation by Gram-positive and Gram-negative bacteria. Biochim Biophys Acta. 1998;1406:251–259. doi: 10.1016/s0925-4439(98)00010-6. [DOI] [PubMed] [Google Scholar]
- Doring G. The role of neutrophil elastase in chronic inflammation. Am J Respir Crit Care Med. 1994;150:S114–S117. doi: 10.1164/ajrccm/150.6_Pt_2.S114. [DOI] [PubMed] [Google Scholar]
- Doumas S, Kolokotronis A, Stefanopoulos P. Anti-inflammatory and antimicrobial roles of secretory leukocyte protease inhibitor. Infect Immun. 2005;73:1271–1274. doi: 10.1128/IAI.73.3.1271-1274.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferry G, Lonchampt M, Pennel L, de Nanteuil G, Canet E, Tucker GC. Activation of MMP-9 by neutrophil elastase in an in vivo model of acute lung injury. FEBS Lett. 1997;402:111–115. doi: 10.1016/s0014-5793(96)01508-6. [DOI] [PubMed] [Google Scholar]
- Fischer BM, Voynow JA. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol. 2002;26:447–452. doi: 10.1165/ajrcmb.26.4.4473. [DOI] [PubMed] [Google Scholar]
- Gaggar A, Jackson PL, Noerager BD, O'Reilly PJ, McQuaid DB, Rowe SM, et al. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180:5662–5669. doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraghty P, Greene CM, O'Mahony M, O'Neill SJ, Taggart CC, McElvaney NG. Secretory leucocyte protease inhibitor inhibits interferon-gamma-induced cathepsin S expression. J Biol Chem. 2007a;282:33389–33395. doi: 10.1074/jbc.M706884200. [DOI] [PubMed] [Google Scholar]
- Geraghty P, Rogan MP, Greene CM, Boxio RM, Poiriert T, O'Mahony M, et al. Neutrophil elastase up-regulates cathepsin B and matrix metalloprotease-2 expression. J Immunol. 2007b;178:5871–5878. doi: 10.4049/jimmunol.178.9.5871. [DOI] [PubMed] [Google Scholar]
- Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell. 1997;88:553–560. doi: 10.1016/s0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- Goldstein W, Doring G. Lysosomal enzymes from polymorphonuclear leukocytes and proteinase inhibitors in patients with cystic fibrosis. Am Rev Respir Dis. 1986;134:49–56. doi: 10.1164/arrd.1986.134.1.49. [DOI] [PubMed] [Google Scholar]
- Greene CM, McElvaney NG, O'Neill SJ, Taggart CC. Secretory leucoprotease inhibitor impairs toll-like receptor 2- and 4-mediated responses in monocytic cells. Infect Immun. 2004;72:3684–3687. doi: 10.1128/IAI.72.6.3684-3687.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene CM, Carroll TP, Smith SG, Taggart CC, Devnaey J, Griffin S, et al. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J Immunol. 2005;174:1638–1646. doi: 10.4049/jimmunol.174.3.1638. [DOI] [PubMed] [Google Scholar]
- Griese M, Latzin P, Kappler M, Weckerle K, Heinzlmaier T, Bernhardt T, et al. alpha1-antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur Respir J. 2007;29:240–250. doi: 10.1183/09031936.00047306. [DOI] [PubMed] [Google Scholar]
- Griese M, Kappler M, Gaggar A, Hartl D. Inhibition of airway proteases in cystic fibrosis lung disease. Eur Respir J. 2008;32:783–795. doi: 10.1183/09031936.00146807. [DOI] [PubMed] [Google Scholar]
- Grimbert D, Vecellio L, Delepine P, Attucci S, Boissinot E, Poncin A, et al. Characteristics of EPI-hNE4 aerosol: a new elastase inhibitor for treatment of cystic fibrosis. J Aerosol Med. 2003;16:121–129. doi: 10.1089/089426803321919889. [DOI] [PubMed] [Google Scholar]
- Guilbault C, Saeed Z, Downey GP, Radzioch D. Cystic fibrosis mouse models. Am J Respir Cell Mol Biol. 2007;36:1–7. doi: 10.1165/rcmb.2006-0184TR. [DOI] [PubMed] [Google Scholar]
- Guyot N, Zani ML, Berger P, Dallet-Choisy S, Moreau T. Proteolytic susceptibility of the serine protease inhibitor trappin-2 (pre-elafin): evidence for tryptase-mediated generation of elafin. Biol Chem. 2005;386:391–399. doi: 10.1515/BC.2005.047. [DOI] [PubMed] [Google Scholar]
- Guyot N, Butler MW, Kessler E, Greene CM, Levine RL, O'Neill SJ, et al. American Thoracic Society. Toronoto, Canada: 2008a. The neutrophil serine protease inhibitor elafin is susceptible to Pseudomonas aeruginosa proteases; p. A36. [Google Scholar]
- Guyot N, Butler MW, McNally P, Weldon S, Greene CM, Levine RL, et al. Elafin, an elastase-specific inhibitor, is cleaved by its cognate enzyme neutrophil elastase in sputum from individuals with cystic fibrosis. J Biol Chem. 2008b;283:32377–32385. doi: 10.1074/jbc.M803707200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl D, Latzin P, Hordijk P, Marcos V, Rudolph C, Woischnik M, et al. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med. 2007;13:1423–1430. doi: 10.1038/nm1690. [DOI] [PubMed] [Google Scholar]
- Hauber HP, Tsicopoulos A, Wallaert B, Griffin S, McElvaney NG, Daigneault P, et al. Expression of HCLCA1 in cystic fibrosis lungs is associated with mucus overproduction. Eur Respir J. 2004;23:846–850. doi: 10.1183/09031936.04.00096504. [DOI] [PubMed] [Google Scholar]
- Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- Henriksen PA, Hitt M, Xing Z, Wang J, Haslett C, Riemersma RA, et al. Adenoviral gene delivery of elafin and secretory leukocyte protease inhibitor attenuates NF-kappa B-dependent inflammatory responses of human endothelial cells and macrophages to atherogenic stimuli. J Immunol. 2004;172:4535–4544. doi: 10.4049/jimmunol.172.7.4535. [DOI] [PubMed] [Google Scholar]
- Hiemstra PS. Novel roles of protease inhibitors in infection and inflammation. Biochem Soc Trans. 2002;30:116–120. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Hiemstra PS, Maassen RJ, Stolk J, Heinzel-Wieland R, Steffens GJ, Dijkman JH. Antibacterial activity of antileukoprotease. Infect Immun. 1996;64:4520–4524. doi: 10.1128/iai.64.11.4520-4524.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser K, Albrecht GJ, Schonberger OL, Rasche B, Lempart K. An elastase-specific inhibitor from human bronchial mucus. Isolation and characterization. Hoppe Seylers Z Physiol Chem. 1981;362:1369–1375. doi: 10.1515/bchm2.1981.362.2.1369. [DOI] [PubMed] [Google Scholar]
- Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- van Houwelingen AH, Weathington NM, Verweij V, Blalock JE, Nijkamp FP, Folkerts G. Induction of lung emphysema is prevented by L-arginine-threonine-arginine. FASEB J. 2008;22:3403–3408. doi: 10.1096/fj.07-096230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Perlmutter DH. Cell-specific involvement of HNF-1beta in alpha(1)-antitrypsin gene expression in human respiratory epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;282:L757–L765. doi: 10.1152/ajplung.00271.2001. [DOI] [PubMed] [Google Scholar]
- Hubbard RC, McElvaney NG, Sellers SE, Healy JT, Czerski DB, Crystal RG. Recombinant DNA-produced alpha 1-antitrypsin administered by aerosol augments lower respiratory tract antineutrophil elastase defenses in individuals with alpha 1-antitrypsin deficiency. J Clin Invest. 1989;84:1349–1354. doi: 10.1172/JCI114305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huovila AP, Turner AJ, Pelto-Huikko M, Karkkainen I, Ortiz RM. Shedding light on ADAM metalloproteinases. Trends Biochem Sci. 2005;30:413–422. doi: 10.1016/j.tibs.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Ikari Y, Mulvihill E, Schwartz SM. Alpha 1-proteinase inhibitor, alpha 1-antichymotrypsin, and alpha 2-macroglobulin are the antiapoptotic factors of vascular smooth muscle cells. J Biol Chem. 2001;276:11798–11803. doi: 10.1074/jbc.M008503200. [DOI] [PubMed] [Google Scholar]
- Ikebe N, Akaike T, Miyamoto Y, Hayashida K, Yoshitake J, Ogawa M, et al. Protective effect of S-nitrosylated alpha(1)-protease inhibitor on hepatic ischemia-reperfusion injury. J Pharmacol Exp Ther. 2000;295:904–911. [PubMed] [Google Scholar]
- Imai K, Yokohama Y, Nakanishi I, Ohuchi E, Fujii Y, Nakai N, et al. Matrix metalloproteinase 7 (matrilysin) from human rectal carcinoma cells. Activation of the precursor, interaction with other matrix metalloproteinases and enzymic properties. J Biol Chem. 1995;270:6691–6697. doi: 10.1074/jbc.270.12.6691. [DOI] [PubMed] [Google Scholar]
- Jacquot J, Tournier JM, Puchelle E. In vitro evidence that human airway lysozyme is cleaved and inactivated by Pseudomonas aeruginosa elastase and not by human leukocyte elastase. Infect Immun. 1985;47:555–560. doi: 10.1128/iai.47.2.555-560.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin FY, Nathan C, Radzioch D, Ding A. Secretory leukocyte protease inhibitor: a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell. 1997;88:417–426. doi: 10.1016/s0092-8674(00)81880-2. [DOI] [PubMed] [Google Scholar]
- Johnson D, Travis J. Structural evidence for methionine at the reactive site of human alpha-1-proteinase inhibitor. J Biol Chem. 1978;253:7142–7144. [PubMed] [Google Scholar]
- Johnson DA, Carter-Hamm B, Dralle WM. Inactivation of human bronchial mucosal proteinase inhibitor by Pseudomonas aeruginosa elastase. Am Rev Respir Dis. 1982;126:1070–1073. doi: 10.1164/arrd.1982.126.6.1070. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Barrett AJ, Mason RW. Cathepsin L inactivates alpha 1-proteinase inhibitor by cleavage in the reactive site region. J Biol Chem. 1986;261:14748–14751. [PubMed] [Google Scholar]
- Knappstein S, Ide T, Schmidt MA, Heusipp G. Alpha 1-antitrypsin binds to and interferes with functionality of EspB from atypical and typical enteropathogenic Escherichia coli strains. Infect Immun. 2004;72:4344–4350. doi: 10.1128/IAI.72.8.4344-4350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koff JL, Shao MX, Ueki IF, Nadel JA. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1068–L1075. doi: 10.1152/ajplung.00025.2008. [DOI] [PubMed] [Google Scholar]
- Kohri K, Ueki IF, Nadel JA. Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L531–L540. doi: 10.1152/ajplung.00455.2001. [DOI] [PubMed] [Google Scholar]
- Lemjabbar H, Basbaum C. Platelet-activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat Med. 2002;8:41–46. doi: 10.1038/nm0102-41. [DOI] [PubMed] [Google Scholar]
- Lin M, Jackson P, Tester AM, Diaconu E, Overall CM, Blalock JE, et al. Matrix metalloproteinase-8 facilitates neutrophil migration through the corneal stromal matrix by collagen degradation and production of the chemotactic peptide pro-gly-pro. Am J Pathol. 2008;173:144–153. doi: 10.2353/ajpath.2008.080081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma G, Greenwell-Wild T, Lei K, Jin W, Swisher J, Hardegen N, et al. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J Exp Med. 2004;200:1337–1346. doi: 10.1084/jem.20041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElvaney NG, Crystal RG. The Lung. New York: Lippincott-Raven; 1997. [Google Scholar]
- McElvaney NG, Hubbard RC, Birrer P, Chernick MS, Caplan DB, Frank MM, et al. Aerosol alpha 1-antitrypsin treatment for cystic fibrosis. Lancet. 1991;337:392–394. doi: 10.1016/0140-6736(91)91167-s. [DOI] [PubMed] [Google Scholar]
- McElvaney NG, Nakamura H, Birrer P, Hebert CA, Wong WL, Alphonso M, et al. Modulation of airway inflammation in cystic fibrosis. In vivo suppression of interleukin-8 levels on the respiratory epithelial surface by aerosolization of recombinant secretory leukoprotease inhibitor. J Clin Invest. 1992;90:1296–1301. doi: 10.1172/JCI115994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElvaney NG, Doujaiji B, Moan MJ, Burnham MR, Wu MC, Crystal RG. Pharmacokinetics of recombinant secretory leukoprotease inhibitor aerosolized to normals and individuals with cystic fibrosis. Am Rev Respir Dis. 1993;148:1056–1060. doi: 10.1164/ajrccm/148.4_Pt_1.1056. [DOI] [PubMed] [Google Scholar]
- McNeely TB, Dealy M, Dripps DJ, Orenstein JM, Eisenberg SP, Wahl SM. Secretory leukocyte protease inhibitor: a human saliva protein exhibiting anti-human immunodeficiency virus 1 activity in vitro. J Clin Invest. 1995;96:456–464. doi: 10.1172/JCI118056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeely TB, Shugars DC, Rosendahl M, Tucker C, Eisenberg SP, Wahl SM. Inhibition of human immunodeficiency virus type 1 infectivity by secretory leukocyte protease inhibitor occurs prior to viral reverse transcription. Blood. 1997;90:1141–1149. [PubMed] [Google Scholar]
- Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- Mariencheck WI, Alcorn JF, Palmer SM, Wright JR. Pseudomonas aeruginosa elastase degrades surfactant proteins A and D. Am J Respir Cell Mol Biol. 2003;28:528–537. doi: 10.1165/rcmb.2002-0141OC. [DOI] [PubMed] [Google Scholar]
- Mason DY, Cramer EM, Masse JM, Crystal R, Bassot JM, Breton-Gorius J. Alpha 1-antitrypsin is present within the primary granules of human polymorphonuclear leukocytes. Am J Pathol. 1991;139:623–628. [PMC free article] [PubMed] [Google Scholar]
- Merlos-Suarez A, Ruiz-Paz S, Baselga J, Arribas J. Metalloprotease-dependent protransforming growth factor-alpha ectodomain shedding in the absence of tumor necrosis factor-alpha-converting enzyme. J Biol Chem. 2001;276:48510–48517. doi: 10.1074/jbc.M103488200. [DOI] [PubMed] [Google Scholar]
- Mihaila A, Tremblay GM. Human alveolar macrophages express elafin and secretory leukocyte protease inhibitor. Z Naturforsch C. 2001;56:291–297. doi: 10.1515/znc-2001-3-420. [DOI] [PubMed] [Google Scholar]
- Molmenti EP, Perlmutter DH, Rubin DC. Cell-specific expression of alpha 1-antitrypsin in human intestinal epithelium. J Clin Invest. 1993;92:2022–2034. doi: 10.1172/JCI116797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morihara K, Tsuzuki H, Harada M, Iwata T. Purification of human plasma alpha 1-proteinase inhibitor and its inactivation by Pseudomonas aeruginosa elastase. J Biochem. 1984;95:795–804. doi: 10.1093/oxfordjournals.jbchem.a134671. [DOI] [PubMed] [Google Scholar]
- Mulgrew AT, Taggart CC, Lawless MW, Greene CM, Brantly ML, O'Neill SJ, et al. Z alpha1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest. 2004;125:1952–1957. doi: 10.1378/chest.125.5.1952. [DOI] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Yoshimura K, McElvaney NG, Crystal RG. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J Clin Invest. 1992;89:1478–1484. doi: 10.1172/JCI115738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara K, Ito S, Ito T, Suzuki Y, Ghoneim MA, Tachibana S, et al. Elastase inhibitor elafin is a new type of proteinase inhibitor which has a transglutaminase-mediated anchoring sequence termed ‘cementoin.’. J Biochem. 1994;115:441–448. doi: 10.1093/oxfordjournals.jbchem.a124357. [DOI] [PubMed] [Google Scholar]
- Nita I, Hollander C, Westin U, Janciauskiene SM. Prolastin, a pharmaceutical preparation of purified human alpha1-antitrypsin, blocks endotoxin-mediated cytokine release. Respir Res. 2005;6:12. doi: 10.1186/1465-9921-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonomura K, Yamanishi K, Yasuno H, Nara K, Hirose S. Up-regulation of elafin/SKALP gene expression in psoriatic epidermis. J Invest Dermatol. 1994;103:88–91. doi: 10.1111/1523-1747.ep12391802. [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291:C1–C10. doi: 10.1152/ajpcell.00620.2005. [DOI] [PubMed] [Google Scholar]
- Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L, et al. alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol. 2006a;169:1155–1166. doi: 10.2353/ajpath.2006.060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache I, Fijalkowska I, Zhen L, Medler TR, Brown E, Cruz P, et al. A novel antiapoptotic role for alpha1-antitrypsin in the prevention of pulmonary emphysema. Am J Respir Crit Care Med. 2006b;173:1222–1228. doi: 10.1164/rccm.200512-1842OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfundt R, van Ruissen F, van Vlijmen-Willems IM, Alkemade HA, Zeeuwen PL, Jap PH, et al. Constitutive and inducible expression of SKALP/elafin provides anti-elastase defense in human epithelia. J Clin Invest. 1996;98:1389–1399. doi: 10.1172/JCI118926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey BW. Management of pulmonary disease in patients with cystic fibrosis. N Engl J Med. 1996;335:179–188. doi: 10.1056/NEJM199607183350307. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Tolle DP, Barrett AJ. Evolutionary families of peptidase inhibitors. Biochem J. 2004;378:705–716. doi: 10.1042/BJ20031825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DD, Brain JD, Wohl ME, Humes JL, Mumford RA. Inhibition of neutrophil elastase in CF sputum by L-658,758. J Pharmacol Exp Ther. 1997;283:1201–1206. [PubMed] [Google Scholar]
- Reid PT, Marsden ME, Cunningham GA, Haslett C, Sallenave JM. Human neutrophil elastase regulates the expression and secretion of elafin (elastase-specific inhibitor) in type II alveolar epithelial cells. FEBS Lett. 1999;457:33–37. doi: 10.1016/s0014-5793(99)01004-2. [DOI] [PubMed] [Google Scholar]
- Rogan MP, Taggart CC, Greene CM, Murphy PG, O'Neill SJ, McElvaney NG. Loss of microbicidal activity and increased formation of biofilm due to decreased lactoferrin activity in patients with cystic fibrosis. J Infect Dis. 2004;190:1245–1253. doi: 10.1086/423821. [DOI] [PubMed] [Google Scholar]
- Rogers J, Kalsheker N, Wallis S, Speer A, Coutelle CH, Woods D, et al. The isolation of a clone for human alpha 1-antitrypsin and the detection of alpha 1-antitrypsin in mRNA from liver and leukocytes. Biochem Biophys Res Commun. 1983;116:375–382. doi: 10.1016/0006-291x(83)90532-6. [DOI] [PubMed] [Google Scholar]
- Rose MC, Nickola TJ, Voynow JA. Airway mucus obstruction: mucin glycoproteins, MUC gene regulation and goblet cell hyperplasia. Am J Respir Cell Mol Biol. 2001;25:533–537. doi: 10.1165/ajrcmb.25.5.f218. [DOI] [PubMed] [Google Scholar]
- Sallenave JM, Marsden MD, Ryle AP. Isolation of elafin and elastase-specific inhibitor (ESI) from bronchial secretions. Evidence of sequence homology and immunological cross-reactivity. Biol Chem Hoppe Seyler. 1992;373:27–33. doi: 10.1515/bchm3.1992.373.1.27. [DOI] [PubMed] [Google Scholar]
- Sallenave JM, Shulmann J, Crossley J, Jordana M, Gauldie J. Regulation of secretory leukocyte proteinase inhibitor (SLPI) and elastase-specific inhibitor (ESI/elafin) in human airway epithelial cells by cytokines and neutrophilic enzymes. Am J Respir Cell Mol Biol. 1994;11:733–741. doi: 10.1165/ajrcmb.11.6.7946401. [DOI] [PubMed] [Google Scholar]
- Sallenave JM, Si Tahar M, Cox G, Chignard M, Gauldie J. Secretory leukocyte proteinase inhibitor is a major leukocyte elastase inhibitor in human neutrophils. J Leukoc Biol. 1997;61:695–702. doi: 10.1002/jlb.61.6.695. [DOI] [PubMed] [Google Scholar]
- Schlondorff J, Lum L, Blobel CP. Biochemical and pharmacological criteria define two shedding activities for TRANCE/OPGL that are distinct from the tumor necrosis factor alpha convertase. J Biol Chem. 2001;276:14665–14674. doi: 10.1074/jbc.M010741200. [DOI] [PubMed] [Google Scholar]
- Schmidtchen A, Frick IM, Andersson E, Tapper H, Bjorck L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol. 2002;46:157–168. doi: 10.1046/j.1365-2958.2002.03146.x. [DOI] [PubMed] [Google Scholar]
- Shamamian P, Schwartz JD, Pocock BJ, Monea S, Whiting D, Marcus SG, et al. Activation of progelatinase A (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: a role for inflammatory cells in tumor invasion and angiogenesis. J Cell Physiol. 2001;189:197–206. doi: 10.1002/jcp.10014. [DOI] [PubMed] [Google Scholar]
- Shao MX, Nadel JA. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci USA. 2005a;102:767–772. doi: 10.1073/pnas.0408932102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-alpha-converting enzyme. J Immunol. 2005b;175:4009–4016. doi: 10.4049/jimmunol.175.6.4009. [DOI] [PubMed] [Google Scholar]
- Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. 2003;163:2329–2335. doi: 10.1016/S0002-9440(10)63589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson AJ, Maxwell AI, Govan JR, Haslett C, Sallenave JM. Elafin (elastase-specific inhibitor) has anti-microbial activity against gram-positive and gram-negative respiratory pathogens. FEBS Lett. 1999;452:309–313. doi: 10.1016/s0014-5793(99)00670-5. [DOI] [PubMed] [Google Scholar]
- Simpson AJ, Cunningham GA, Porteous DJ, Haslett C, Sallenave JM. Regulation of adenovirus-mediated elafin transgene expression by bacterial lipopolysaccharide. Hum Gene Ther. 2001;12:1395–1406. doi: 10.1089/104303401750298553. [DOI] [PubMed] [Google Scholar]
- Sires UI, Murphy G, Baragi VM, Fliszar CJ, Welgus HG, Senior RM. Matrilysin is much more efficient than other matrix metalloproteinases in the proteolytic inactivation of alpha 1-antitrypsin. Biochem Biophys Res Commun. 1994;204:613–620. doi: 10.1006/bbrc.1994.2503. [DOI] [PubMed] [Google Scholar]
- Sørensen O, Follin P, Johnsen A, Calafat J, Tjabringa G, Hiemstra P, et al. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. 2001;97:3951–3959. doi: 10.1182/blood.v97.12.3951. [DOI] [PubMed] [Google Scholar]
- Streit V, Wiedow O, Bartels J, Christophers E. Antiprotease activity in urine of patients with inflammatory skin disorders. J Invest Dermatol. 1995;105:562–566. doi: 10.1111/1523-1747.ep12323460. [DOI] [PubMed] [Google Scholar]
- Suter S. The role of bacterial proteases in the pathogenesis of cystic fibrosis. Am J Respir Crit Care Med. 1994;150:S118–S122. doi: 10.1164/ajrccm/150.6_Pt_2.S118. [DOI] [PubMed] [Google Scholar]
- Taggart C, Cervantes-Laurean D, Kim G, McElvaney NG, Wehr N, Moss J, et al. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem. 2000;275:27258–27265. doi: 10.1074/jbc.M004850200. [DOI] [PubMed] [Google Scholar]
- Taggart CC, Lowe GJ, Greene CM, Mulgrew AT, O'Neill SJ, Levine RL, et al. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J Biol Chem. 2001;276:33345–33352. doi: 10.1074/jbc.M103220200. [DOI] [PubMed] [Google Scholar]
- Taggart CC, Greene CM, McElvaney NG, O'Neill S. Secretory leucoprotease inhibitor prevents lipopolysaccharide-induced IkappaBalpha degradation without affecting phosphorylation or ubiquitination. J Biol Chem. 2002;277:33648–33653. doi: 10.1074/jbc.M203710200. [DOI] [PubMed] [Google Scholar]
- Taggart CC, Greene CM, Smith SG, Levine RL, McCray PB, Jr, O'Neill S, et al. Inactivation of human beta-defensins 2 and 3 by elastolytic cathepsins. J Immunol. 2003;171:931–937. doi: 10.4049/jimmunol.171.2.931. [DOI] [PubMed] [Google Scholar]
- Taggart CC, Cryan SA, Weldon S, Gibbons A, Greene CM, Kelly E, et al. Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J Exp Med. 2005;202:1659–1668. doi: 10.1084/jem.20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis J, Owen M, George P, Carrell R, Rosenberg S, Hallewell RA, et al. Isolation and properties of recombinant DNA produced variants of human alpha 1-proteinase inhibitor. J Biol Chem. 1985;260:4384–4389. [PubMed] [Google Scholar]
- Turk V, Stoka V, Turk D. Cystatins: biochemical and structural properties, and medical relevance. Front Biosci. 2008;13:5406–5420. doi: 10.2741/3089. [DOI] [PubMed] [Google Scholar]
- Venembre P, Boutten A, Seta N, Dehoux MS, Crestani B, Aubier M, et al. Secretion of alpha 1-antitrypsin by alveolar epithelial cells. FEBS Lett. 1994;346:171–174. doi: 10.1016/0014-5793(94)80695-0. [DOI] [PubMed] [Google Scholar]
- Voynow JA, Selby DM, Rose MC. Mucin gene expression (MUC1, MUC2, and MUC5/5AC) in nasal epithelial cells of cystic fibrosis, allergic rhinitis, and normal individuals. Lung. 1998;176:345–354. doi: 10.1007/pl00007616. [DOI] [PubMed] [Google Scholar]
- Walsh DE, Greene CM, Carroll TP, Taggart CC, Gallagher PM, O'Neill SJ, et al. Interleukin-8 up-regulation by neutrophil elastase is mediated by MyD88/IRAK/TRAF-6 in human bronchial epithelium. J Biol Chem. 2001;276:35494–35499. doi: 10.1074/jbc.M103543200. [DOI] [PubMed] [Google Scholar]
- Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, et al. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol. 2007;9:1162–1171. doi: 10.1111/j.1462-5822.2006.00857.x. [DOI] [PubMed] [Google Scholar]
- Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12:317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- van Wetering S, van der Linden AC, van Sterkenburg MA, de Boer WI, Kuijpers AL, Schalkwijk J, et al. Regulation of SLPI and elafin release from bronchial epithelial cells by neutrophil defensins. Am J Physiol Lung Cell Mol Physiol. 2000;278:L51–L58. doi: 10.1152/ajplung.2000.278.1.L51. [DOI] [PubMed] [Google Scholar]
- Wiedow O, Luademann J, Utecht B. Elafin is a potent inhibitor of proteinase 3. Biochem Biophys Res Commun. 1991;174:6–10. doi: 10.1016/0006-291x(91)90476-n. [DOI] [PubMed] [Google Scholar]
- Wiedow O, Harder J, Bartels J, Streit V, Christophers E. Antileukoprotease in human skin: an antibiotic peptide constitutively produced by keratinocytes. Biochem Biophys Res Commun. 1998;248:904–909. doi: 10.1006/bbrc.1998.9069. [DOI] [PubMed] [Google Scholar]
- Williams SE, Brown TI, Roghanian A, Sallenave JM. SLPI and elafin: one glove, many fingers. Clin Sci (Lond) 2006;110:21–35. doi: 10.1042/CS20050115. [DOI] [PubMed] [Google Scholar]
- Witko-Sarsat V, Halbwachs-Mecarelli L, Schuster A, Nusbaum P, Ueki I, Canteloup S, et al. Proteinase 3, a potent secretagogue in airways, is present in cystic fibrosis sputum. Am J Respir Cell Mol Biol. 1999;20:729–736. doi: 10.1165/ajrcmb.20.4.3371. [DOI] [PubMed] [Google Scholar]
- Woods DE, Cantin A, Cooley J, Kenney DM, Remold-O'Donnell E. Aerosol treatment with MNEI suppresses bacterial proliferation in a model of chronic Pseudomonas aeruginosa lung infection. Pediatr Pulmonol. 2005;39:141–149. doi: 10.1002/ppul.20167. [DOI] [PubMed] [Google Scholar]
- Zanetti M. The role of cathelicidins in the innate host defenses of mammals. Curr Issues Mol Biol. 2005;7:179–196. [PubMed] [Google Scholar]
- Zanetti M, Gennaro R, Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995;374:1–5. doi: 10.1016/0014-5793(95)01050-o. [DOI] [PubMed] [Google Scholar]
- Zani ML, Nobar SM, Lacour SA, Lemoine S, Boudier C, Bieth JG, et al. Kinetics of the inhibition of neutrophil proteinases by recombinant elafin and pre-elafin (trappin-2) expressed in Pichia pastoris. Eur J Biochem. 2004;271:2370–2378. doi: 10.1111/j.1432-1033.2004.04156.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, DeWitt DL, McNeely TB, Wahl SM, Wahl LM. Secretory leukocyte protease inhibitor suppresses the production of monocyte prostaglandin H synthase-2, prostaglandin E2, and matrix metalloproteinases. J Clin Invest. 1997;99:894–900. doi: 10.1172/JCI119254. [DOI] [PMC free article] [PubMed] [Google Scholar]