

Abstract
Background and purpose:
ABC multidrug transporters (MDR-ABC proteins) cause multiple drug resistance in cancer and may be involved in the decreased anti-cancer efficiency and modified pharmacological properties of novel specifically targeted agents. It has been documented that ABCB1 and ABCG2 interact with several first-generation, small-molecule, tyrosine kinase inhibitors (TKIs), including the Bcr-Abl fusion kinase inhibitor imatinib, used for the treatment of chronic myeloid leukaemia. Here, we have investigated the specific interaction of these transporters with nilotinib, dasatinib and bosutinib, three clinically used, second-generation inhibitors of the Bcr-Abl tyrosine kinase activity.
Experimental approach:
MDR-ABC transporter function was screened in both membrane- and cell-based (K562 cells) systems. Cytotoxicity measurements in Bcr-Abl-positive model cells were coupled with direct determination of intracellular TKI concentrations by high-pressure liquid chromatography-mass spectrometry and analysis of the pattern of Bcr-Abl phosphorylation. Transporter function in membranes was assessed by ATPase activity.
Key results:
Nilotinib and dasatinib were high-affinity substrates of ABCG2, and this protein mediated an effective resistance in cancer cells against these compounds. Nilotinib and dasatinib also interacted with ABCB1, but this transporter provided resistance only against dasatinib. Neither ABCB1 nor ABCG2 induced resistance to bosutinib. At relatively higher concentrations, however, each TKI inhibited both transporters.
Conclusions and implications:
A combination of in vitro assays may provide valuable preclinical information for the applicability of novel targeted anti-cancer TKIs, even in multidrug-resistant cancer. The pattern of MDR-ABC transporter–TKI interactions may also help to understand the general pharmacokinetics and toxicities of new TKIs.
Keywords: CML, Bcr-Abl, TKI, ABCB1, ABCG2, MDR, ADME-Tox
Introduction
Chronic myeloid leukaemia (CML) is a haematopoietic stem cell disorder, whose initiation, maintenance and progression is driven by the Bcr-Abl oncoprotein, exhibiting unregulated tyrosine kinase activity (Ren, 2005). Identification of imatinib as a potent inhibitor of the Abl kinase and the subsequent findings that this compound displays growth inhibitory and pro-apoptotic effects in Bcr-Abl+ cells has revolutionized trends in cancer treatment (Ren, 2005). Despite the remarkable success of imatinib therapy, it was soon realized that imatinib resistance was a major obstacle in CML treatment (Ren, 2005; Quintas-Cardama et al., 2007; Weisberg et al., 2007b). Analysis of the resistance mechanisms has led to the design of a wide array of second-generation Bcr-Abl inhibitors with improved inhibitory potential, broader target spectrum or diverse mechanisms of action (Quintas-Cardama et al., 2007; Weisberg et al., 2007b). Second-generation inhibitors include nilotinib, a close structural analogue of imatinib and the dual Abl-Src kinase inhibitors dasatinib and bosutinib (Quintas-Cardama et al., 2007; Weisberg et al., 2007b). While nilotinib and dasatinib have already been granted FDA approval for treatment of CML in patients resistant to or intolerant of imatinib (Quintas-Cardama et al., 2007; Weisberg et al., 2007b; Hazarika et al., 2008), the efficacy of bosutinib is being evaluated in clinical trials, with promising results (Quintas-Cardama et al., 2007; Weisberg et al., 2007b).
Multidrug resistance ATP-binding cassette (MDR-ABC) transporters are membrane glycoproteins that play a key role in the energy-dependent cellular efflux of toxic agents. MDR-ABC transporters are capable of recognizing and extruding a broad range of compounds, unrelated to chemical structure or cellular target, including tyrosine kinase inhibitors (TKIs). By means of this promiscuous character and overlapping substrate specificities, human MDR-ABC transporters are believed to form a chemo-immunity defence network, providing broad protection against xenobiotics in various tissues (Sarkadi et al., 2006; Szakacs et al., 2008). These proteins might also have a significant impact on the pharmacokinetics and toxicities of clinically used drugs (Szakacs et al., 2008), and MDR-ABC transporters are known to account for the multidrug-resistant phenotype of various cancer cells (Gottesman et al., 2002).
ABCB1 (also known as MDR1/Pgp) and ABCG2 [also known as breast cancer resistance protein (BCRP)/mitoxantrone resistance protein (MXR)/placenta specific ABC transporter (ABCP)] are two important examples of human MDR-ABC proteins. ABCB1, and especially ABCG2, are abundantly expressed in the so-called side-population of haematopoietic progenitor cells (Smeets et al., 1997; Scharenberg et al., 2002), representing pluripotent stem cells (Zhou et al., 2001,Kim et al., 2002). These transporters were also reported to be overexpressed in various normal and cancer stem cells (Dean et al., 2005) and thus they may represent key components in the development of resistance to anti-cancer agents.
ABCB1 displays high-affinity interactions with imatinib (Hegedus et al., 2002; Hamada et al., 2003) and can confer resistance to imatinib in vitro (Kotaki et al., 2003; Mahon et al., 2003) by extruding imatinib from the cells (Thomas et al., 2004). Following the initial controversial findings about the interaction of ABCG2 with imatinib (Burger et al., 2004; Houghton et al., 2004; Ozvegy-Laczka et al., 2004), we now know that ABCG2 can mediate in vitro imatinib resistance as well (Nakanishi et al., 2006; Brendel et al., 2007). Importantly, both transporters show elevated expression in CML stem cells (Jordanides et al., 2006; Jiang et al., 2007). However, there are few detailed in vitro studies of the interaction between these transporters and second-generation Bcr-Abl inhibitors (see Discussion).
In the present study, we sought to characterize the interactions of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2. We applied various membrane- and cell-based model systems to gain a better insight into the nature of the interaction. For direct and quantitative evaluation of drug transport by ABCB1/ABCG2, we also developed a novel high-pressure liquid chromatography-mass spectrometry (HPLC-MS) assay. We demonstrated that these second-generation Bcr-Abl inhibitors exhibited distinctly different interactions with ABCB1 and ABCG2, coupled with a specific protection of the tumour cells against these agents. These findings, exploring drug–transporter interactions over a wide concentration range of the TKIs, provide important information regarding both the anti-cancer effect and the pharmacokinetics and toxicities of these novel agents.
Methods
Cell lines, cell growth and propagation
The K562/ABCB1 cell line was generated by selection of parental K562 cells with doxorubicin as described previously (Hollo et al., 1996). The K562/ABCG2 cell line stably expressing wild type ABCG2 (ABCG2 R482) was generated by using a retroviral transduction system as reported earlier (Elkind et al., 2005). In each cell line, copy number of the bcr-abl chimeric gene was determined by fluorescent in situ hybridization (FISH). Expression of ABCB1 and ABCG2 was checked by immunostaining and subsequent flow cytometry analysis using MRK-16 and 5D3 antibodies respectively (Figure S1A,B). In order to test ABCB1 and ABCG2 function, calcein-AM and Hoechst 33342 uptake experiments were performed in both parental and transporter-expressing K562 cell lines, as described earlier (Hollo et al., 1994; Ozvegy et al., 2002) (Figure S1C). Parental K562 cells showed no endogenous ABCB1 and ABCG2 expression and function. Cells were maintained in RPMI medium without nucleosides supplemented with 10% fetal bovine serum, 50 units·mL−1 penicillin, 50 units·mL−1 streptomycin and 5 mM glutamine, at 37°C in 5% CO2.
Cellular toxicity assays
For the cellular toxicity assay 1 × 105 per millilitre parental K562, K562/ABCB1 and K562/ABCG2 cells were seeded in 24-well plates. Following 15 min incubation with 1 µM PSC-833 (selective inhibitor of ABCB1) or 5 µM fumitremorgin C (FTC) (selective inhibitor of ABCG2), cells were treated with increasing doses of TKIs. After 48 h, the number of living cells in 500 µL samples was determined by TOPRO-3 staining (50 nM) and subsequent flow cytometry analysis. The assay was carried out in duplicate.
TKI-mediated Bcr-Abl dephosphorylation
A total of 2 × 106 parental K562, K562/ABCB1 and K562/ABCG2 cells in a final volume of 5 mL were treated with 25 nM nilotinib, 2.5 nM dasatinib or 25 nM bosutinib in the presence or in the absence of 1 µM PSC-833 or 5 µM FTC. After 24 h, total soluble protein lysates were prepared from the samples as described previously (Brozik et al., 2006). Forty micrograms proteins were resolved by SDS-PAGE and transferred to PVDF membrane using the Mini-Protean II System (Bio-Rad, Budapest, Hungary). In order to study phospho-Bcr-Abl patterns, Western blots were probed with the polyclonal phospho-Bcr (Tyr177) antibody (Cell Signaling Technology, Danvers, MA, USA) and the monoclonal c-Abl (24-11) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Rabbit and mouse IgG HRP conjugates were used as secondary antibodies (Jackson Immunoresearch, West Grove, PA, USA). To visualize immunostaining we used the ECL detection system (Amersham Biosciences/GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Quantitative determination of intracellular amount of TKIs by HPLC-MS
A total of 2 × 106 parental K562, K562/ABCB1 and K562/ABCG2 cells in a final volume of 5 mL were seeded in Petri dishes. Following 15 min pre-incubation with 1 µM PSC-833 (selective inhibitor of ABCB1) or 5 µM FTC (selective inhibitor of ABCG2), cells were incubated with 25 nM nilotinib, 5 nM dasatinib or 10 nM bosutinib (all final concentrations in 5 mL volume) at 37°C for 60 min. After 60 min, transport reaction was stopped by addition of 5 mL ice-cold phosphate-buffered saline. Harvested cells were kept on ice throughout the sample preparation procedure. Following three washing steps using 5 mL phosphate-buffered saline, cells were precipitated with 500 µL acetonitrile, containing imatinib at 30 nM as an internal standard. After centrifugation at 8000×g for 5 min, clear supernatant was transferred into Eppendorf tubes and acetontirile was evaporated in a heated vacuum concentrator centrifuge (UNIVAPO 100 H, UniEquip). Nilotinib, dasatinib, bosutinib and imatinib (internal standard) were separated using a RP-18 column on a XLC binary HPLC pump system (Jasco International, Tokyo, Japan). Twenty microlitres of reconstituted sample was injected; flow rate of HPLC eluent was set to 200 µL·min−1. Mobile phases used were: (A) 0.1% acetic acid in 100 mM ammonium acetate buffer and (B) 0.1% acetic acid in acetonitrile. The total HPLC run time was 9 min, using the following gradient: 0–1 min: 80% A, 1–6 min: 5% A, 6–7 min: 5% A, 7–9 min: 80% A. TKIs were detected using a TSQ Quantum Discovery (Thermo Finnigan, San Jose, CA, USA) triple quadrupole mass spectrometer operated in positive ion electrospray mode. Protonated molecular ions of analytes were detected in multiple reaction monitoring mode using m/z 488→232 and m/z 488→401 fragmentation channel for dasatinib, m/z 530→289 and m/z 530→261 for nilotinib, m/z 530→141 and m/z 530→113 for bosutinib and m/z 494→217 and m/z 494→394 for imatinib. TKI content of the samples were calculated from the respective TKI calibration curves and were normalized to the amount of imatinib. Experiments were carried out in duplicate.
Membrane ATPase measurements
Spodoptera frugiperda (Sf9) ovarian cell membranes enriched in ABCB1 were prepared as described earlier (Hegedus et al., 2002). Cholesterol content of the membrane significantly modulates ABCG2 function (Telbisz et al., 2007), therefore in ABCG2 ATPase measurements we used cholesterol-loaded Sf9 membranes expressing human wild-type ABCG2 prepared according to Telbisz et al. (2007). Vanadate-sensitive ATPase activity was measured by determining the liberation of inorganic phosphate from ATP with a colorimetric reaction (Hegedus et al., 2002; Ozvegy-Laczka et al., 2004).
Fluorescent dye extrusion studies
The effect of TKIs on the fluorescent dye transport capacities of ABCB1 and ABCG2 was followed as published previously (Ozvegy-Laczka et al., 2004). Briefly, cellular fluorescence in K562/ABCB1 and K562/ABCG2 cells was determined in the absence of inhibitors (F0), in the presence of the TKI (Fx) and in the presence of the ABCB1 inhibitor verapamil (50 µM) or ABCG2 inhibitor FTC (2 µM) (F100). Data representing the inhibitory effect of the respective TKI were calculated as (Fx−F0)/(F100−F0) × 100.
Data analysis
Results are shown as means with SD. Significance of differences between means was calculated by Student's t-test, at 95% confidence interval.
Materials
The Bcr-Abl inhibitors imatinib, nilotinib, dasatinib and bosutinib were synthesized by VICHEM (Budapest, Hungary). Verapamil was purchased from Sigma (Budapest, Hungary). Calcein-AM, Hoechst 33342 and TOPRO-3 were obtained from Molecular Probes (Eugene, OR, USA). FTC was kindly provided by Lee M Greenberger (Wyeth-Ayerst Research). 5D3 was purified and labelled with Alexa647 as described previously (Ozvegy-Laczka et al., 2008). MRK-16 was obtained from Alexis Biochemicals (San Diego, CA, USA). PSC-833 was provided by Novartis Pharmaceuticals (East Hanover, NJ, USA). Acetonitrile and water were HPLC grade and were purchased from Sigma (Budapest, Hungary); acetic acid was obtained from Reanal (Budapest, Hungary). Purospher STAR RP-18 endcapped columns (3 µm, 2 × 55 mm) was obtained from Merck (Budapest, Hungary).
Results
Investigation of the anti-cancer effect of TKIs in a multidrug-resistant background
Multidrug resistance-ABC transporter function might modify the anti-cancer effect of TKIs. In order to analyse the in vitro anti-cancer potential of these three Bcr-Abl inhibitors in a multidrug-resistant background, we used the CML-derived human Bcr-Abl+ K562 cell line engineered to overexpress ABCB1 or ABCG2. Selectivity and stability of the expression and function of the relevant MDR-ABC transporter was confirmed as described in Methods (also see Figure S1). Parental K562 cells showed no endogenous ABCB1 and ABCG2 expression and function.
Cellular TKI toxicity assays in K562, K562/ABCB1 and K562/ABCG2 cells
In order to examine the direct effect of ABCB1 and ABCG2 function on the cytotoxic effects of nilotinib, dasatinib and bosutinib, parental K562, K562/ABCB1 and K562/ABCG2 cells were treated with increasing concentrations of the drugs. After 48 h, cells were harvested and the relative number of living cells in the samples was determined by TOPRO-3 staining and subsequent flow cytometry analysis. As shown in Figure 1, in parental K562 cells all three TKIs were cytotoxic at very low concentrations (also see Table 1).
Figure 1.
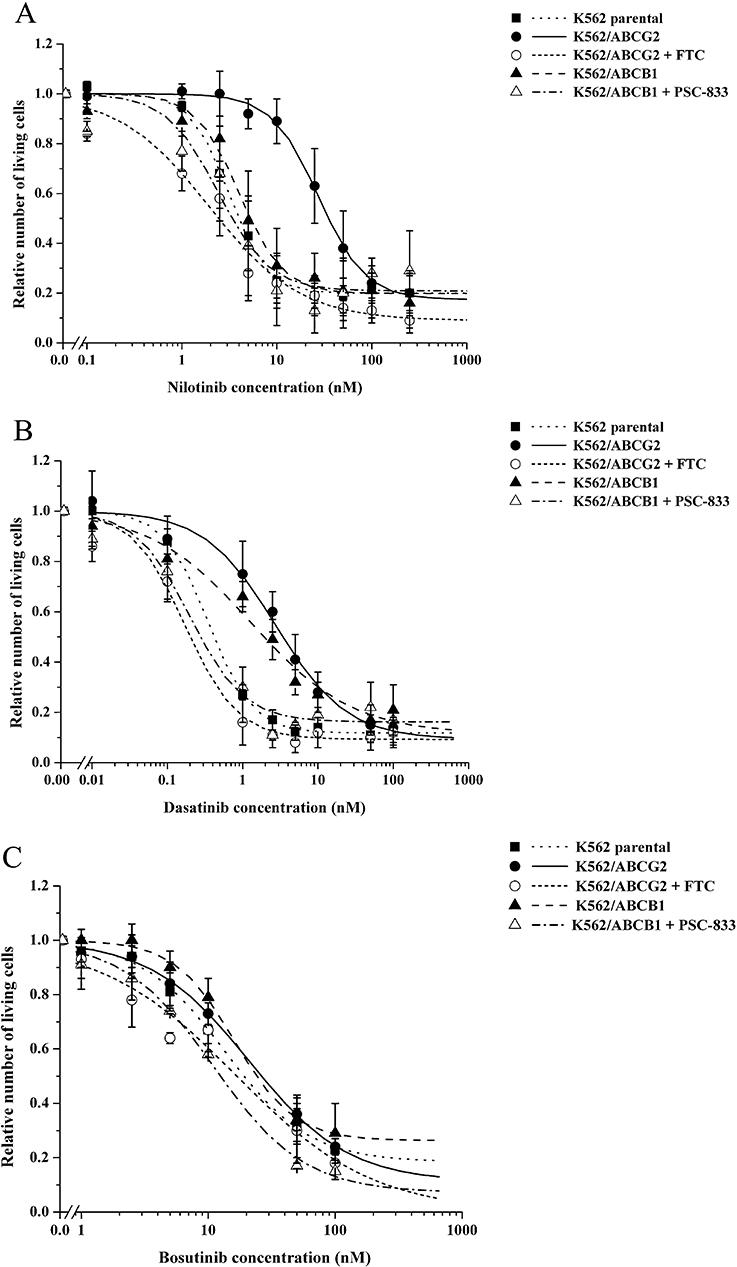
Cytotoxic effect of nilotinib, dasatinib and bosutinib in parental K562, K562/ABCB1 and K562/ABCG2 cells. Parental K562, K562/ABCB1 and K562/ABCG2 cells were incubated with increasing concentration of nilotinib (A), dasatinib (B) or bosutinib (C) in the presence or in the absence of the specific multidrug resistance ATP-binding cassette inhibitors 1 µM PSC-833 or 5 µM fumitremorgin C (FTC) for 48 h. Cells were then harvested, stained with TOPRO-3 and counted by flow cytometry. The number of living cells relative to those measured in vehicle-treated control samples was plotted against the log of tyrosine kinase inhibitor (TKI) concentrations, and exponential fit was applied. Data represent the mean of at least three independent experiments each in duplicate; bars represent SD.
Table 1.
IC50 values of the tyrosine kinase inhibitors (TKIs) in the different K562 cell lines
| Cell line |
IC50 (nM) of TKI |
||
|---|---|---|---|
| Nilotinib | Dasatinib | Bosutinib | |
| K562 parental | 3.1 ± 0.1 | 0.35 ± 0.02 | 13.5 ± 1.8 |
| K562/ABCB1 | 4.0 ± 0.4 (1.3) | 1.3 ± 0.4 (3.7) | 16.0 ± 1.7 n.s. |
| K562/ABCG2 | 27.4 ± 1.5 (8.8) | 2.7 ± 0.5 (7.8) | 21.0 ± 4.4 (1.6) |
All IC50 values are significantly different from the corresponding IC50 values measured in parental K562 cells unless marked otherwise (n.s.). Statistical analysis was performed using Student's t-test. Values in brackets represent fold-resistance compared with parental K562 cells.
Figure 1A shows the cytotoxic effect of nilotinib in parental K562, K562/ABCB1 and K562/ABCG2 cells. The presence of functional ABCG2 effectively protected K562/ABCG2 cells from the cytotoxic effects of nilotinib, resulting in 8.8-fold resistance (Table 1). This protection was ABCG2-specific, as simultaneous addition of FTC (a selective inhibitor of ABCG2) completely abolished the cytoprotective effect (FTC alone had no effect on the proliferation of parental K562 or K562/ABCG2 cells – data not shown). Expression of ABCB1 had only minor effects on nilotinib cytotoxicity in K562/ABCB1 cells (1.3-fold resistance), and when co-treated with nilotinib and PSC-833 (selective inhibitor of ABCB1), K562/ABCB1 cells showed survival similar to that of parental K562 cells or K562/ABCB1 cells treated with nilotinib alone (PSC-833 alone had no effect on the proliferation of parental K562 or K562/ABCB1 cells – data not shown).
As demonstrated in Figure 1B, K562/ABCB1 and K562/ABCG2 cells also exhibited 3.7-fold and 7.8-fold dasatinib resistance respectively (Table 1). The protective effect of the two different MDR-ABC transporters was specific, as it could be fully reversed by the addition of PSC-833 and FTC respectively.
Figure 1C documents the effect of bosutinib in the different K562 cell lines. We found that, over this concentration range of bosutinib, ABCB1 had no significant effect on the cellular toxicity of this TKI and even ABCG2 exerted only a minor protective effect, resulting only in a small (albeit significant) 1.6-fold resistance (Table 1).
Taken together, these results indicate that ABCB1 and ABCG2 function have different effects on the cytotoxic potential of the three investigated compounds. Whereas K562 cells were efficiently protected from nilotinib cytotoxicity, mainly by ABCG2, both ABCB1 and ABCG2 conferred dasatinib resistance. Importantly, ABCB1 and ABCG2 only slightly modified the cytotoxic effect of bosutinib.
Determination of the phospho-Bcr-Abl status in K562, K562/ABCB1 and K562/ABCG2 cells treated with TKIs
CML cell proliferation depends on the uncontrolled tyrosine kinase activity of Bcr-Abl (Ren, 2005), and K562 cells have constitutively phosphorylated Bcr-Abl (Brozik et al., 2006). In order to assess the molecular consequences of treatment with Bcr-Abl inhibitors, we determined the phosphorylation status of Bcr-Abl in the K562 cell lines exposed to cytotoxic concentrations of TKIs, namely 25 nM nilotinib, 2.5 nM dasatinib or 25 nM bosutinib for 24 h in the presence or in the absence of 1 µM PSC-833 or 5 µM FTC. For these experiments, TKI concentrations were chosen based on their IC50 values measured in the different K562 cell lines (see Table 1). Bcr-Abl phosphorylation was detected by using the polyclonal phospho-Bcr (Tyr177) antibody, while the total amount of Bcr-Abl was estimated by the monoclonal c-Abl (24-11) antibody. Data obtained by densitometry analysis of the phospho-Bcr-Abl lanes were normalized to those of the corresponding Bcr-Abl lanes (Figure S2.).
In agreement with published data, K562, K562/ABCB1 and K562/ABCG2 cells revealed a constitutively phosphorylated Bcr-Abl (Figure 2). Figure 2A shows that, in parental K562 cells, all three TKIs caused a significant decrease in phospho-Bcr-Abl levels, and this decrease was more pronounced in the case of nilotinib and dasatinib. Simultaneous TKI and PSC-833 or FTC treatment resulted in similar phospho-Bcr-Abl patterns as in TKI-treated parental K562 cells.
Figure 2.
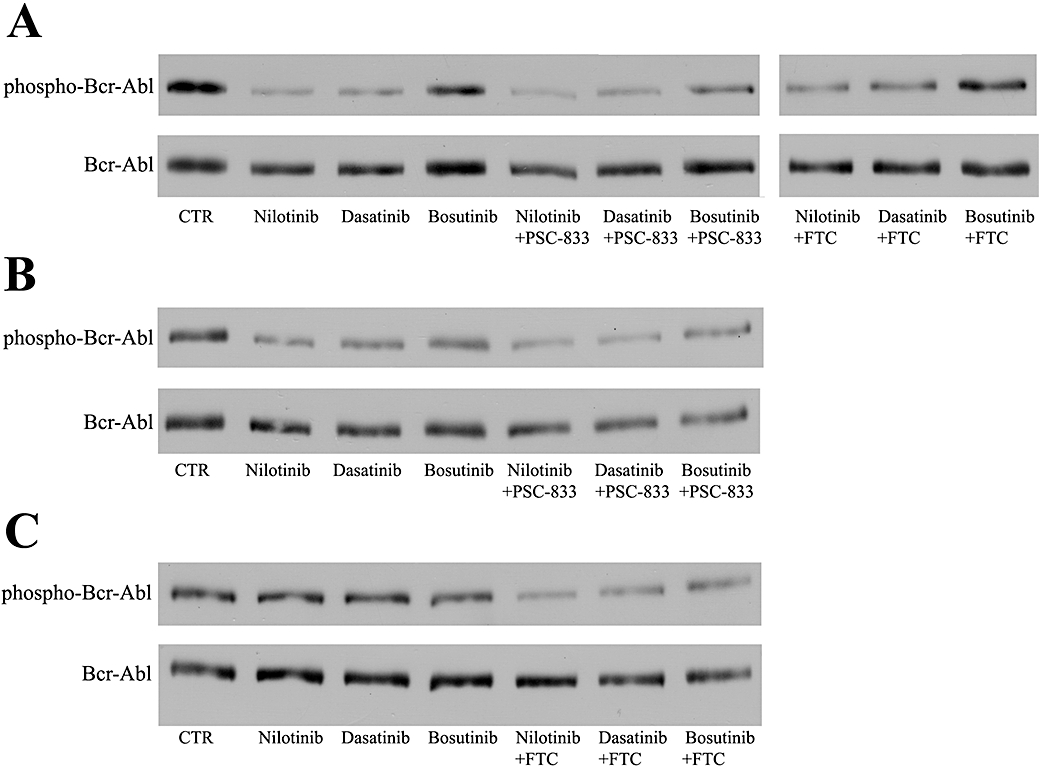
Bcr-Abl phosphorylation pattern in tyrosine kinase inhibitor-treated parental K562, K562/ABCB1 and K562/ABCG2 cells. Parental K562 (A), K562/ABCB1 (B) and K562/ABCG2 (C) cells were exposed to 25 nM nilotinib, 2.5 nM dasatinib or 25 nM bosutinib in the presence or in the absence of the specific multidrug resistance ATP-binding cassette inhibitors PSC-833 (1 µM) or fumitremorgin C (FTC) (5 µM) for 24 h. Bcr-Abl phosphorylation status in 40 µg protein of cell lysates was probed by Western blot analysis using the p-Bcr (Tyr177) antibody. Total amount of the Bcr-Abl protein (210 kDa) was estimated by c-Abl staining. The figure shows the result of one of two independent experiments.
The phospho-Bcr-Abl levels in the K562/ABCB1 cells (Figure 2B) were still decreased by nilotinib and dasatinib, compared with the untreated controls. However, for both these TKIs cases, the simultaneous addition of the specific ABCB1 inhibitor PSC-833 could further lower phospho-Bcr-Abl protein levels, indicating a specific interaction of ABCB1 with these TKIs. Exposure of K562/ABCB1 cells to bosutinib resulted in a phospho-Bcr-Abl pattern, similar to that after simultaneous treatment with bosutinib and PSC-833.
As shown in Figure 2C, K562/ABCG2 cells incubated with either nilotinib or dasatinib displayed very strong phospho-Bcr-Abl signals that were comparable to that measured in untreated K562/ABCG2 cells. In both cases, this high level of phospho-Bcr Abl could be diminished by the addition of the specific ABCG2 inhibitor FTC. These data suggest that ABCG2 strongly protects these cells from the inhibitory effect of nilotinib and dasatinib on Bcr-Abl phosphorylation. In contrast, in the K562/ABCG2 cells, addition of bosutinib still decreased the phospho-Bcr-Abl levels, and the co-administration of FTC caused only a slight additional drop in phospho-Bcr-Abl signal.
Collectively, these data indicate that specific extrusion of the TKIs by ABCB1 and ABCG2 can be clearly observed by the modulation of downstream molecular effects at the target kinase sites. Both ABCB1 and ABCG2 transporter function can effectively prevent the respective TKIs from reaching and reducing the phosphorylation of their intracellular target in cases when cytoprotective effect of the respective transporter was also observed.
Direct and quantitative measurement of TKI accumulation in K562, K562/ABCB1 and K562/ABCG2 cells
The cellular toxicity assays revealed that ABCG2 efficiently protected K562 cells from the cytotoxic effect of nilotinib, whereas both ABCB1 and ABCG2 rescued K562 cells from dasatinib-induced cell death. In order to further confirm that these protective phenomena are mediated by specific transporter-dependent elimination of these TKIs from the cells, we applied a novel HPLC-MS assay for direct and quantitative evaluation of drug transport by ABCB1/ABCG2.
This was a challenging task, as the relevant cellular drug concentrations had to be measured in the nanomolar range corresponding to the IC50 values obtained in the cytotocicity experiments (Table 1). Figure 3 shows the intracellular amounts of nilotinib, dasatinib and bosutinib, measured in K562/ABCB1 and K562/ABCG2 cells, relative to the respective TKI levels detected in the parental K562 cells. Although at these very low TKI concentrations the standard error of the multiple measurements was relatively large, the results were in good accordance with the cellular toxicity data. ABCG2 function resulted in a significant decrease in intracellular nilotinib levels, while ABCB1 did not significantly modify the intracellular accumulation of this compound. Dasatinib was effectively extruded by both ABCB1 and ABCG2, as we detected significantly lower dasatinib levels in K562/ABCB1 and K562/ABCG2 cells, compared with parental K562 cells. There was no significant effect of either ABCB1 or ABCG2 expression on cellular bosutinib concentrations. Nilotinib extrusion by ABCG2 and dasatinib extrusion by ABCB1 and ABCG2 were all reversed by the addition of the ABCB1 inhibitor PSC-833 and the ABCG2 inhibitor FTC, while these inhibitors had no effect on cellular bosutinib accumulation (data not shown).
Figure 3.
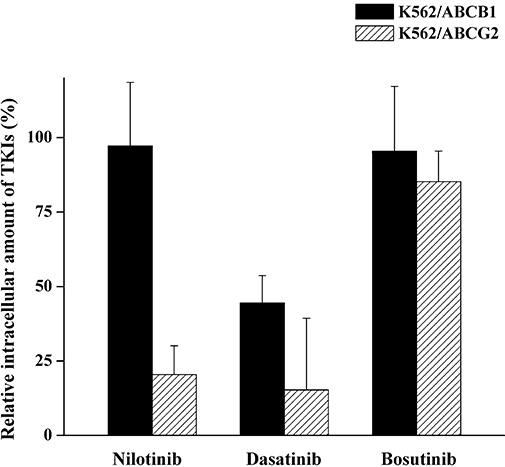
Effect of ABCB1 and ABCG2 on the intracellular accumulation of nilotinib, dasatinib and bosutinib. Parental K562, K562/ABCB1 and K562/ABCG2 cells were incubated with 25 nM nilotinib, 5 nM dasatinib or 10 nM bosutinib at 37°C for 60 min. Following precipitation of cells with acetonitrile containing 30 nM imatinib as an internal standard, the amount of intracellularly accumulated tyrosine kinase inhibitors (TKIs) was determined by high-pressure liquid chromatography-mass spectrometry. The figure shows the amount of nilotinib, dasatinib and bosutinib measured in K562/ABCB1 and K562/ABCG2 cells relative to those detected in parental K562 cells (100%). Columns represent the mean of at least two independent experiments, each in duplicate; bars represent SD.
Exploration of the MDR-ABC–TKI interactions in a wide concentration range
Membrane ATPase measurements
Because transport by MDR-ABC proteins is driven by ATP, the ATPase activity of cell membranes enriched in functional MDR-ABC transporters provides a model for the characterization of transporter–drug interactions over a very wide drug concentration range. We therefore tested the modulatory effect of nilotinib, dasatinib and bosutinib on the vanadate-sensitive ATPase activity in Sf9 insect cell membranes overexpressing ABCB1 or ABCG2. As the cholesterol content of the membrane significantly modulates ABCG2-ATPase activity (Telbisz et al., 2007), ABCG2 ATPase measurements were performed in cholesterol-loaded Sf9 membranes (see Methods).
As illustrated in Figure 4A, nilotinib, dasatinib and bosutinib had markedly different effects on the ATPase activity of ABCB1. Nilotinib increased the ATPase activity of ABCB1 in submicromolar concentrations, reaching a maximum stimulatory effect at about 500 nM. At nilotinib concentrations above 1 µM, this stimulatory effect was lost and ABCB1–ATPase activity returned to its basal level. Dasatinib or bosutinib did not show measurable activation of ABCB1-ATPase up to 1 µM concentrations, while higher concentrations of both drugs produced a modest increase in ABCB1–ATPase activity (Figure 4A).
Figure 4.
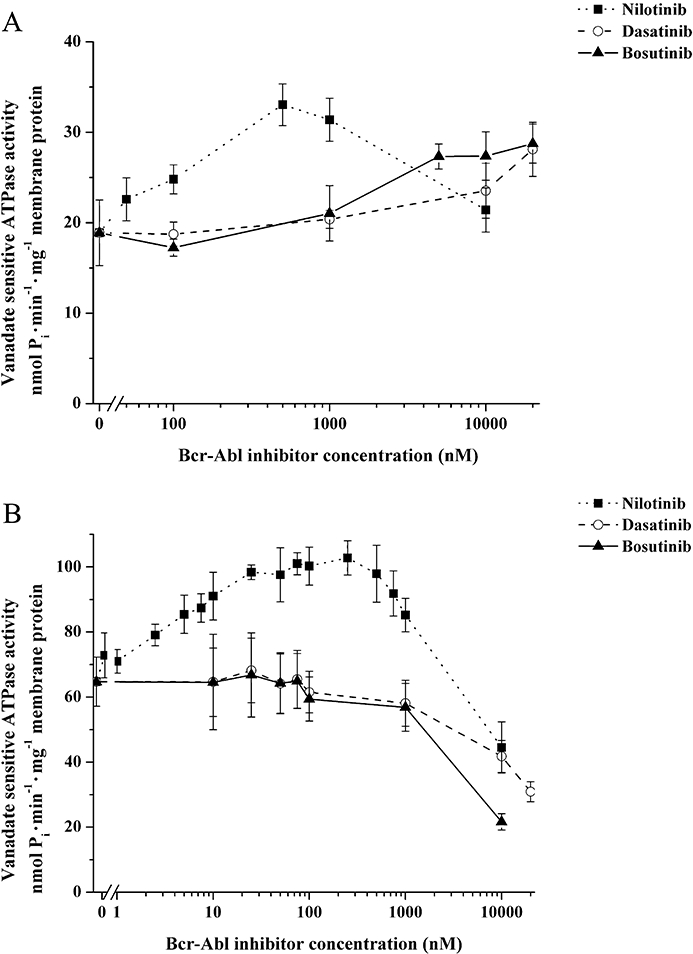
Modulatory effect of nilotinib, dasatinib and bosutinib on the ATPase activity of ABCB1 and ABCG2. The modulatory effects of tyrosine kinase inhibitors on the vanadate-sensitive ATPase activity (nmol Pi·min−1·mg−1 membrane protein) of human ABCB1 (A) and human wild-type ABCG2 (B) were tested using 10 µg insect Spodoptera frugiperda (Sf9) ovarian cell membranes expressing the respective multidrug resistance ATP-binding cassette transporter. For ABCG2 ATPase activity measurements, cholesterol-loaded Sf9 membrane vesicles were used. Expressed in Sf9 membranes, both transporters exhibit a relatively high basal ATPase activity. Full ABCB1 and ABCG2 transporter function in the membrane vesicles was tested by maximal ATPase stimulation with verapamil (50 µM) and prazosin (50 µM) or quercetin (1 µM) respectively (data not shown). Data represent the mean of at least three independent experiments each in duplicate; bars represent SD.
Figure 4B shows the effects of nilotinib, dasatinib and bosutinib on the ATPase activity of membranes containing ABCG2. Nilotinib, already at low nanomolar concentrations, greatly increased the ABCG2 ATPase activity, reaching a maximum stimulatory effect at about 25 nM. Above 1 µM, nilotinib was found to inhibit the ATPase activity of ABCG2. As shown in Figure 4B, in submicromolar concentrations neither dasatinib nor bosutinib could significantly activate the ABCG2-ATPase, whereas at high micromolar concentrations, both dasatinib and bosutinib inhibited the ABCG2-ATPase.
Inhibition of calcein-AM and Hoechst 33342 dye efflux from K562/ABCB1 and K562/ABCG2 cells by TKIs
As MDR-ABC transporters have a wide range of substrates, they can contribute to drug–drug interactions, when several drugs are given together. In order to model this condition we assessed the effects of the TKIs on the transport of two established MDR-ABC substrates, the dyes calcein-AM and Hoechst 33342, in K562/ABCB1 and K562/ABCG2 cells respectively.
Figure 5A summarizes the concentration-dependent inhibition of calcein-AM efflux by nilotinib, dasatinib and bosutinib in K562/ABCB1 cells. Nilotinib inhibited ABCB1-mediated calcein-AM efflux in submicromolar concentrations (half maximum inhibition was achieved by about 100 nM nilotinib), whereas dasatinib and bosutinib exhibited an inhibitory effect on the active transport of the dye only when applied above 1–2 µM. Figure 5B shows the effects of nilotinib, dasatinib and bosutinib on the efflux of Hoechst 33342 dye in K562/ABCG2 cells. It should be mentioned that Hoechst 33342 is also a substrate for ABCB1 (Sarkadi et al., 2006) but, in our K562/ABCG2 cells, the ABCB1 transporter is not expressed at measurable levels (data not shown). Also, ABCG2 can be selectively examined using the selective ABCG2 inhibitor FTC (see Methods). In K562/ABCG2 cells, nilotinib effectively inhibited the outward transport of the Hoechst 33342 dye in low nanomolar concentrations (Figure 5B). Half-maximum inhibition of ABCG2-mediated dye transport could be obtained at about 50 nM nilotinib. In contrast, dasatinib and bosutinib were ineffective up to 1 µM concentration, and inhibition of Hoechst 33342 efflux could be detected only when the compounds were applied at 2–20 µM concentrations (Figure 5B). None of these TKIs, up to a concentration of 20 µM, displayed any effect on the accumulation of the reporter dyes in parental K562 cells, suggesting that the increase of cellular fluorescence in the MDR-ABC-expressing cell lines is a result of drug–transporter interaction.
Figure 5.
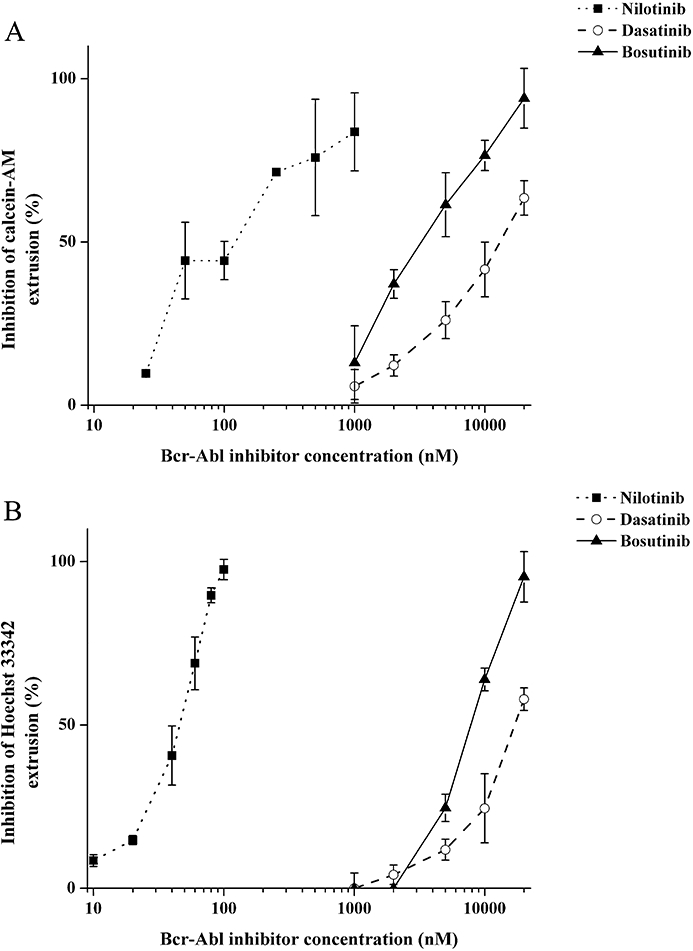
Inhibition of transporter-dependent fluorescent reporter dye extrusion from K562/ABCB1 and K562/ABCG2 cells by nilotinib, dasatinib and bosutinib. Concentration-dependent inhibitory potential of tyrosine kinase inhibitors (TKIs) on the ABCB1-dependent extrusion of calcein-AM (A) and ABCG2-dependent extrusion of Hoechst 33342 (B) was tested using K562/ABCB1 and K562/ABCG2 intact cells respectively. Cellular fluorescence was measured by using a spectrofluorometer, and TKI-mediated relative inhibition of dye extrusion was calculated as described in Methods. Data represent the mean of at least three independent experiments; bars represent SD.
Discussion and conclusions
Chronic myeloid leukaemia is a haematopoietic stem cell disorder characterized by the expression and abnormal tyrosine kinase activity of the Bcr-Abl protein. Imatinib, the prototype of Bcr-Abl inhibitors is successfully applied for the treatment of the majority of CML patients; however, resistance is a major impediment to successful treatment of CML. The MDR-ABC transporters ABCB1 and ABCG2 show elevated expression in CML stem cells (Jordanides et al., 2006; Jiang et al., 2007), and transporter-mediated TKI efflux has been implicated as a possible mechanism for resistance to imatinib (Thomas et al., 2004; Jiang et al., 2007). There are only a few studies investigating the interaction of second-generation Bcr-Abl inhibitors with MDR-ABC transporters. Nilotinib has been implied to interact with ABCB1; however, this interaction has not been without conjectures and controversies (White et al., 2006; 2007; Jorgensen et al., 2007; Weisberg et al., 2007a; Deguchi et al., 2008). Very recently, Mahon et al. have demonstrated that ABCB1 can confer nilotinib resistance (Mahon et al., 2008). Only one report has been published as yet describing the interaction of nilotinib with ABCG2, suggesting that nilotinib is an ABCG2 substrate (Brendel et al., 2007). ABCB1 and ABCG2 have both been shown to confer decreased intracellular uptake and retention and increased IC50 of dasatinib (Hiwase et al., 2008) suggesting that this TKI is also a substrate for both transporters. ABCB1-mediated dasatinib transport has also been demonstrated in a cell monolayer culture (Giannoudis et al., 2008). Still, no detailed and comparative in vitro analysis is currently available on the interaction of nilotinib or dasatinib with ABCB1 and ABCG2. Moreover, to our current knowledge, no data have been published yet regarding MDR-ABC and bosutinib interactions. In this report, we describe the detailed and comparative in vitro characterization of the interaction of these second-generation Bcr-Abl inhibitors with ABCB1 and ABCG2.
Interactions between MDR-ABC proteins and possible substrates might influence the properties of the TKIs at two different types of locations. As ABCB1 and ABCG2 are physiologically expressed in various stem cells (Dean et al., 2005) and show elevated expression in CML stem cells (Jordanides et al., 2006; Jiang et al., 2007), these proteins might modify the anti-cancer properties of Bcr-Abl inhibitors at their target, namely CML stem cells, potentially mediating the development of a TKI-resistant phenotype. On the other hand, as ABCB1 and ABCG2 are abundantly expressed at important pharmacological barriers, they may also influence the pharmacokinetics and toxicities of TKIs and affect drug–drug interactions (Szakacs et al., 2008). In our current study we combined several in vitro systems that might provide useful information about both of these possible outcomes. On one hand, we investigated the emergence of a TKI-resistant phenotype and direct modulation of the anti-cancer effect of the TKIs in CML model cells and their parental counterparts. In order to explore the mechanisms of drug resistance we also studied modifications of the target kinase and determined the transporter-modified intracellular concentrations of the respective TKIs. On the other hand, by using the membrane ATPase and dye transport models, we were able to screen a wide concentration range of TKIs for their possible transport and MDR-ABC inhibition. Interactions observed in these assays, even if at relatively high concentrations, may become relevant at tissue barriers, that is, at the site of absorption or at the site of drug excretion (in the liver or kidney) or at the blood–brain barrier.
In our first set of experiments using CML-derived Bcr-Abl+ K562 model cells and their transporter-expressing counterparts, we sought to study if ABCB1 or ABCG2 function could alter the cytotoxic potential of the investigated TKIs. We found that K562/ABCG2 cells were 8.8-fold more resistant to nilotinib than parental K562 cells, whereas ABCB1 function only slightly modified the cytotoxic effect of this drug, resulting in a small (albeit significant) resistance of K562/ABCB1 cells. As demonstrated by our Western blot experiments, marked resistance to nilotinib, mediated by ABCG2, was a result of the unaltered constitutive phosphorylation of Bcr-Abl, even in the presence of this TKI. Very recently Mahon et al. (2008) demonstrated an increased number of viable ABCB1-overexpressing K562 cells in the presence of increasing concentrations of nilotinib. Such an effect may be due to a higher level of ABCB1 expression than in our cells.
In the case of dasatinib, we observed 3.7-fold and 7.8-fold resistance, specifically caused by ABCB1 and ABCG2 functions, respectively, in the 1–10 nM dasatinib concentration range. We showed that ABCB1 and ABCG2 conferred cellular dasatinib resistance via preventing dasatinib from reaching its intracellular target and blocking Bcr-Abl phosphorylation. Our findings are compatible with two recently published reports showing by different methods that dasatinib is indeed pumped out of cells by both ABCB1 (Giannoudis et al., 2008; Hiwase et al., 2008) and ABCG2 (Hiwase et al., 2008). Interestingly, neither ABCB1 nor ABCG2 exerted noticeable protective effects in the bosutinib cellular toxicity assays. Also, phospho-Bcr-Abl patterns were similar in bosutinib-treated K562, K562/ABCB1 and K562/ABCG2 cells, implying no clear interaction of ABCB1 or ABCG2 with this compound.
For direct exploration of ABCB1- and ABCG2-mediated transport of cytotoxic concentrations of TKIs, we used a novel HPLC-MS method. By employing this sensitive and quantitative system, we were able to measure TKI accumulation in K562, K562/ABCB1 and K562/ABCG2 cells even with very low concentrations of TKIs present. One advantage of this method is that it does not require radioactively or fluorescently labelled compounds. Moreover, this experimental procedure might provide direct indications regarding the nature of drug–transporter interactions, as decreased accumulation of the drug clearly indicates that it is a substrate of the investigated transporter. Our HPLC-MS method proved to be highly sensitive, as the detection threshold of all three TKIs fell in the 0.1–1 ng range. By this assay, we showed that in relevant, cytotoxic concentrations, nilotinib was recognized by ABCG2, while dasatinib was effectively extruded by both ABCB1 and ABCG2. These findings agree with previous studies performed with radiolabelled nilotinib (Brendel et al., 2007) and dasatinib (Hiwase et al., 2008). In keeping with the cytotoxicity data, HPLC-MS measurements revealed only slight differences between bosutinib accumulations in these K562 cell lines. Overall, we show here for the first time that bosutinib did not noticeably interact with ABCB1 and ABCG2 in relevant cytotoxic concentrations.
By assaying membrane ATPase activity, we were able to analyse the MDR-ABC–TKI interaction profiles over a much wider TKI concentration range. Additionally, in fluorescent dye efflux studies, we aimed to determine the effective inhibitory concentration range of the TKIs in the context of drug–drug interactions. We found that nilotinib greatly stimulated both ABCB1 and ABCG2 ATPase activities. However, while 500 nM nilotinib was needed to reach maximum ABCB1 ATPase stimulation, the maximum stimulation of ABCG2 ATPase was reached at 25 nM nilotinib. These data strongly suggest that nilotinib is a high-affinity substrate for ABCG2 and a somewhat poorer substrate for ABCB1, which could be one possible explanation for the observed difference in cytoprotective effect of the two transporters against this TKI. In higher concentrations, nilotinib reduced both ABCB1 and ABCG2 ATPase activities. In the presence of an additional transporter substrate compound, nilotinib also displayed an inhibitory effect on both ABCB1 and ABCG2. For dasatinib, the ATPase assays did not show a measurable effect either on the ABCB1 or on the ABCG2 ATPase activities up to 1 µM concentration. The ability of dasatinib to inhibit the transporter-substrate dye efflux correlated well with the data obtained in the ATPase assays, as effective inhibition could be observed only when 2–20 µM dasatinib was used. Bosutinib displayed a very similar interaction profile to dasatinib in terms of its effects on ABCB1 and ABCG2 ATPase activity and its inhibitory potential on transporter-mediated dye efflux. We measured no significant effect on ABCB1 or ABCG2 transporter function for either of these assays up to 1 µM bosutinib concentration. The inhibitory effects of TKIs, observed at higher than cytotoxic concentrations imply that at important pharmacological barriers and in the presence of additional drugs, MDR-ABC transporter function might significantly alter the pharmacokinetics and toxicities of these compounds.
In summary, our current study demonstrates that nilotinib, dasatinib and bosutinib exhibit distinct interaction profiles with the ABCB1 and ABCG2 multidrug transporters, which might be a key determinant in their clinical efficacy. These interactions may significantly affect cellular TKI resistance, as well as the pharmacokinetics and toxicities of these compounds. Nilotinib and dasatinib act both as transported substrates and, at high concentrations, inhibitors of ABCB1 and ABCG2. We show here for the first time, that neither ABCB1 nor ABCG2 can confer bosutinib resistance; however, this TKI efficiently inhibits both transporters at higher concentrations. These properties might contribute to the improved anti-cancer effects and pharmacokinetics and toxicities of bosutinib.
Acknowledgments
The technical help by Éva Krizsán, Zsuzsanna Andrási and Judit Kis is greatly appreciated. We thank András Kozma for the FISH analyses.
This work has been supported by research grants from OTKA (K 46965, AT48986, and NK72057), NKTH (Stemkill), ETT and KKK (Hungary). Csilla Özvegy-Laczka is a recipient of the Postdoctoral Fellowship (PD45957) of OTKA, Hungary and the János Bolyai Scholarship of the Hungarian Academy of Sciences. Gergely Szakács is a Bolyai fellow and a Special Fellow of the leukaemia and Lymphoma Society.
Glossary
Abbreviations:
- ABCP
placenta specific ABC transporter
- ABC transporters
ATP-binding cassette transporters
- BCRP
breast cancer resistance protein
- CML
chronic myeloid leukaemia
- HPLC-MS
high-pressure liquid chromatography-mass spectrometry
- MDR
multidrug resistance
- MXR
mitoxantrone resistance protein
- Sf9 cells
Spodoptera frugiperda ovarian cells
- TKI
tyrosine kinase inhibitor
Conflicts of interest
The authors state no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Characterization of the different K562 cell lines. In order to test the cell surface expression of ABCB1 and ABCG2 in the different K562 cells lines, (A) parental K562 (red) and K562/ABCB1 (blue) cells were labelled with anti-ABCB1 MRK-16 antibody and phycoerythrin-conjugated secondary antibody; or (B) parental K562 (red) and K562/ABCG2 (blue) cells were labelled with anti-ABCG2 Alexa647-conjugated 5D3 antibody. Staining of parental K562 cells (red) with either of the transporter-specific antibodies corresponded to that with the appropriate isotype controls (data not shown). Proper ABCB1 and ABCG2 transporter function was evaluated by determining the transport activity factors of the different K562 cell lines in calcein-AM and Hoechst 33342 dye uptake experiments (C). Parental K562 cells showed no endogenous ABCB1 or ABCG2 expression.
Figure S2 Graphical representation of the phospho-Bcr-Abl patterns in tyrosine kinase inhibitor (TKI)-treated parental K562 (A), K562/ABCB1 (B) and K562/ABCG2 (C) cells. Phospho-Bcr-Abl patterns of the TKI-treated K562 cell lines were probed by Western blotting as described under Methods. The figure shows the phospho-Bcr-Abl/Bcr-Abl ratios in TKI- or TKI + PSC-833/fumitremorgin C-treated cells as determined by densitometry analysis, relative to that measured in untreated control cells (100%). Data represent the mean of at least two experiments, bars represent SD.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Brendel C, Scharenberg C, Dohse M, Robey RW, Bates SE, Shukla S, et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia. 2007;21:1267–1275. doi: 10.1038/sj.leu.2404638. [DOI] [PubMed] [Google Scholar]
- Brozik A, Casey NP, Hegedus C, Bors A, Kozma A, Andrikovics H, et al. Reduction of Bcr-Abl function leads to erythroid differentiation of K562 cells via downregulation of ERK. Ann N Y Acad Sci. 2006;1090:344–354. doi: 10.1196/annals.1378.038. [DOI] [PubMed] [Google Scholar]
- Burger H, van Tol H, Boersma AW, Brok M, Wiemer EA, Stoter G, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104:2940–2942. doi: 10.1182/blood-2004-04-1398. [DOI] [PubMed] [Google Scholar]
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Deguchi Y, Kimura S, Ashihara E, Niwa T, Hodohara K, Fujiyama Y, et al. Comparison of imatinib, dasatinib, nilotinib and INNO-406 in imatinib-resistant cell lines. Leuk Res. 2008;32:980–983. doi: 10.1016/j.leukres.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Elkind NB, Szentpetery Z, Apati A, Ozvegy-Laczka C, Varady G, Ujhelly O, et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib) Cancer Res. 2005;65:1770–1777. doi: 10.1158/0008-5472.CAN-04-3303. [DOI] [PubMed] [Google Scholar]
- Giannoudis A, Davies A, Lucas CM, Harris RJ, Pirmohamed M, Clark RE. Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood. 2008;112:3348–3354. doi: 10.1182/blood-2007-10-116236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Hamada A, Miyano H, Watanabe H, Saito H. Interaction of imatinib mesilate with human P-glycoprotein. J Pharmacol Exp Ther. 2003;307:824–828. doi: 10.1124/jpet.103.055574. [DOI] [PubMed] [Google Scholar]
- Hazarika M, Jiang X, Liu Q, Lee SL, Ramchandani R, Garnett C, et al. Tasigna for chronic and accelerated phase Philadelphia chromosome-positive chronic myelogenous leukemia resistant to or intolerant of imatinib. Clin Cancer Res. 2008;14:5325–5331. doi: 10.1158/1078-0432.CCR-08-0308. [DOI] [PubMed] [Google Scholar]
- Hegedus T, Orfi L, Seprodi A, Varadi A, Sarkadi B, Keri G. Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim Biophys Acta. 2002;1587:318–325. doi: 10.1016/s0925-4439(02)00095-9. [DOI] [PubMed] [Google Scholar]
- Hiwase DK, Saunders V, Hewett D, Frede A, Zrim S, Dang P, et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin Cancer Res. 2008;14:3881–3888. doi: 10.1158/1078-0432.CCR-07-5095. [DOI] [PubMed] [Google Scholar]
- Hollo Z, Homolya L, Davis CW, Sarkadi B. Calcein accumulation as a fluorometric functional assay of the multidrug transporter. Biochim Biophys Acta. 1994;1191:384–388. doi: 10.1016/0005-2736(94)90190-2. [DOI] [PubMed] [Google Scholar]
- Hollo Z, Homolya L, Hegedus T, Sarkadi B. Transport properties of the multidrug resistance-associated protein (MRP) in human tumour cells. FEBS Lett. 1996;383:99–104. doi: 10.1016/0014-5793(96)00237-2. [DOI] [PubMed] [Google Scholar]
- Houghton PJ, Germain GS, Harwood FC, Schuetz JD, Stewart CF, Buchdunger E, et al. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004;64:2333–2337. doi: 10.1158/0008-5472.can-03-3344. [DOI] [PubMed] [Google Scholar]
- Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–935. doi: 10.1038/sj.leu.2404609. [DOI] [PubMed] [Google Scholar]
- Jordanides NE, Jorgensen HG, Holyoake TL, Mountford JC. Functional ABCG2 is overexpressed on primary CML CD34+ cells and is inhibited by imatinib mesylate. Blood. 2006;108:1370–1373. doi: 10.1182/blood-2006-02-003145. [DOI] [PubMed] [Google Scholar]
- Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109:4016–4019. doi: 10.1182/blood-2006-11-057521. [DOI] [PubMed] [Google Scholar]
- Kim M, Turnquist H, Jackson J, Sgagias M, Yan Y, Gong M, et al. The multidrug resistance transporter ABCG2 (breast cancer resistance protein 1) effluxes Hoechst 33342 and is overexpressed in hematopoietic stem cells. Clin Cancer Res. 2002;8:22–28. [PubMed] [Google Scholar]
- Kotaki M, Motoji T, Takanashi M, Wang YH, Mizoguchi H. Anti-proliferative effect of the abl tyrosine kinase inhibitor STI571 on the P-glycoprotein positive K562/ADM cell line. Cancer Lett. 2003;199:61–68. doi: 10.1016/s0304-3835(03)00338-0. [DOI] [PubMed] [Google Scholar]
- Mahon FX, Belloc F, Lagarde V, Chollet C, Moreau-Gaudry F, Reiffers J, et al. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood. 2003;101:2368–2373. doi: 10.1182/blood.V101.6.2368. [DOI] [PubMed] [Google Scholar]
- Mahon FX, Hayette S, Lagarde V, Belloc F, Turcq B, Nicolini F, et al. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008;68:9809–9816. doi: 10.1158/0008-5472.CAN-08-1008. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Shiozawa K, Hassel BA, Ross DD. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood. 2006;108:678–684. doi: 10.1182/blood-2005-10-4020. [DOI] [PubMed] [Google Scholar]
- Ozvegy C, Varadi A, Sarkadi B. Characterization of drug transport, ATP hydrolysis, and nucleotide trapping by the human ABCG2 multidrug transporter. Modulation of substrate specificity by a point mutation. J Biol Chem. 2002;277:47980–47990. doi: 10.1074/jbc.M207857200. [DOI] [PubMed] [Google Scholar]
- Ozvegy-Laczka C, Hegedus T, Varady G, Ujhelly O, Schuetz JD, Varadi A, et al. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol. 2004;65:1485–1495. doi: 10.1124/mol.65.6.1485. [DOI] [PubMed] [Google Scholar]
- Ozvegy-Laczka C, Laczko R, Hegedus C, Litman T, Varady G, Goda K, et al. Interaction with the 5D3 monoclonal antibody is regulated by intramolecular rearrangements but not by covalent dimer formation of the human ABCG2 multidrug transporter. J Biol Chem. 2008;283:26059–26070. doi: 10.1074/jbc.M803230200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- Sarkadi B, Homolya L, Szakacs G, Varadi A. Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol Rev. 2006;86:1179–1236. doi: 10.1152/physrev.00037.2005. [DOI] [PubMed] [Google Scholar]
- Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99:507–512. doi: 10.1182/blood.v99.2.507. [DOI] [PubMed] [Google Scholar]
- Smeets M, Raymakers R, Vierwinden G, Pennings A, van de Locht L, Wessels H, et al. A low but functionally significant MDR1 expression protects primitive haemopoietic progenitor cells from anthracycline toxicity. Br J Haematol. 1997;96:346–355. doi: 10.1046/j.1365-2141.1997.d01-2024.x. [DOI] [PubMed] [Google Scholar]
- Szakacs G, Varadi A, Ozvegy-Laczka C, Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox) Drug Discov Today. 2008;13:379–393. doi: 10.1016/j.drudis.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Telbisz A, Muller M, Ozvegy-Laczka C, Homolya L, Szente L, Varadi A, et al. Membrane cholesterol selectively modulates the activity of the human ABCG2 multidrug transporter. Biochim Biophys Acta. 2007;1768:2698–2713. doi: 10.1016/j.bbamem.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Thomas J, Wang L, Clark RE, Pirmohamed M. Active transport of imatinib into and out of cells: implications for drug resistance. Blood. 2004;104:3739–3745. doi: 10.1182/blood-2003-12-4276. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Catley L, Wright RD, Moreno D, Banerji L, Ray A, et al. Beneficial effects of combining nilotinib and imatinib in preclinical models of BCR-ABL+ leukemias. Blood. 2007a;109:2112–2120. doi: 10.1182/blood-2006-06-026377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007b;7:345–356. doi: 10.1038/nrc2126. [DOI] [PubMed] [Google Scholar]
- White DL, Saunders VA, Dang P, Engler J, Zannettino AC, Cambareri AC, et al. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108:697–704. doi: 10.1182/blood-2005-11-4687. [DOI] [PubMed] [Google Scholar]
- White DL, Saunders VA, Quinn SR, Manley PW, Hughes TP. Imatinib increases the intracellular concentration of nilotinib, which may explain the observed synergy between these drugs. Blood. 2007;109:3609–3610. doi: 10.1182/blood-2006-11-058032. [DOI] [PubMed] [Google Scholar]
- Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.