

Abstract
Background and purpose:
The proteasome inhibitor model of Parkinson's disease (PD) appears to reproduce many of the important behavioural, imaging, pathological and biochemical features of the human disease. However, the mechanisms involved in the lactacystin-induced, mitochondria-mediated apoptotic pathway remain poorly defined.
Experimental approach:
We have used lactacystin as a specific inhibitor of the 20S proteasome in the dopaminergic neuroblastoma cell line SH-SY5Y. We over-expressed a green fluorescent protein (GFP)–Bax fusion protein in these cells to study localization of Bax. Free radical scavengers were used to assess the role of reactive oxygen species (ROS) in these pathways.
Key results:
Lactacystin triggered a concentration-dependent increase in cell death mediated by the mitochondrial apoptotic pathway, and induced a change in mitochondrial membrane permeability accompanied by cytochrome c release. The participation of Bax protein was more critical than the formation of the permeability transition pore in mitochondria. GFP–Bax over-expression demonstrated Bax redistribution from the cytosol to mitochondria after the addition of lactacystin. ROS, but not p38 mitogen-activated protein kinase, participated in lactacystin-induced mitochondrial Bax translocation. Lactacystin disrupted the intracellular redox state by increasing ROS production and depleting endogenous antioxidant systems such as glutathione (GSH). Pharmacological depletion of GSH, using l-buthionine sulphoxide, potentiated lactacystin-induced cell death. Lactacystin sensitized neuroblastoma cells to oxidative damage, induced by subtoxic concentrations of 6-hydroxydopamine.
Conclusions and implications:
The lactacystin-induced, mitochondrial-mediated apoptotic pathway involved interactions between ROS, GSH and Bax. Lactacystin could constitute a potential factor in the development of sporadic PD.
Keywords: proteasome, Parkinson's disease, mitochondria, Bax, reactive oxygen species, glutathione
Introduction
One of the main goals of neuroscience is to shed light upon the cellular pathways involved in neurodegenerative processes. Once the pathophysiology is understood, we can then offer new pharmacological targets to develop effective treatments. The aetiology of neurodegenerative diseases may be diverse. However, a common pathological feature is impaired protein degradation machinery, leading to the formation of inclusion bodies or aggregates. The formation of aberrant protein conformers and the occurrence of pathognomonic proteinaceous deposits (Winklhofer et al., 2008) are characteristic features of several neurodegenerative disorders including Alzheimer's, Huntington's and Parkinson's disease (PD) (Forman et al., 2004). PD is caused by both genetic and environmental factors with a characteristic appearance of cytoplasmic inclusion bodies, termed Lewy bodies. Lewy bodies contain a heterogeneous mixture of insoluble filamentous lipids and proteins including α-synuclein, ubiquitin, neurofilaments and oxidized or nitrated proteins (Forno, 1996). Impairment of the proteolytic activities of the 26/20S proteasomes in the substantia nigra pars compacta in sporadic PD has been demonstrated through post mortem examinations (McNaught and Jenner, 2001; McNaught et al., 2002). The proteasome inhibitor model of PD appears to reproduce many of the important behavioural, imaging, pathological and biochemical features of the human condition (McNaught and Olanow, 2006).
Proteasome inhibitors, such as lactacystin, MG132 and ALLN, target the 20S proteasome, a component of the ubiquitin–proteasome complex responsible for the degradation of unwanted cellular proteins. The effects of these inhibitors on cell viability have been unclear. Pharmacological inhibitors of proteasomes induced neuronal apoptosis in a variety of primary neuron culture models (Qiu et al., 2000; Piccioli et al., 2001), although other authors have demonstrated that the inhibition of proteasome activity prevented apoptosis in some neuronal death models, such as cultured sympathetic neurons deprived of nerve growth factor (Sadoul et al., 1996), cerebellar granule neurons exposed to low potassium (Canu et al., 2000) and cortical neurons exposed to β-amyloid fragments (Favit et al., 2000).
Lactacystin-induced cell death involves both mitochondria-mediated apoptosis and cytochrome c release as a consequence of the inhibition of proteasome activity. Two predominant, but opposing, theories on mitochondrial membrane permeability and cytochrome c release have been reported (Martinou and Green, 2001). The first theory, initially described in ischaemia–reperfusion injury, is based on the development of a permeability transition pore (PTP) that opens under conditions of oxidative stress, high Ca2+ or low ATP concentrations. The opened pore allows influx of low-molecular weight solutes, leading to mitochondrial swelling, rupture and leakage of cytochrome c. The second theory relating to cytochrome c release contends that Bax and other pro-apoptotic Bcl-2 family member proteins polymerize and insert into the outer mitochondrial membrane, and allow cytochrome c release without disrupting mitochondrial function or the mitochondrial potential (Martinou and Green, 2001).
In our present work, we have examined the pathways leading to lactacystin-induced activation of the mitochondrial apoptotic pathway in SH-SY5Y cells. We found that lactacystin induced mitochondrial cytochrome c release by translocation of Bax protein into the mitochondria through a mechanism involving reactive oxygen species (ROS).
Methods
Cell culture and drug treatment procedures
SH-SY5Y cultures were grown as described previously (Jordan et al., 2004) in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM l-glutamine, 20 units·mL−1 penicillin, 5 µg·mL−1 streptomycin and 15% (v/v) fetal bovine serum (Invitrogen, Carlsbad, CA, USA). Mouse embryonic fibroblast (MEF) Bax−/− and +/+ cells, a gift from Dr M. Serrano (CNIO, Madrid, Spain), were grown in DMEM supplemented with 2 mM l-glutamine, penicillin (20 units·mL−1), streptomycin (5 µg·mL−1) and 15% (v/v) fetal bovine serum. Cells were grown in a humidified cell incubator at 37°C under a 5% CO2 atmosphere. For treatments, lactacystin in dimethyl sulphoxide (DMSO) was directly added to the culture medium. The corresponding controls were treated with the same concentration of DMSO, which was always below 0.1% (final concentration).
Lactate dehydrogenase activity
Cell viability after lactacystin additions was assessed by measurement of lactate dehydrogenase activity according to the protocol provided by the manufacturer (Promega, Madison, WI, USA). Briefly, the reaction mixture was added to conditioned media, and removed from 24-well plates after centrifugation at 250×g for 10 min. Absorbance of samples at 490 nm was measured in a microplate reader (Bio-Rad, Hercules, CA, USA) after 30 min of incubation at room temperature.
3-(-4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cell viability was measured by the ability to reduce MTT to the blue formazan product. After removal of the culture medium, the cells were incubated with 1 mg·mL−1 MTT in normal culture medium for 2 h at 37°C. The medium was then aspirated, and the formazan was dissolved in 200 µL DMSO. Absorbance at 540 nm (A540) was measured in a VERSAmax Lunable microplate reader (Molecular Devices, Sunnyvale, CA, USA), and the A540 in control conditions was used as 100% cell culture viability.
Chromatin state
For assessment of chromatin state, the SH-SY5Y cells were plated on poly(l-lysine)-coated glass slides. Nuclei were stained with 0.5 µg·mL−1 Hoechst 33342. Uniformly stained nuclei were scored as healthy, viable cells. Condensed or fragmented nuclei were scored as apoptotic.
Measurement of levels of ROS
We used the oxidation-sensitive fluorescent dye 5-(and-6)-chloromethyl-2,7 dichlorodihydrofluorescein diacetate to measure the production of ROS, mainly hydrogen peroxide and hydroxyl radicals. 2,7-Dichlorofluorescein diacetate is deacetylated by esterases. This non-fluorescent product is then converted by ROS into dichlorofluorescein, which can easily be visualized by fluorescence at 530 nm when excited at 485 nm. The SH-SY5Y cells seeded in 96-well culture plates were incubated with dichlorodihydrofluorescein diacetate (10 µg·mL−1) for 5 min, and fluorescence intensity was measured in a Spectra Max Gemini XS (Molecular Devices). The average ROS production (relative to levels in vehicle-treated controls) was calculated from four individual wells in at least three independent platings.
Measurement of glutathione (GSH) levels
Levels of GSH were determined by using monochlorobimane [syn-(ClCH2, CH3)-1,5-diazabicyclo-(3,3,0)-octa-3,6-dione-2,8-dione; mBCl] fluorescence. GSH is specifically conjugated with mBCl to form a fluorescent bimane–GSH adduct in a reaction catalysed by GSH S-transferases (Shrieve et al., 1998). The concentration of the bimane–GSH adducts increases during the initial 10–12 min period of this reaction with first-order kinetics before levelling off (Young et al., 1994). The culture medium was removed, and cells were washed twice with 0.2 mL Krebs solution, and then incubated for 1 h at 37°C in 0.2 mL fresh Krebs solution containing 160 µM mBCl. After incubation, fluorescence was measured at an excitation wavelength of 340 nm and emission wavelength of 460 nm in a Spectra Max Gemini XS (Molecular Devices). The mean GSH levels from at least three separate cultures were determined and expressed as a percentage of the control values. Experiments were performed in 96-multi-well plates, allowing four samples for each experimental point. Results are expressed as the mean ± SE values.
Evaluation of the mitochondrial translocation of green fluorescent protein (GFP)–Bax construct
The transient transfection of SH-SY5Y cells with a GFP–Bax construct and confocal microscopy was based on a previously described procedure (Gomez-Lazaro et al., 2008). In brief, the SH-SY5Y cells were plated on poly(l-lysine)-coated glass slides for 24 h before transfection at a density of 5.3 × 104 cells·cm−2. Transfection was performed with a plasmid containing GFP–Bax (Gomez-Lazaro et al., 2007) and the Lipofectamine reagent (Invitrogen, Barcelona, Spain). After 4 h of co-incubation, the transfection mixture was removed and replaced with fresh complete medium. The cells were then incubated for 24 h to allow for sufficient GFP–Bax expression, and finally treated for 12 h with the different compounds to be tested. The cells were fixed in 4% paraformaldehyde, and subsequent confocal microscopy was performed. Mitochondrial translocation of the construct resulted in a change from a diffusely green fluorescent cytoplasm to a brighter spotted one. The total number of transfected cells and those displaying the spotted pattern were counted, and the number of spotted pattern cells expressed as a percentage of all transfected cells. The mean ± SD of five independent experiments was obtained, and statistical significance was assessed by a Student's two-tailed, unpaired t-test. Values of P < 0.05 are considered significant.
Mitochondrial isolation
Mitochondria were isolated from rat liver as previously described (Galindo et al., 2003a). Briefly, the tissue was manually homogenized by four strokes with a Teflon pestle in solution I containing 230 mM mannitol, 70 mM sucrose, 1 mM ethylene glycol tetraacetic acid (EGTA) and 5 mM HEPES (pH 7.4) on ice. After centrifugation (100×g for 80 s at 4°C), the supernatant was layered in solution II [460 mM mannitol, 14 mM sucrose, 1 mM EGTA and 10 mM HEPES (pH 7.4)], and centrifuged at 800×g for 3 min at 4°C. The top layer was then centrifuged at 2000×g for 5 min at 4°C. The mitochondrial pellet was resuspended in 215 mM mannitol, 71 mM sucrose, 10 mM succinate and 10 mM HEPES (pH 7.4), and kept on ice until mitochondrial PTP determinations.
PTP activity
Opening of PTPs was assayed spectrophotometrically as previously described (Galindo et al., 2003b). Specifically, the mitochondria were suspended to reach a protein concentration of 1 mg·mL−1 in 200 µL of solution containing 125 mM KCl, 20 mM HEPES, 2 mM KH2PO4, 1 µM EGTA, 1 mM MgCl2, 5 mM malate and 5 mM glutamate with the pH adjusted to 7.4 with KOH. After the addition of different compounds, mitochondrial swelling due to PTP opening was indicated by changes in absorbance at 540 nm (A540) using a microplate reader (Bio-Rad). The initial A540 values were 0.8, and minor differences in the loading of the wells were compensated by representing the data as a fraction of the initial absorbance determination remaining at a given time. Experiments were performed in 96-multi-well plates, allowing four samples for each experimental point. Mean ± SD of six independent experiments was obtained.
Preparation of cytosolic and mitochondrial extracts
Immunoblot analysis was performed on cytosolic extracts from control and lactacystin-treated cultures, as previously described (Fernandez-Gomez et al., 2005). The cells were washed once with phosphate-buffered saline (PBS) and collected by centrifugation (2000×g; 5 min). Cell pellets were resuspended in 100 µL of extraction buffer containing (in mM): sucrose, 250; Tris–HCl, 50; EGTA, 1; EDTA, 1; DTT, 1; PMSF, 0.1 (pH 7.4), then homogenized in a Teflon-glass homogenizer (five strokes). After 20 min on ice, it was centrifuged (15 000×g; 20 min). The supernatants (i.e. cytosolic fractions) were removed and stored at −80°C until analysed by gel electrophoresis.
Western blots
The protein from each condition was quantified spectrophotometrically (Micro BCA Protein Reagent Kit, Pierce, Rockford, IL, USA), and an equal amount of protein (30 µg) was loaded onto 15% SDS–PAGE gels. After electrophoresis, the proteins were transferred to polyvinylidene difluoride membranes (Immobilon, Millipore Corporation, Billerica, MA, USA). Non-specific protein binding was blocked with Blotto (4% w/v non-fat dried milk, 4% bovine serum albumin (Sigma, St Louis, MO, USA), and 0.1% Tween 20 Sigma) in PBS for 1 h. The membranes were incubated overnight at 4°C with anti-cytochrome c (1:1000 dilution of mouse monoclonal IgG, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) or with anti-cytochrome c oxidase subunit IV (COX-IV; BD Biosciences, San Jose, CA, USA), p38MAPK and anti-phospho-p38 (1:1000 dilution of polyclonal antibody, Cell Signaling, Beverly, MA, USA), anti-Bax (1:1000 dilution of polyclonal antibody, Cell Signaling) and a monoclonal anti-α-tubulin antibody (Sigma). After washing with Blotto, the membranes were incubated with a secondary antibody (1:5000 dilution of peroxidase-labelled antibody, Promega) in Blotto. The signal was detected using an enhanced chemiluminescence detection kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Immunoblots were developed by exposure to X-ray film (Eastman Kodak, Rochester, NY, USA).
Statistics
The results were expressed as the mean ± SD of at least three independent experiments. Student's two-tailed, unpaired t-test was used, and values of P < 0.05 were considered statistically significant. When comparing more than two conditions, statistically significant differences between groups were determined by analysis of variance followed by a post hoc Tukey analysis. The level of statistical significance was set at P < 0.05.
Materials
Most chemicals, including lactacystin (L6785), buthionine sulphoxide (BSO), N-acetyl-l-cysteine (NAC), (S)-6-methoxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) and 6-hydroxydopamine (6-OHDA) were obtained from Sigma; 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPOL) was from Calbiochem (San Diego, CA, USA), while 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, mBcl and Hoechst 33342 were purchased from Molecular Probes, Inc. (Eugene, OR, USA). The Micro BCA Protein Reagent Kit came from Pierce.
Results
Lactacystin induces cell death in SH-SY5Y cultures
Consistent with previous reports, proteasome inhibition by lactacystin was toxic to SH-SY5Y cell cultures (Lev et al., 2006). We used lactate dehydrogenase, MTT and Hoechst 33342 to analyse viability of SH-SY5Y cell cultures challenged with or without lactacystin (0.01 to 10 µM). As Figure 1A illustrates, lactacystin triggered an increase in cell death of SH-SY5Y cells in a concentration-dependent manner. The concentration of lactacystin used was within a range that provided graded inhibition of proteasome activity (Fenteany et al., 1994; Figueiredo-Pereira et al., 1994). Lower concentrations of lactacystin, up to 0.5 µM, did not compromise cell viability. Cells began to demonstrate morphological changes at higher concentrations of lactacystin (1–10 µM). Cells challenged with lactacystin tended to shrink and round up, displaying a high nucleus : cytoplasm ratio, and morphology typical of apoptotic cells. Furthermore, analysis of the nuclear morphology with Hoechst 33342 DNA staining clearly showed chromatin condensation and pyknotic nuclei in cells challenged with lactacystin (Figure 1D). Apoptotic nuclei were minimal or absent in control cells (Figure 1C).
Figure 1.
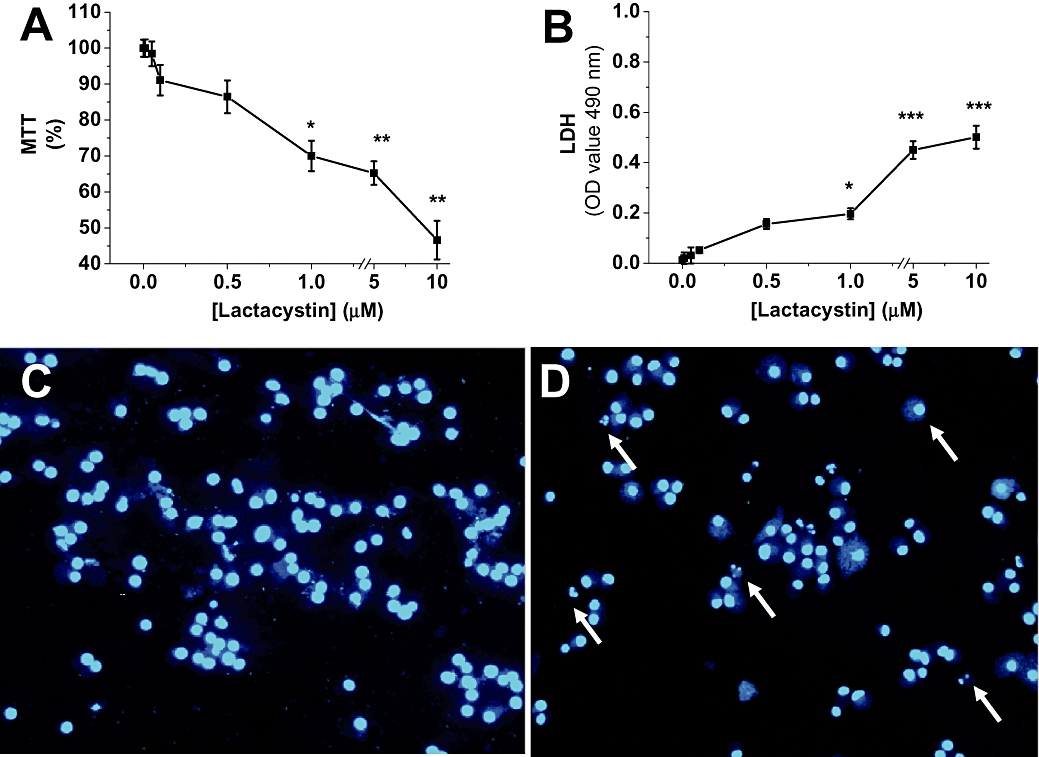
Lactacystin-induced cytotoxicity in SH-SY5Y cell cultures. (A) Cells were treated with lactacystin (0.01–10 µM) or dimethyl sulphoxide (control; 0.1%) as a solvent. A 3-(-4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) survival assay was performed 24 h after lactacystin addition. Data were expressed as the percentage of live cells relative to total cells. Data were normalized to vehicle-treated cells (taken as 100%), and values were reported as the mean ± SEM; they are representative of at least five experiments, each performed in quadruplicate. Statistically significant differences from respective controls: *P < 0.05, **P < 0.01 (one-way analysis of variance; Tukey's test). (B) Lactacystin induced loss of viability in SH-SY5Y cell cultures. Cell viability was assayed 24 h after lactacystin treatment. Culture medium was collected and analysed for lactate dehydrogenase release as an indicator of cell death. Data are presented as mean ± SEM of percentage of untreated SH-SY5Y cells of three different experiments (***P < 0.01). (C,D) SH-SY5Y cells were stained with the dye Hoechst 33342 as indicated in Methods. Normal nuclei without signs of chromatin fragmentation can be observed in untreated cultures (C), while in cell cultures exposed to lactacystin (10 µM, D), chromatin fragmentation is evident by 24 h (arrows). Images of representative fields are captured at a magnification of 40×. Similar morphological changes were found in at least five different experiments.
Lactacystin induces changes in mitochondrial membrane permeability
Mitochondrial cytochrome c release to the cytosol is considered one of the key steps in cell death pathways. In order to ascertain if this occurred in our working system, levels of cytochrome c were determined in cytosolic fractions, derived from cell cultures treated with lactacystin. Figure 2 shows that cell cultures challenged with 1–10 µM lactacystin for 12 h were accompanied by cytochrome c release from the mitochondria.
Figure 2.
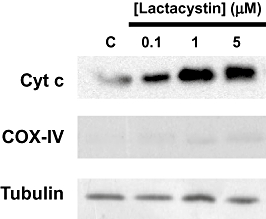
Lactacystin induced mitochondrial cytochrome c release. A representative immunoblot from SH-SY5Y cell cultures showing levels of cytochrome c in cytosolic fractions. Cells were treated for 12 h with different lactacystin concentrations ranging from 1–10 µM. Cytochrome c oxidase subunit IV (COX-IV) protein levels were used as an index of mitochondrial contamination. Equal amounts of protein (30 µg per lane) were loaded onto the gels (tubulin). The record shown is representative of three experiments.
To better understand the effect of lactacystin on PTP formation, we added lactacystin to isolated mitochondrial suspensions. As shown in Figure 3, lactacystin din not induce any significant mitochondrial swelling at any of the tested concentrations (1–10 µM). As a positive control (Galindo et al., 2003b), Ca2+ was used as a PTP activator to induce a rapid decrease in A540.
Figure 3.
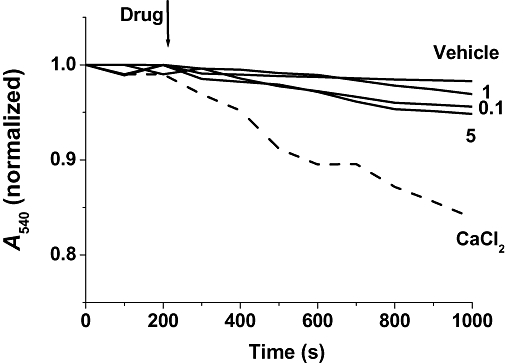
Lactacystin failed to induce mitochondrial swelling. Lactacystin was added (1–10 µM) to isolated mitochondria. Mitochondrial swelling was then noted by changes in absorbance at 540 nm (A540). Ca2+ was used as a positive control for mitochondrial swelling. Data are the mean values obtained from one experiment performed in triplicate. Similar data were found in five different mitochondrial preparations.
Bax is involved in mitochondrial membrane permeability, and we have recently reported how it translocates to mitochondria after a cytotoxic stimulus (Gomez-Lazaro et al., 2007; 2008;). The SH-SY5Y cell cultures were transfected with a GFP–Bax fusion construct, and 24 h later challenged with lactacystin. Under a confocal microscope, GFP–Bax-transfected cells showed a diffuse distribution of green fluorescence in the absence of lactacystin (Figure 4A). In contrast, cell cultures challenged with lactacystin for 12 h showed an increased punctate pattern of GFP–Bax distribution (Figure 4C). Consistent with the mentioned concentration-dependent effect, GFP–Bax translocation reached its higher values with 10 µM lactacystin (Figure 4E). When we treated cell cultures over-expressing a vector that contains GFP alone, no clusters of GFP fluorescence were noted in lactacystin-treated cells (data not shown). Thus, GFP–Bax aggregation was not a consequence of alteration in GFP protein distribution. In later experiments, we consistently observed that cells with a GFP–Bax punctate distribution had chromatin condensation (Figure 4C,D; red squares), while diffusely patterned cells had none (white circles).
Figure 4.
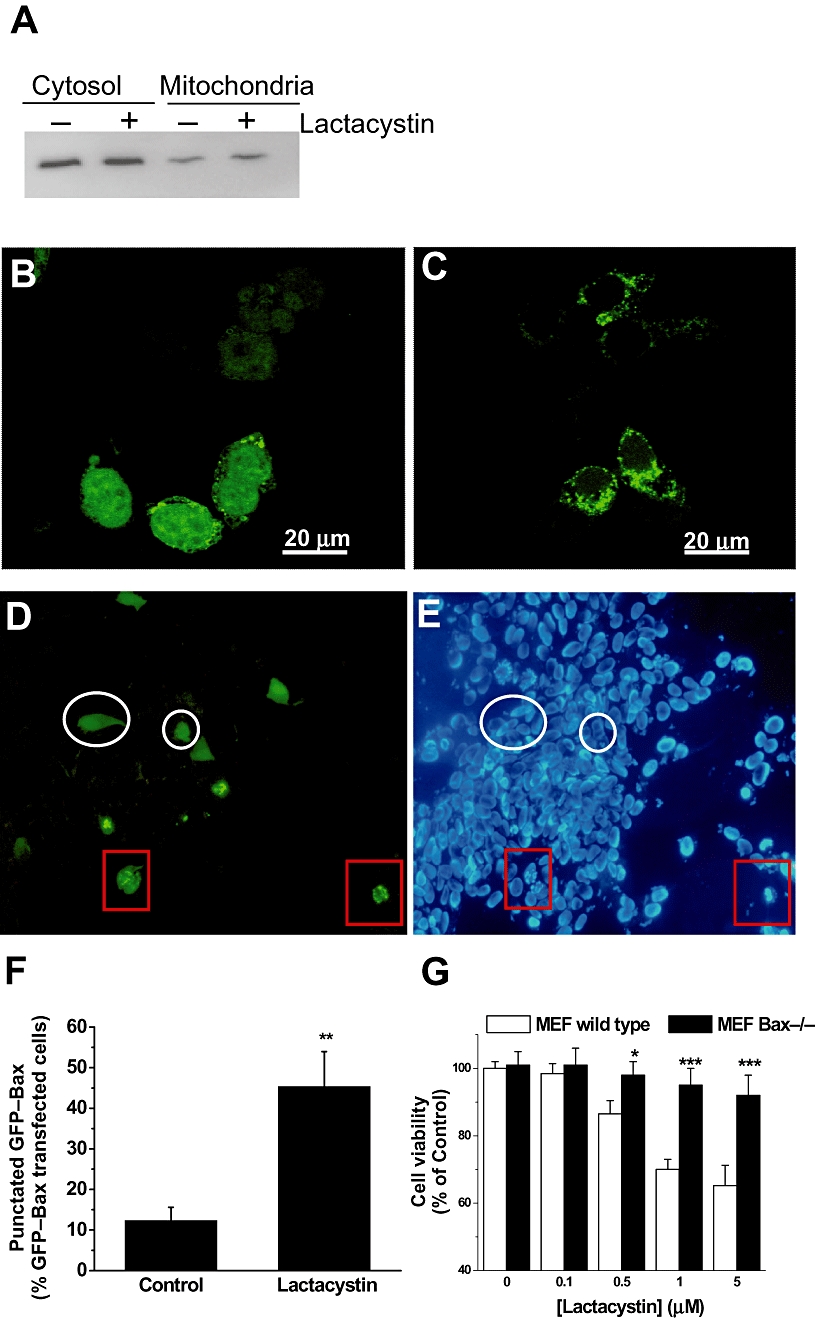
Lactacystin induced Bax translocation to the mitochondria. (A) Cytosolic and mitochondrial extracts from SH-SY5Y cells treated with or without lactacystin (10 µM, 12 h) were subjected to Western blotting and probed with anti-Bax antibody. Similar results were obtained in two independent experiments. (B–E) SH-SY5Y cells were transfected with green fluorescent protein (GFP)–Bax, incubated for 24 h to allow for sufficient GFP–Bax expression. After 12 h of lactacystin exposure, the cells were fixed in 4% p-formaldehyde, and confocal images were captured using a 63× oil immersion lens. GFP–Bax primarily demonstrated diffuse staining under control settings (B), while a punctate pattern (C) was evident, 12 h after 10 µM lactacystin treatment. In (D) and (E), after 12 h of 10 µM lactacystin treatment, GFP–Bax cells with a punctate pattern presented fragmented chromatin (red squares), while those with a diffuse pattern exhibited no fragmented chromatin (white circles). Hoechst 33342 dye was added to study the state of chromatin. The images shown are representative of results obtained in four separate experiments, each performed in triplicate. (F) Twelve hours after treatment, the number of cells with punctate Bax distribution was expressed as a percentage of the total number of cells expressing GFP–Bax protein. Results are presented as the mean ± SEM; they are representative of at least five experiments, each performed in triplicate. Statistically significant difference from respective controls: **P < 0.01 versus control conditions. Significance was determined by Student's t-test. (G) Bax protein expression is required for lactacystin-induced cell death. Cultures of mouse embryonic fibroblast (MEF) cells from wild type and Bax−/− mice were treated with 10 µM lactacystin. By 24 h after addition, the proportion of apoptotic nuclei was determined using Hoechst 33348. Results are presented as mean ± SD; they are representative of at least three experiments, each performed in triplicate. ***P < 0.001 versus wild-type MEF.
The next set of experiments were designed to find out if endogenous Bax protein behaved as the over-expressed GFP–Bax. To do so, we measured by Western blot, endogenous Bax levels in the mitochondrial fraction after lactacystin treatment. As shown in Figure 4A, by 12 h after lactacystin (10 µM) addition, we found an increase of Bax levels in mitochondrial fraction. Furthermore, we addressed the question whether Bax was required for lactacystin-induced apoptosis. We used MEF cultures from Bax-deficient mice. The lack of Bax protein conferred resistance to lactacystin toxicity, as Bax−/− MEFs were protected against lactacystin-induced apoptosis (Figure 4F).
To see whether the proposed mechanisms for lactacystin can be generalized to other proteasome inhibitors, we also challenged cell cultures with MG132 (1 and 10 µM) and ALLN (20 and 40 µM). These two inhibitors were toxic to SH-SY5Y cells (Figure 5A) and induced the translocation of GFP–Bax into the mitochondria (Figure 5B).
Figure 5.
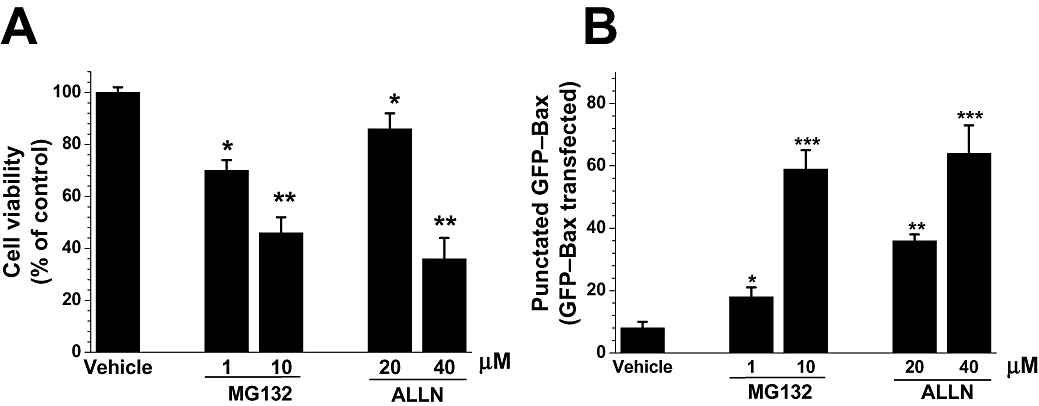
Effect of other proteasome inhibitors on SH-SY5Y cells. Cells were treated with MG132 (1 and 10 µM), ALLN (20 and 40 µM) or dimethyl sulphoxide (vehicle; 0.1%) as a solvent. (A) Cell viability assayed 24 h after drug additions by assessing the state of chromatin using Hoechst 33342. Data were normalized to vehicle-treated cells (taken as 100%), and values were reported as the mean ± SEM; they are representative of at least three experiments, each performed in quadruplicate. Statistically significant differences from respective controls: *P < 0.05, **P < 0.01 (one way analysis of variance; Tukey's test). (B) Proteasome inhibitors induced green fluorescent protein (GFP)–Bax translocation. SH-SY5Y cells were transfected with GFP–Bax, incubated for 24 h to allow for sufficient GFP–Bax expression. After 12 h of exposure to proteasome inhibitors, the cells were fixed in 4% p-formaldehyde, and the number of cells with punctate Bax distribution was counted and expressed as a percentage of the total number of cells expressing GFP–Bax protein. Results are presented as the mean ± SEM; they are representative of at least four experiments, each performed in triplicate. Statistically significant difference from respective controls: *P < 0.05; **P < 0.01; ***P < 0.001 versus control conditions. Significance was determined by Student's t-test.
Lactacystin disrupts intracellular redox state
To determine whether the lactacystin-induced cytotoxic effects were mediated through oxidative stress, we measured the production of ROS and the levels of GSH in cell cultures challenged with lactacystin. By 4 h after treatment with lactacystin, SH-SY5Y cells demonstrated an increased level of ROS production (Figure 6A). To evaluate the participation of second messengers in lactacystin-induced cell death, we pretreated cell cultures with Trolox (0.75 mM), TEMPOL (0.2 µM) and the porphyrin MnTBAP (10 µM), 30 min before the addition of 10 µM lactacystin. By 24 h later, cell viability assays revealed that TEMPOL and MnTBAP, but not Trolox, afforded cytoprotection from lactacystin (10 µM) in the SH-SY5Y cells (Figure 6B).
Figure 6.
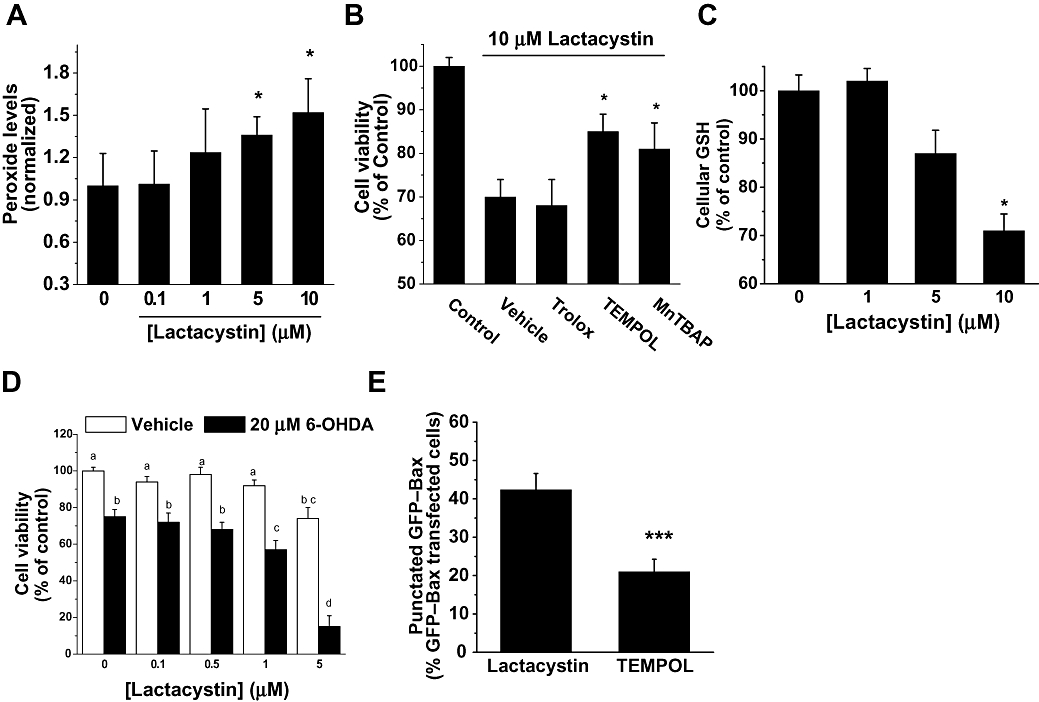
Effect of lactacystin on cellular redox state. (A) The production of reactive oxygen species was monitored 4 h after treatment of SH-SY5Y cells with lactacystin at different concentrations (0–10 µM), by fluorescence of dichlorofluorescein. Data were normalized to vehicle-treated (0 µM lactacystin) cells, and values were reported as the mean ± SD of five independent experiments performed in quadruplicate. *P < 0.05; versus control conditions (0 µM lactacystin). (B) Antioxidant drug effects on lactacystin-induced toxicity. Cell cultures were pretreated with either Trolox (0.75 mM), TEMPOL (0.2 µM) or MnTBAP (10 µM) for 30 min before the addition of 10 µM lactacystin. Cell viability was determined at 24 h by 3-(-4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Data are expressed as the mean ± SD of at least four different cultures. Student's two-tailed, unpaired t-test. *P < 0.05. (C) Levels of glutathione in SH-SY5Y cell cultures were determined 12 h after lactacystin treatment. The values are expressed as a percentage of monochlorobimane fluorescence from untreated conditions. Data are expressed as the mean ± SD of four different cultures, performed in quadruplicate. Student's two-tailed, unpaired t-test. *P < 0.05. (D) Lactacystin potentiated 6-hydroxydopamine (6-OHDA)-induced cell death. Cell cultures were co-treated with lactacystin (0–5 µM) and 20 µM 6-OHDA. Cell viability was determined by MTT assays 24 h later. Data are expressed as the mean ± SD of at least four different cultures. Treatments with the same lowercase letters are not significantly different (one-way analysis of variance; Tukey's test, P > 0.05). (E) SH-SY5Y cells were transfected with green fluorescent protein (GFP)–Bax and were incubated for 24 h to allow sufficient GFP–Bax expression. Cells were pretreated with TEMPOL (0.2 µM) for 30 min, prior to the addition of lactacystin (10 µM) for an additional 12 h. The number of cells with punctate Bax distribution was expressed as a percentage of the total number of cells expressing GFP–Bax protein. Data are expressed as the mean ± SD of at least four different cultures. Student's two-tailed, unpaired t-test. *P < 0.05.
GSH is a ubiquitous, non-enzymic, reducing tri-peptide used by cells to alleviate oxidative stress, and it is depleted in patients with PD (Jenner, 1993). Lactacystin (10 µM, 12 h) modified the intracellular levels of GSH in SH-SY5Y cells (Figure 6C). Further experiments were performed to ascertain whether a correlation exists between GSH concentration and lactacystin-induced cell death. Cell cultures were pretreated with BSO (100 mM) or NAC (100 µM) for 12 h to deplete or to increase GSH concentration, respectively (see Jordan et al., 2004). Consistent with the cytoprotective antioxidant role for GSH in these cells, BSO potentiates lactacystin-induced cell death (54 ± 5% survival in the presence of BSO vs. 74 ± 5% in the absence of BSO, P < 0.05, n= 5 different cultures). Nevertheless, NAC failed to afford protection to SH-SY5Y cell cultures (79 ± 5% survival in the presence of NAC vs. 74 ± 5% in the absence of NAC; P > 0.05, n= 5 different cultures).
Lactacystin sensitizes SH-SY5Y cells to 6-OHDA-induced cell death
Depletion of GSH can be a stressor condition. Thus, in the next set of experiments, we analysed whether lactacystin could sensitize cell cultures to cytotoxic stimuli. Cells were treated for 24 h with vehicle (DMSO), lactacystin (1–5 µM), 6-OHDA or both lactacystin and 6-OHDA to determine whether lactacystin would sensitize SH-SY5Y cells to 6-OHDA-induced cell death. As a single treatment of 6-OHDA (20 µM) alone did not induce marked cellular toxicity, this concentration was used in our experiments (Figure 6D). Adding lactacystin to 6-OHDA increased the loss of cell viability induced by 6-OHDA, reaching a significant effect at the threshold concentration of 1 µM (Figure 6D). The concentration-dependent manner in which lactacystin acted upon cell cultures made clear its additive effects in the context of 6-OHDA-induced toxicity.
Participation of ROS in lactacystin-induced mitochondrial Bax translocation
Previous studies from our group have shown that p38 MAPK and ROS are crucial for Bax translocation after cytotoxic stimuli, including malonate (Gomez-Lazaro et al., 2007) and 6-OHDA (Gomez-Lazaro et al., 2008). By using Western blots and a specific antibody against the phosphorylated form of p38 MAPK, we found that lactacystin did not induce the levels of the phosphorylated form of this MAPK at any of the assayed times (0.5, 1, 3, 6, 12 and 24 h) (data not shown). To elucidate whether ROS are upstream mediators of Bax translocation in the lactacystin model, we pretreated cells with the broadly antioxidant drug TEMPOL for cytoprotection. We did not use MnTBAP, as this compound modified GFP distribution under control conditions (data not shown). As is shown in Figure 6E, GFP–Bax redistribution was inhibited in SH-SY5Y cells pretreated with 0.2 µM TEMPOL for 30 min prior to exposure to 10 µM lactacystin for 24 h. These findings support the involvement of ROS in lactacystin-mediated toxicity in SH-SY5Y cells.
Discussion
The main goal of this study was to obtain a better understanding of the mechanism responsible for the activation of the mitochondrial pathway in lactacystin-induced cell death. Our current results clearly highlighted the key role for Bax protein in triggering the mitochondria-mediated apoptotic pathway, extending earlier studies (Lang-Rollin et al., 2005, 2008). Lactacystin induced cell death in SH-SY5Y cell cultures in a concentration-dependent manner. We used this drug up to a concentration of 10 µM, as it has been shown in numerous neuronal cellular systems to lead to a graded inhibition of proteasome activity, accompanied by graded biological consequences (Fenteany et al., 1994; Figueiredo-Pereira et al., 1994). We do not propose that lactacystin will universally inhibit proteasome activity. Rather, it serves as a good or an acceptable approximation, as shown by the moderate inhibition of proteasome activity (33–42%) in the substantia nigra of PD patients (McNaught and Jenner, 2001). Moreover, a near-complete blockade of proteasome activity interfered with the progression of the mitochondrial apoptotic pathway, possibly through mechanisms involving preservation of inhibitors of apoptosis proteins with resultant cytoprotection of the cells (Suh et al., 2005).
Consistent with previous studies (Forman et al., 2004; Suh et al., 2005), we found that lactacystin treatment induced cytochrome c release from the mitochondria. Our experiments have elucidated the manner in which lactacystin activated this particular event. At the present time, several models have been proposed including activation of the mitochondrial PTP and insertion of proteins in the mitochondrial outer membrane. Our data revealed that cytochrome c release occurred indirectly, via the redistribution of Bax, rather than via mitochondrial PTP formation or direct mitochondrial damage. We observed that the addition of lactacystin to a suspension of isolated mitochondria failed to induce a reduction in light absorbance used an indicator of mitochondrial swelling as a consequence of PTP formation. Interestingly, a lack of PTP formation has been shown in other experimental models for PD such as those using 6-OHDA (Galindo et al., 2003a; Lee et al., 2006; Gomez-Lazaro et al., 2008). Using confocal microscopy and the over-expression of a fusion GFP–Bax protein, we found a change in the localization of Bax after lactacystin treatment, a question that remained unclear in other studies (Lang-Rollin et al., 2004). As Figure 4 illustrates, cell cultures challenged with lactacystin showed a significant translocation of GFP–Bax to mitochondria during the first 24 h of lactacystin treatments. The translocation of Bax from the cytosol to the mitochondria might be responsible for mitochondrial membrane permeability changes in this model of neurotoxicity. Lang-Rollin et al. (2004) found that deletion of this protein prevented the release of cytochrome c and, in their conditions, completely prevented lactacystin-induced sympathetic neuron death. So, we can conclude that Bax is a pivotal element responsible for lactacystin-induced changes in mitochondrial membrane permeability.
The question then arises as to the nature of the mechanism involved in lactacystin-induced Bax activation. While p38 MAPK is responsible for phosphorylation of critical amino acid residues (De et al., 2006) and has been shown to be involved in other models, including 6-OHDA and malonate (Gomez-Lazaro et al., 2007; 2008;), our data exclude a primary role for p38 MAPK, as lactacystin did not increase the levels of phosphorylated forms of this MAPK.
We consider ROS as more relevant second messengers in this lactacystin model. Lactacystin induced an increase in ROS formation after 4 h. Antioxidant agents, such as TEMPOL and MnTBAP, inhibited lactacystin-induced cell death. Indeed, we have found that GSH was depleted in cell cultures challenged with this proteasome inhibitor, indicating a collapse in antioxidant defences. A key role for this endogenous antioxidant was demonstrated when intracellular levels of GSH were depleted with BSO, pre-disposing cell cultures to lactacystin-induced toxicity. Thus, depletion of GSH may explain why lactacystin sensitized neuroblastoma cells to oxidative damage. This explanation is supported by what happened in our cell cultures challenged with lactacystin and exposed to subtoxic concentrations of 6-OHDA. This oxidative event markedly decreased cell viability and, in other models, increased aggregate formation in cells expressing mutated α-synuclein (Lev et al., 2006). This hypothesis is of major importance in PD, where neurotoxicity was also associated with the production of ROS and severe oxidative stress. In light of those results, lactacystin could constitute a potential factor in the development of sporadic PD. It has been suggested that ROS produced during the normal metabolism of dopamine in the substantia nigra pars compacta might initiate the degenerative processes in PD (Offen et al., 1999). In fact, post mortem brain studies of these patients revealed several chemical fingerprints of damaging oxidative events, including carbonyl group formation in cellular proteins (Alam et al., 1997). Finally, it is noteworthy that antioxidant treatment ameliorated the effects of lactacystin on redistribution of Bax. This finding suggests that lactacystin increases ROS production, which subsequently yields Bax translocation that leads to the release of cytochrome c from the mitochondria.
In conclusion, our data revealed that lactacystin caused mitochondrial cell death, mediated by increased ROS production and GSH depletion. These deleterious conditions resulted in translocation of Bax and the release of cytochrome c. These new observations provide further elucidation of the mechanism of cell death induced by the proteasome inhibitor lactacystin, and will help to improve our understanding of the underlying pathology in PD.
Acknowledgments
We thank Dr Francisco J. Fernandez-Gomez for his careful reading and comments on the manuscript. We are grateful to Vanesa Guijarro and Cristina Sepulcre for technical assistance. This work was supported by SAF2008-05143-C03-1 from Ministerio de Ciencia e Innovación; Investigación sobre drogodependiencias, Ministerio de Sanidad y Consumo and 04005-00 and PI2007/55 Consejería de Sanidad from Junta de Comunidades de Castilla-La Mancha (to J.J.) and by ‘CCM Obra Social y Cultural-FISCAM’ and ‘Incorporación de grupos emergentes’ FIS CARLOS III (to M.F.G.). S.P-A. is a fellow from the Spanish Ministerio de Sanidad y Consumo.
Glossary
Abbreviations:
- 6-OHDA
6-hydroxydopamine
- BSO
l-buthionine sulphoxide
- COX IV
cytochrome c oxidase subunit IV
- GFP
green fluorescent protein
- GSH
glutathione
- LDH
lactate dehydrogenase
- mBcl
monochlorobimane, syn-(ClCH2,CH3)-1,5-diazabicyclo-[3,3,0]-octa-3,6-dione-2,8-dione
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAC
N-acetyl-l-cysteine
- PD
Parkinson's disease
- p38 MAPK
p38 mitogen-activated protein kinase
- PTP
permeability transition pore
- ROS
reactive oxygen species
- TEMPOL
4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl
- Trolox
(S)-6-methoxy-2,5,7,8-tetramethylchromane-2-carboxylic acid
Conflicts of interest
None.
References
- Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- Canu N, Barbato C, Ciotti MT, Serafino A, Dus L, Calissano P. Proteasome involvement and accumulation of ubiquitinated proteins in cerebellar granule neurons undergoing apoptosis. J Neurosci. 2000;20:589–599. doi: 10.1523/JNEUROSCI.20-02-00589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De CG, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, et al. Bcl-2 phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- Favit A, Grimaldi M, Alkon DL. Prevention of beta-amyloid neurotoxicity by blockade of the ubiquitin–proteasome proteolytic pathway. J Neurochem. 2000;75:1258–1263. doi: 10.1046/j.1471-4159.2000.0751258.x. [DOI] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Reichard GA, Corey EJ, Schreiber SL. A beta-lactone related to lactacystin induces neurite outgrowth in a neuroblastoma cell line and inhibits cell cycle progression in an osteosarcoma cell line. Proc Natl Acad Sci USA. 1994;91:3358–3362. doi: 10.1073/pnas.91.8.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gomez FJ, Galindo MF, Gómez-Lázaro M, Yuste VJ, Comella JX, Aguirre N, et al. Malonate induces cell death via mitochondrial potential collapse and delayed swelling through an ROS-dependent pathway. Br J Pharmacol. 2005;144:528–537. doi: 10.1038/sj.bjp.0706069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo-Pereira ME, Berg KA, Wilk S. A new inhibitor of the chymotrypsin-like activity of the multicatalytic proteinase complex (20S proteasome) induces accumulation of ubiquitin-protein conjugates in a neuronal cell. J Neurochem. 1994;63:1578–1581. doi: 10.1046/j.1471-4159.1994.63041578.x. [DOI] [PubMed] [Google Scholar]
- Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10:1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- Galindo MF, Jordan J, Gonzalez-Garcia C, Cena V. Chromaffin cell death induced by 6-hydroxydopamine is independent of mitochondrial swelling and caspase activation. J Neurochem. 2003a;84:1066–1073. doi: 10.1046/j.1471-4159.2003.01592.x. [DOI] [PubMed] [Google Scholar]
- Galindo MF, Jordan J, Gonzalez-Garcia C, Ceña V. Reactive oxygen species induce swelling and cytochrome c release but not transmembrane depolarization in isolated rat brain mitochondria. Br J Pharmacol. 2003b;139:797–804. doi: 10.1038/sj.bjp.0705309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Lazaro M, Galindo MF, Melero-Fernandez de Mera RM, Fernandez-Gomez FJ, Concannon CG, Segura MF, et al. Reactive oxygen species and p38 mitogen-activated protein kinase activate Bax to induce mitochondrial cytochrome c release and apoptosis in response to malonate. Mol Pharmacol. 2007;71:736–743. doi: 10.1124/mol.106.030718. [DOI] [PubMed] [Google Scholar]
- Gomez-Lazaro M, Galindo MF, Concannon CG, Segura MF, Fernandez-Gomez FJ, Llecha N, et al. 6-Hydroxydopamine activates the mitochondrial apoptosis pathway through p38 MAPK-mediated, p53-independent activation of Bax and PUMA. J Neurochem. 2008;104:1599–1612. doi: 10.1111/j.1471-4159.2007.05115.x. [DOI] [PubMed] [Google Scholar]
- Jenner P. Altered mitochondrial function, iron metabolism and glutathione levels in Parkinson's disease. Acta Neurol Scand Suppl. 1993;146:6–13. [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Tornero D, Gonzalez-Garcia C, Cena V. Bcl-x L blocks mitochondrial multiple conductance channel activation and inhibits 6-OHDA-induced death in SH-SY5Y cells. J Neurochem. 2004;89:124–133. doi: 10.1046/j.1471-4159.2003.02299.x. [DOI] [PubMed] [Google Scholar]
- Lang-Rollin I, Vekrellis K, Wang Q, Rideout HJ, Stefanis L. Application of proteasomal inhibitors to mouse sympathetic neurons activates the intrinsic apoptotic pathway. J Neurochem. 2004;90:1511–1520. doi: 10.1111/j.1471-4159.2004.02684.x. [DOI] [PubMed] [Google Scholar]
- Lang-Rollin I, Maniati M, Jabado O, Vekrellis K, Papantonis S, Rideout HJ, et al. Apoptosis and the conformational change of Bax induced by proteasomal inhibition of PC12 cells are inhibited by bcl-xL and bcl-2. Apoptosis. 2005;10:809–820. doi: 10.1007/s10495-005-0378-5. [DOI] [PubMed] [Google Scholar]
- Lang-Rollin I, Dermentzaki G, Vekrellis K, Xilouri M, Rideout HJ, Stefanis L. A novel cell death pathway that is partially caspase dependent, but morphologically non-apoptotic, elicited by proteasomal inhibition of rat sympathetic neurons. J Neurochem. 2008;105:653–665. doi: 10.1111/j.1471-4159.2007.05165.x. [DOI] [PubMed] [Google Scholar]
- Lee CS, Park WJ, Ko HH, Han ES. Differential involvement of mitochondrial permeability transition in cytotoxicity of 1-methyl-4-phenylpyridinium and 6-hydroxydopamine. Mol Cell Biochem. 2006;289:193–200. doi: 10.1007/s11010-006-9164-0. [DOI] [PubMed] [Google Scholar]
- Lev N, Melamed E, Offen D. Proteasomal inhibition hypersensitizes differentiated neuroblastoma cells to oxidative damage. Neurosci Lett. 2006;399:27–32. doi: 10.1016/j.neulet.2005.09.086. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001;297:191–194. doi: 10.1016/s0304-3940(00)01701-8. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Olanow CW. Proteasome inhibitor-induced model of Parkinson's disease. Ann Neurol. 2006;60:243–247. doi: 10.1002/ana.20936. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Belizaire R, Jenner P, Olanow CW, Isacson O. Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson's disease. Neurosci Lett. 2002;326:155–158. doi: 10.1016/s0304-3940(02)00296-3. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2:63–67. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- Offen D, Gorodin S, Melamed E, Hanania J, Malik Z. Dopamine–melanin is actively phagocytized by PC12 cells and cerebellar granular cells: possible implications for the etiology of Parkinson's disease. Neurosci Lett. 1999;260:101–104. doi: 10.1016/s0304-3940(98)00950-1. [DOI] [PubMed] [Google Scholar]
- Piccioli P, Porcile C, Stanzione S, Bisaglia M, Bajetto A, Bonavia R, et al. Inhibition of nuclear factor-kappaB activation induces apoptosis in cerebellar granule cells. J Neurosci Res. 2001;66:1064–1073. doi: 10.1002/jnr.1251. [DOI] [PubMed] [Google Scholar]
- Qiu JH, Asai A, Chi S, Saito N, Hamada H, Kirino T. Proteasome inhibitors induce cytochrome c–caspase-3-like protease-mediated apoptosis in cultured cortical neurons. J Neurosci. 2000;20:259–265. doi: 10.1523/JNEUROSCI.20-01-00259.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoul R, Fernandez PA, Quiquerez AL, Martinou I, Maki M, Schroter M, et al. Involvement of the proteasome in the programmed cell death of NGF-deprived sympathetic neurons. EMBO J. 1996;15:3845–3852. [PMC free article] [PubMed] [Google Scholar]
- Shrieve DC, Bump EA, Rice GC. Heterogeneity of cellular glutathione among cells derived from a murine fibrosarcoma or a human renal cell carcinoma detected by flow cytometric analysis. J Biol Chem. 1998;263:14107–14114. [PubMed] [Google Scholar]
- Suh J, Lee YA, Gwag BJ. Induction and attenuation of neuronal apoptosis by proteasome inhibitors in murine cortical cell cultures. J Neurochem. 2005;95:684–694. doi: 10.1111/j.1471-4159.2005.03393.x. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27:336–349. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young PR, Connor S, White AL, Dzido GA. Kinetic analysis of the intracellular conjugation of monochlorobimane by IC-21 murine macrophage glutathione-S-transferase. Biochim Biophys Acta. 1994;1201:461–465. doi: 10.1016/0304-4165(94)90077-9. [DOI] [PubMed] [Google Scholar]