

Abstract
ADAR1 (adenosine deaminase acting on RNA) catalyzes the conversion of adenosine to inosine, a process known as A-to-I editing. Extensive A-to-I editing has been described in viral RNAs isolated from the brains of patients persistently infected with measles virus, although the precise role of ADAR during measles virus infection remains unknown. We generated human HeLa cells stably deficient in ADAR1 (“ADAR1kd cells”) through short hairpin RNA-mediated knockdown, and using these cells, we tested the effect of ADAR1 deficiency on measles virus (MVvac strain) growth and virus-induced cell death. We found that the growth of mutant viruses lacking expression of the viral accessory proteins V and C (Vko and Cko, respectively) was decreased in ADAR1-deficient cells compared with ADAR1-sufficient cells. In addition, apoptosis was enhanced in ADAR1-deficient cells following infection with wild type and Vko virus but not following infection with Cko virus or treatment with tumor necrosis factor-α or staurosporine. Furthermore, in Cko-infected ADAR1-sufficient cells when ADAR1 did not protect against apoptosis, caspase cleavage of the ADAR1 p150 protein was detected. Finally, enhanced apoptosis in ADAR1kd cells following infection with wild type and Vko virus correlated with enhanced activation of PKR kinase and interferon regulatory factor IRF-3. Taken together, these results demonstrate that ADAR1 is a proviral, antiapoptotic host factor in the context of measles virus infection and suggest that the antiapoptotic activity of ADAR1 is achieved through suppression of activation of proapoptotic and double-stranded RNA-dependent activities, as exemplified by PKR and IRF-3.
ADAR1 (adenosine deaminase acting on RNA) is an RNA-specific C-6 adenosine deaminase that catalyzes the conversion of adenosine (A) to inosine (I) on RNAs with double-stranded character (1, 2). Such “A-to-I editing” by ADAR1 is of broad biologic importance, because I is recognized as G instead of A by ribosomes and polymerases (2, 3). For example, ADAR1 plays an important role in the nervous system, where site-specific editing of glutamate receptor and serotonin-2C receptor pre-mRNAs changes their coding capacity, thereby leading to neurotransmitter receptor protein products with altered physiological properties (4–6). In addition, ADAR1 is involved in the RNA interference pathway and is known to alter both the targeting and the processing of microRNAs (7, 8). Furthermore, RNA editing patterns characteristic of ADAR enzymes have been detected in several viral RNAs, including those of measles virus (9, 10), influenza virus (11), lymphocytic choriomeningitis virus (12), polyomavirus (13), hepatitis delta virus (14), and hepatitis C virus (15).
ADAR1 is encoded by a single-copy gene that maps to human chromosome 1q21 (16, 17). The domain structure of the ADAR1 protein product includes the C-terminal deaminase catalytic domain, three centrally located dsRNA2 binding domains, and one or two N-terminal Z-DNA binding domains (18–20). Two size forms of the ADAR1 protein are known (18, 21, 22). One, p110, is constitutively expressed and found predominantly in the nucleus of cells; the other, p150, is interferon (IFN)-inducible and is found in both the nucleus and the cytoplasm. Compared with p110, the p150 form of human ADAR1 possesses an additional 295 N-terminal amino acids. The function of the N-terminal extension of p150 is not entirely understood, but the region contains a nuclear export signal (23) and an additional Z-DNA binding domain (19). Because of its regulation by IFN and cytoplasmic localization, the p150 version of ADAR1 is thought to be the form responsible for the A-to-I editing of viral RNAs produced by viruses that replicate in the cytoplasm of infected cells (2, 3).
Measles virus (MV), a member of the Morbillivirus genus of the family Paramyxoviridae, is a negative-stranded RNA virus whose replication cycle takes place entirely in the host cell cytoplasm (24). MV causes an acute respiratory illness in humans, and despite effective vaccination, MV continues to be a pathogen of major global importance (24). In rare cases, the acute infection can lead to a persistent central nervous system infection, resulting in a chronic and fatal disease known as subacute sclerosing panencephalitis (SSPE) (25). Analysis of viral RNA from SSPE autopsies reveals extensive A-to-I hypermutation in the matrix gene and several other virus genes (10, 26, 27), implicating a role for ADAR during MV infection. However, the effects of ADAR enzymes on the infectious cycle of MV remain largely unknown.
To investigate the role of ADAR1 during MV infection, we used an shRNA-mediated knockdown strategy to create a human HeLa cell line stably deficient in ADAR1 expression. This cell line, called ADAR1kd, was then used to study the effects of ADAR1 on virus growth and cell killing induced by MVvac, a recombinant MV based on the Moraten vaccine strain. The results obtained with the parental MVvac were compared with those of derived isogenic mutant viruses lacking expression of either the V or C accessory protein. Because ADAR1 is an interferon-inducible enzyme and because biased hypermutation such as that observed in SSPE appears to be detrimental to MV, we predicted that MVvac, or at least the V or C mutants of this virus, would grow to higher titers in the ADAR1kd cells compared with parental controls. However, we found the opposite, that ADAR1 acts as a proviral host factor in the context of MV infection and functions to enhance virus growth and suppress virus-induced cell death. Moreover, the mechanism of ADAR1 enhancement of virus growth and suppression of virus-induced cell death is consistent with impairment of dsRNA-mediated innate antiviral responses, exemplified by those of PKR (protein kinase regulated by RNA) and IRF-3 (interferon regulatory transcription factor-3).
EXPERIMENTAL PROCEDURES
Cells and Viruses
Parental HeLa and Vero cells were maintained in Dulbecco's modified Eagle's medium supplemented with 5% (v/v) fetal bovine serum (HyClone), 100 μg/ml penicillin, and 100 units/ml streptomycin (Invitrogen), as described previously (28). HeLa cells with the stable knockdown of PKR (PKRkd) (29, 30) or ADAR1 (ADAR1kd; see below) by RNA interference were maintained in the above-named medium containing 1 μg/ml puromycin (Sigma). Treatment with TNF-α (Sigma) was carried out at the indicated concentrations and times together with 1 μg/ml cyclohexamide. Treatment with staurosporine (Chemicon) was carried out with the indicated concentrations and times. Treatment with the pancaspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (z-VAD-fmk; EMD/Calbiochem) was with a concentration of 100 μm beginning after the virus inoculum was removed.
The recombinant parental MVvac GFP(H) virus, herein designated as wild type (WT), as well as V-deficient (Vko) and C-deficient (Cko) versions of this virus were constructed based on the Moraten vaccine strain, as described previously (28, 31).
Construction of shRNA Expression Vector Plasmid
The pSUPER.retro.puro vector with H1 promoter (Oligo Engine) was used for construction of short hairpin expression constructs to silence human ADAR1. The successful 21-nucleotide human ADAR1 targeting sequence (5′-GTTGACTAAGTCACATGTAAA-3′) present in the 3′-untranslated region of human ADAR1 (18) was identified using Promega online software. The sense strand oligonucleotide (5′-GATCCCCGTTGACTAAGTCACATGTAAATTCAAGAGATTTACATGTGACTTAGTCAACTTTTTA-3′) and antisense-strand oligonucleotide (5′-AGCTTAAAAAGTTGACTAAGTCACATGTAAATCTCTTGAATTTACATGTGACTTAGTCAACGGG-3′) shown with the targeting sequence underlined were annealed, and the resulting duplex was inserted into the pSUPER.retro.puro vector using the HindIII and BglII restriction sites. Recombinant plasmid constructs were verified by DNA sequence analysis.
Cell Transfection
HeLa cells were transfected at 80–90% confluence using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Briefly, Lipofectamine 2000 and plasmid DNA were diluted and mixed in Opti-MEM (Invitrogen), the DNA-Lipofectamine 2000 complexes were applied to the cultures, and incubation continued for 4 h at 37 °C, after which the transfection medium was replaced with fresh maintenance medium containing 5% (v/v) fetal bovine serum. To select for stable transfectants, cells were trypsinized 24 h after transfection with the shRNA pSUPER.retro.puro construct, seeded at various dilutions, and then carried in the presence of 1 μg/ml puromycin. Puromycin-resistant clones were isolated and screened by Western immunoblot analysis for ADAR1 protein knockdown. HeLa cells transfected with the pSUPER.retro.puro vector with H1 promoter without shRNA-encoded insertion were selected and maintained in the puromycin-containing medium. These cells, called CONkd (formerly called PKRkd-con (28)), were used as an additional PKR- and ADAR1-positive control.
Virus Infections and Growth Assays
ADAR1kd, CONkd, and/or ADAR1+ cells were infected at a multiplicity of infection of 5 TCID50/cell for the indicated amounts of time, as indicated throughout. Virus yields were determined by 50% TCID50 titration on Vero cells according to the Spearman-Karber method (28, 32).
Western Immunoblot Analysis
Whole cell extracts were prepared at the indicated times postinfection, and protein concentrations were determined as described previously (28). Proteins were fractionated by SDS-PAGE and transferred to nitrocellulose, and the membranes were blocked in 5% milk in phosphate-buffered saline. Rabbit polyclonal antibodies against measles virus proteins were described previously (28). Rabbit polyclonal antibody against ADAR1 (K88#2) was described previously (18). Rabbit polyclonal antibodies from the indicated sources were used to detect GFP (Santa Cruz Biotechnology), human PKR (Santa Cruz Biotechnology), IRF-3 (Santa Cruz Biotechnology), phospho-Thr446-PKR (Santa Cruz Biotechnology and Epitomics), and phospho-Ser51-eIF-2α (Cell Signaling). Mouse monoclonal antibodies were used to detect β-actin (Sigma) and human poly(ADP-ribose) polymerase (PARP) (BD Biosciences).
Cell Viability Assays
The MTS colorimetric assay (Promega) was performed according to the manufacturer's instructions. This assay is based on the conversion of the MTS tetrazolium salt into soluble formazan by endogenous dehydrogenase enzymes found in metabolically active cells. Cells were plated at 2.5 × 104 cells/well in 96-well plates. The following day, cells were infected with WT, Vko, or Cko virus or treated with TNF-α/cyclohexamide or staurosporine as indicated. Following infection or treatment, 20 μl of MTS was added, followed by a 1-h incubation at 37 °C, after which the absorbance was measured at 490 nm on a Victor3V plate reader (PerkinElmer Life Sciences).
RESULTS
ADAR1 Enhances the Growth of Measles Virus
To test the effect of ADAR1 on MV infection, we utilized a HeLa clonal line in which ADAR1 expression is stably knocked down by RNA interference. These ADAR1kd cells have less than 15% of the p110 form and less than 10% of the p150 form of ADAR1 found in either parental HeLa cells (ADAR1+) or drug-treated control cells (CONkd) (data not shown) (see Figs. 2C and 3B). We used the recombinant MVvac strain and isogenic Vko and Cko mutants of this virus. All of these viruses encode a GFP gene downstream of the hemagglutinin gene, and since MVvac is a vaccine lineage strain, these viruses can enter through CD46 (cluster of differentiation 46) (33), a receptor expressed on ADAR1-deficient and -sufficient HeLa cell lines.
FIGURE 2.

Measles virus-induced apoptosis is enhanced in ADAR1-deficient cells and ADAR1 is cleaved by caspases during MV infection. ADAR1+, ADAR1kd, and CONkd HeLa cells were left uninfected (UI) or infected with WT, Vko, or Cko recombinant virus for 36 h. A, phase-contrast images taken immediately before harvest. B, results of the colorimetric MTS assay to measure cell viability 36 h postinfection, displayed as percentages of the number of viable uninfected cells with S.D. (n = 4). *, p < 0.005 by Student's t test for comparison of the Cko virus yields in ADAR1kd cells and those in ADAR1+ or CONkd cells. C, Western immunoblot analyses performed on whole cell extracts using antibodies against ADAR1, PARP, and β-actin. The quantity of PARP cleavage based on the immunoblots, expressed as a ratio of the 85-kDa cleavage product to total PARP (85/(85 + p116)), is shown below each lane. Likewise, the quantity of ADAR1 cleavage based on the immunoblots, expressed as a ratio of the 120-kDa cleavage product to total ADAR1 (120/(120 + p150)), is shown below each lane. The arrowheads mark the positions of the PARP 85 and ADAR1 120 cleavage products.
FIGURE 3.

ADAR1 and PKR protein deficiencies do not affect TNF-α or staurosporine-induced apoptosis. A, cells were treated with varying concentrations of TNF-α and 1 μg/ml of cyclohexamide (top) or staurosporine (bottom), and MTS analysis was performed 8 h (TNF-α and cyclohexamide) or 12 h (staurosporine) after treatment. The assay was performed with CONkd cells (dotted line, triangles), ADAR1kd cells (dashed line, diamonds), and PKRkd cells (solid line, circles). The results shown are the means with S.D. values (n = 3 for each time point). B, top, CONkd, PKRkd, and ADAR1kd cells were treated with 10 ng/ml TNF-α and 1 μg/ml cyclohexamide (+) or left untreated (−), and whole cell extracts were prepared 12 h after treatment. Western immunoblot analyses were performed using the indicated antibodies. Bottom, same as the top panel, only cells were treated with 0.25 μm staurosporine (stauro) (+) or left untreated (−). The arrowheads mark the positions of the PARP 85 and ADAR1 120 cleavage products.
We examined the growth of MVvac WT and mutant recombinant MV in ADAR1-deficient (ADAR1kd) and ADAR1-sufficient (ADAR1+ and CONkd) human cell lines, both qualitatively (Fig. 1A) and quantitatively (Fig. 1B). As previously observed for PKR-sufficient cells (28), the GFP reporter signal viewed by fluorescence microscopy was high for WT and Vko but low for Cko in parental HeLa (ADAR1+) and drug control (CONkd) cells (Fig. 1A). However, in ADAR1kd cells, the GFP signal for all three viruses was very low (Fig. 1A), suggesting that these viruses grew poorly in the absence of ADAR1. To quantify the growth of virus, infectious virus yields were determined on Vero cells. When grown in ADAR1+ parental HeLa cells and CONkd drug control cells (28), the yield of the Cko virus was about 30-fold lower than the WT virus, whereas the yield of Vko was similar to the WT virus (Fig. 1B), consistent with earlier findings (28). The yields of the Vko and Cko viruses, however, were significantly reduced in ADAR1kd cells compared with ADAR1-sufficient cells, about 8- and 12-fold, respectively (Fig. 1B). These results suggest that ADAR1 enhances the growth of measles virus.
FIGURE 1.

Growth of wild type and V- and C-mutant measles virus in ADAR1-deficient and ADAR1-sufficient cells. ADAR1+, ADAR1kd, and CONkd HeLa cells were infected with WT, Vko, or Cko recombinant virus at a multiplicity of infection of 5 TCID50/cell for 36 h. A, fluorescence images showing GFP signal in cells taken immediately before harvest. UI, uninfected cells. B, virus yields from ADAR1-sufficient (parental ADAR1+ and CONkd) and stably ADAR1-deficient (ADAR1kd) cells were determined by 50% TCID50 titration on Vero cells. The results shown are the means with S.D. (n = 4). *, p < 0.05 by Student's t test for comparison of Vko and Cko virus yields in ADAR1kd cells versus ADAR1-sufficient controls.
ADAR1 Suppresses WT and Vko Virus-induced Apoptosis
While determining MV growth in ADAR1-sufficient and ADAR1-deficient cells, we observed greater cytopathic effect after infection of ADAR1kd compared with ADAR1+ or CONkd cells with WT and especially Vko virus, as illustrated by the phase-contrast images shown in Fig. 2A.
To quantify the effect, the colorimetric MTS assay was employed. The MTS assay revealed that the number of viable ADAR1kd cells was indeed decreased compared with the numbers of viable ADAR1+ or CONkd cells following infection with WT and Vko virus (Fig. 2B). Although the Cko virus was far more cytopathic than either WT or Vko (Fig. 2, A and B), which is consistent with our prior observations in ADAR1-sufficient cells (28), the cell killing capacity of Cko, however, was not further enhanced in ADAR1kd cells (Fig. 2B). We showed previously that the MV-induced cytopathic effect observed by microscopy correlated well with cleavage of the caspase substrate PARP and was therefore probably apoptotic (28). We thus performed western immunoblot analysis with an anti-PARP antibody to further assess the cell killing in ADAR1kd versus ADAR1-sufficient controls. As reported previously for parental HeLa cells and drug control cells (28), the PARP cleavage was most pronounced following infection with the Cko virus and least with the WT virus (Fig. 2C). In the ADAR1kd cells compared with the ADAR1-sufficient cells, the PARP cleavage following infection with WT and especially Vko virus was greatly enhanced. However, apoptosis following infection with Cko virus was not enhanced by the ADAR1 deficiency.
ADAR1 Does Not Protect against TNF-α- or Staurosporine-induced Apoptosis
Double-stranded RNA is potentially the trigger for apoptosis in MV-infected cells, since MV-induced apoptosis is mediated at least in part by PKR (28), a protein kinase that is activated upon binding structured RNA or dsRNA. Because ADAR1 suppresses MV-induced apoptosis (Fig. 2) and because ADAR1 binds dsRNA (18) and was discovered based on its dsRNA-unwinding activity (34, 35), it is conceivable that ADAR1 protects against dsRNA-induced apoptosis. To determine if ADAR1 is a general suppressor of apoptosis, we also tested whether ADAR1 protects against cell death induced by two other inducers of apoptosis: TNF-α, an inflammatory cytokine that stimulates apoptosis via the FADD adaptor to activate initiator caspase 8 (36), and staurosporine, a broad kinase inhibitor and inducer of apoptosis (37). PKRkd, ADAR1kd, and CONkd cells were treated with TNF-α together with cyclohexamide, and cell viability following treatment was measured by MTS assay and PARP cleavage. The percentage of viable cells (Fig. 3A, top) and the amount of PARP cleavage (Fig. 3B, top) following treatment with TNF-α/cyclohexamide were similar between PKRkd and CONkd cells, consistent with previous reports indicating that PKR does not play a role in TNF-α-induced apoptosis (29, 38). Likewise, the percentage of viable cells and the amount of PARP cleavage following TNF-α/cyclohexamide treatment were similar between ADAR1kd and CONkd cells (Fig. 3, A (top) and B (top)). In addition, cell viability and PARP cleavage following staurosporine treatment (Fig. 3, A (bottom) and B (bottom)) were similar in PKRkd and ADAR1kd compared with CONkd cells. These results suggest that PKR enhances and ADAR1 protects against apoptosis induced by some, but not all, triggers.
Caspase Cleavage of ADAR1 p150
Following infection with the Cko virus and to a lesser extent Vko virus and also upon treatment with TNF-α/cyclohexamide or staurosporine, we detected an additional protein species with our ADAR1 polyclonal antiserum (Figs. 2C and 3B). This protein migrated at ∼120 kDa and is a candidate degradation product of ADAR1 because it was recognized by ADAR1 antiserum and was reduced in amount in the ADAR1kd cells (Figs. 2C and 3B). Since the appearance of this protein correlated precisely with PARP cleavage, we hypothesized that it was a caspase cleavage product of ADAR1 p150. To test this hypothesis, we used the pancaspase inhibitor z-VAD-fmk. Treatment of ADAR1-sufficient cells with this drug prevented accumulation both of the ∼120-kDa ADAR1 product and the ∼85-kDa PARP cleavage product (Fig. 4), consistent with the conclusion that the novel ADAR1 protein species was a caspase-mediated cleavage product.
FIGURE 4.
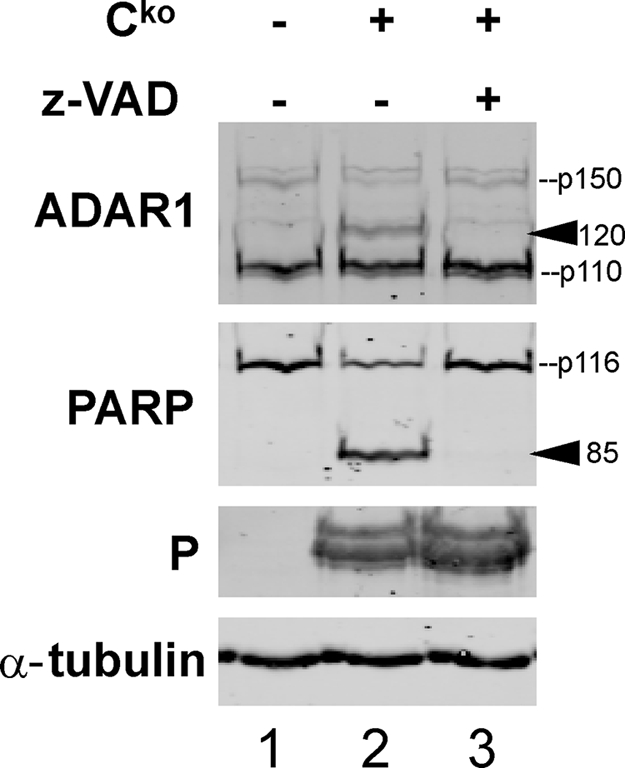
Accumulation of the presumed ADAR p150 caspase cleavage product is inhibited by z-VAD-fmk. CONkd cells were infected with Cko virus or left uninfected and were treated with medium alone or medium containing the caspase inhibitor z-VAD-fmk (100 μm) as indicated. Whole cell extracts were made 36 h postinfection, and Western immunoblot analyses were performed using the indicated antibodies. The arrowheads mark the positions of the PARP 85 and ADAR1 120 cleavage products.
ADAR1 Suppresses WT and Vko Virus-induced PKR Activation
We showed previously that PKR is activated upon infection with Cko virus but not WT or Vko virus and that activation of PKR correlated with PKR-dependent measles virus growth inhibition and apoptosis induction (28). We therefore considered the possibility that the enhanced cell death and decreased virus growth observed in ADAR1kd cells was due to enhanced PKR activation. To test this hypothesis, we measured PKR phosphorylation using an antibody against phosphothreonine 446. As we previously reported for PKR/ADAR1-sufficient cells (28), very low levels of PKR activation were detected following infection with the WT or Vko viruses, but substantial PKR activation by Cko virus was observed by 24 h postinfection in these cells (Fig. 5, lanes 1–7). In the ADAR1kd cells, however, activation of PKR was observed for all three viruses; furthermore, the amount and kinetics of activation were similar to that of the Cko virus-induced activation in the CONkd cells (Fig. 5, lanes 8–14). Activation of PKR correlated with activation of eIF-2α, as measured by phosphorylation of eIF-2α on serine 51, and also with decreased expression of viral proteins hemagglutinin and GFP.
FIGURE 5.

PKR activation is enhanced in ADAR1-deficient cells compared with ADAR1-sufficient cells following infection with WT and Vko but not Cko MV. CONkd and ADAR1kd cells were infected with WT, Vko, or Cko virus or left uninfected (UI). Whole cell extracts were prepared at the indicated times postinfection (hpi) and analyzed by Western immunoblot analysis with phospho-PKR (Thr446), phospho-eIF-2α (Ser51), and β-actin antibodies as well as with antibodies against hemagglutinin (H) and GFP proteins expressed by MV.
ADAR1 Suppresses IRF-3 Activation by WT and Vko Viruses
Because we observed enhanced PKR activation in ADAR1kd cells but not ADAR1-sufficient cells upon infection with WT and Vko, we next asked whether activation of additional dsRNA-dependent pathways might also be enhanced in these cells. To test this possibility, we examined IRF-3 activation. IRF-3 is a key transcription factor for the induction of IFN-β (39). IRF-3 is activated by C-terminal phosphorylation and following activation translocates to the nucleus and mediates transcription (40). IRF-3 is activated by dsRNA-dependent pathways, including those that involve TLR3 (toll-like receptor 3), RIG-I (retinoic-acid-inducible gene I)-like cytoplasmic receptors, and PKR (41–43).
Following infection with the Cko virus, but not the WT or Vko viruses, substantial IRF-3 activation was observed in ADAR1-sufficient CONkd cells (Fig. 6, top, lane 4), as determined by the appearance of slower migrating IRF-3 species that represent activated IRF-3 and correspond to C-terminal phosphorylation of IRF-3 (41, 44). In the ADAR1kd cells, however, IRF-3 was activated by all three viruses, albeit at different levels (Fig. 6, top, lanes 5–8). Enhanced phosphorylation of PKR upon infection with each of the three viruses, WT, Vko, and Cko, was again observed in the ADAR1kd cells (Fig. 6, second panel, lanes 5–8), similar to the findings shown in Fig. 5. These results indicate that ADAR1 enhances IRF-3 activation as well as PKR activation following MV infection.
FIGURE 6.

IRF-3 phosphorylation is enhanced in ADAR1-deficient cells compared with ADAR1-sufficient cells following infection with WT and Vko MV. CONkd and ADAR1kd cells were infected with WT, Vko, and Cko virus or left uninfected (UI). Whole cell extracts were prepared 24 h after infection and analyzed by Western immunoblot analysis with antibody against IRF-3, phospho-PKR (Thr446) PKR, and β-actin. The brackets indicate the mobility position of non-phosphorylated and C-terminally phosphorylated IRF-3 forms (41, 44).
DISCUSSION
Although biased A-to-I hypermutation in SSPE brains implicates ADAR1 in MV replication (1, 10), the role of ADAR1 in measles virus infection has not been characterized. To test the role of ADAR1 during MV infection, we generated a human cell line stably deficient in ADAR1 that expressed levels of ADAR1 protein 10–15-fold lower than those of the parental cell line that fully supports MV replication. We found that ADAR1 enhances the growth of Vko and Cko virus mutants deleted for the V and C pathogenic factors (Fig. 1). In addition, we observed that ADAR1 protected against apoptosis induced by WT and Vko but not Cko virus (Fig. 2). These results indicate a proviral and antiapoptotic role of ADAR1 during MV infection; in contrast, PKR is antiviral and proapoptotic during MV infection (28). Thus, one enzyme may oppose the other's activity. Indeed, we observed enhanced PKR activation in ADAR1kd compared with ADAR1-sufficient control cells in response to infection with WT and Vko virus.
Enhanced PKR activation in the ADAR1kd cells following infection with WT and Vko virus correlated with enhanced eIF-2α phosphorylation and decreased viral protein expression (Fig. 5). Therefore, PKR-mediated translation inhibition might be responsible for the decreased growth of WT and Vko viruses in the ADAR1kd cells. However, PKR-mediated translation inhibition cannot explain the substantially decreased growth of the Cko virus in the ADAR1kd cells (Fig. 1), since enhanced PKR phosphorylation was not seen in these cells following infection with the Cko virus (Fig. 5). Since C protein has several functions in cells, including control of virus transcription and replication (45, 46) as well as virion assembly and release (47), it is conceivable that ADAR1 affects some other step in the MV replication cycle in addition to protein synthesis.
Our results revealing an antiapoptotic activity for ADAR1 in virus-infected cells are consistent with those obtained with mouse Adar1 gene disruption studies. Adar1−/− mouse embryos die at embryonic day ∼12.5 due to severe apoptosis in the liver and defects in hematopoiesis (48, 49). Moreover, mouse embryo fibroblasts derived from Adar−/− mice are highly prone to serum-deprived apoptosis (49). However, we observed that ADAR1 did not block cell death triggered by TNF-α or staurosporine (Fig. 3), suggesting that ADAR1 is not a general suppressor of apoptosis. Because ADAR1 is a dsRNA-binding protein with dsRNA editing and destabilizing activity (1, 34, 35), it is likely that ADAR1 selectively protects against cell death induced by dsRNA or another virus-specific RNA signal. If this is correct, then what might be the inducer of apoptosis in the hematopoietic system during embryogenesis in the Adar1−/− mouse? It is tempting to speculate that a structured RNA, possibly a microRNA, regulates apoptosis in that system, and ADAR1 impairs the activating activity of this RNA through editing (1, 50).
Upon infection with Cko but not WT measles virus, we observed an ADAR1-related protein product of ∼120 kDa. We also observed this product following infection with E3L deletion vaccinia virus but not WT vaccinia virus (data not shown). Since the Cko virus is highly apoptotic compared with WT measles, and likewise E3L deletion vaccinia virus is highly apoptotic compared with WT vaccinia virus (30), we hypothesized that this ADAR1 species might be a caspase cleavage product of ADAR1 p150. Fully consistent with this idea, the appearance of the 120-kDa species correlated with a decrease in ADAR1 p150 levels and with PARP cleavage (Figs. 2C, 3B, and 4) and furthermore was inhibited by the caspase inhibitor z-VAD-fmk (Fig. 4). The cleavage of ADAR1 by caspases further supports the idea that ADAR1 plays an important role during apoptosis, but the biological relevance of the presumed caspase cleavage product remains entirely unknown.
In addition to PKR activation, we also observed increased IRF-3 activation in the ADAR1-deficient cells following infection with WT and Vko virus (Fig. 6). IRF-3 is a pivotal component of the host antiviral innate response leading to the induction of IFN-β (39, 40). IRF-3 is activated through TLR3 and RLR (RIG-I-like cytosolic receptor) pathways (41–43), which, like PKR, are activated in response to dsRNA. IRF-3 activation, in some cases, is enhanced by PKR (41). Enhanced activation of IRF-3 in ADAR1kd cells, therefore, might indicate that ADAR1 suppresses dsRNA-dependent antiviral pathways, such as TLR3 or RIG pathways. The observation that IRF-3 is enhanced in ADAR1kd cells is intriguing, since enhanced IFN-stimulated gene expression was reported in Adar−/− mice during embryogenesis (51).
The p150 form of ADAR1 is an IFN-inducible enzyme, and since IFNs are cytokines with antiviral activity, our results showing enhanced virus growth and reduced activation of antiviral pathways in the presence of ADAR1 seem paradoxical. However, a proviral role for ADAR1 is not unprecedented, since others have found that ADAR1 increases susceptibility to infection with other viruses, including vesicular stomatitis virus (52), human immunodeficiency virus (53–55), and hepatitis delta virus (56). Together with our findings with MV, ADAR1 thus has a positive role in the replication of RNA viruses of three different genome classes, negative-strand linear RNA (rhabdoviridae, paramyxoviridae), positive-strand linear RNA that undergoes reverse transcription (retroviridae), and the single-strand circular RNA delta virus agent. Furthermore, a prominent role for ADAR1 during MV infection is consistent with the frequent detection of regions of A-to-I hypermutation in MV genomes amplified from brains of SSPE patients (27). It is conceivable that these regions of biased hypermutation in MV are the consequence of routine use of ADAR1 by MV as a replication factor. Presumably, MV not only takes advantage of the ability of ADAR to inhibit antiviral pathways, such as PKR and IRF3, but also has mechanisms to avoid frequent A-to-I modification of its genome RNA by ADAR1 during replication.
In conclusion, we report an antiapoptotic and proviral role for ADAR1 during MV infection, and our results imply that these activities of ADAR1 are achieved through suppression of dsRNA-dependent pathways, such as PKR and IRF-3. Our observations suggest that viruses, including MV, might use ADAR1 as a positive replication factor by selectively exploiting the ability of ADAR to suppress dsRNA-dependent and antiviral pathways.
This work was supported, in whole or in part, by National Institutes of Health, NIAID, Grants AI-12520 and AI-20611 (to C. E. S.) and AI-63476 (to R. C.).
- dsRNA
- double-stranded RNA
- ADAR1kd
- ADAR1 knockdown cells
- Cko
- C knockout measles virus
- CONkd
- control knockdown cells
- eIF-2α
- eukaryotic translation initiation factor-2α
- IFN
- interferon
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H-tetrazolium
- MV
- measles virus
- PARP
- poly(ADP-ribose) polymerase
- PKRkd
- PKR knockdown cells
- shRNA
- short hairpin RNA
- SSPE
- subacute sclerosing panencephalitis
- TCID50
- 50% tissue culture infectious dose
- TNF-α
- tumor necrosis factor-α
- Vko
- V knockout measles virus
- WT
- wild type
- GFP
- green fluorescent protein
- z
- benzyloxycarbonyl
- fmk
- fluoromethylketone.
REFERENCES
- 1.Toth A. M., Zhang P., Das S., George C. X., Samuel C. E. (2006) Prog. Nucleic Acid Res. Mol. Biol. 81, 369–434 [DOI] [PubMed] [Google Scholar]
- 2.Samuel C. E. (2001) Clin. Microbiol. Rev. 14, 778–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bass B. L. (2002) Annu. Rev. Biochem. 71, 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seeburg P. H., Hartner J. (2003) Curr. Opin. Neurobiol. 13, 279–283 [DOI] [PubMed] [Google Scholar]
- 5.Liu Y., Samuel C. E. (1999) J. Biol. Chem. 274, 5070–5077 [DOI] [PubMed] [Google Scholar]
- 6.Liu Y., Emeson R. B., Samuel C. E. (1999) J. Biol. Chem. 274, 18351–18358 [DOI] [PubMed] [Google Scholar]
- 7.Yang W., Chendrimada T. P., Wang Q., Higuchi M., Seeburg P. H., Shiekhattar R., Nishikura K. (2006) Nat. Struct. Mol. Biol. 13, 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawahara Y., Zinshteyn B., Sethupathy P., Iizasa H., Hatzigeorgiou A. G., Nishikura K. (2007) Science 315, 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baczko K., Lampe J., Liebert U. G., Brinckmann U., ter Meulen V., Pardowitz I., Budka H., Cosby S. L., Isserte S., Rima B. K. (1993) Virology 197, 188–195 [DOI] [PubMed] [Google Scholar]
- 10.Cattaneo R., Schmid A., Eschle D., Baczko K., ter Meulen V., Billeter M. A. (1988) Cell 55, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tenoever B. R., Ng S. L., Chua M. A., McWhirter S. M., García-Sastre A., Maniatis T. (2007) Science 315, 1274–1278 [DOI] [PubMed] [Google Scholar]
- 12.Zahn R. C., Schelp I., Utermöhlen O., von Laer D. (2007) J. Virol. 81, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar M., Carmichael G. G. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 3542–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo G. X., Chao M., Hsieh S. Y., Sureau C., Nishikura K., Taylor J. (1990) J. Virol. 64, 1021–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor D. R., Puig M., Darnell M. E., Mihalik K., Feinstone S. M. (2005) J. Virol. 79, 6291–6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y., George C. X., Patterson J. B., Samuel C. E. (1997) J. Biol. Chem. 272, 4419–4428 [DOI] [PubMed] [Google Scholar]
- 17.Weier H. U., George C. X., Greulich K. M., Samuel C. E. (1995) Genomics 30, 372–375 [DOI] [PubMed] [Google Scholar]
- 18.Patterson J. B., Samuel C. E. (1995) Mol. Cell. Biol. 15, 5376–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herbert A., Alfken J., Kim Y. G., Mian I. S., Nishikura K., Rich A. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 8421–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim U., Wang Y., Sanford T., Zeng Y., Nishikura K. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 11457–11461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patterson J. B., Thomis D. C., Hans S. L., Samuel C. E. (1995) Virology 210, 508–511 [DOI] [PubMed] [Google Scholar]
- 22.George C. X., Samuel C. E. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 4621–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poulsen H., Nilsson J., Damgaard C. K., Egebjerg J., Kjems J. (2001) Mol. Cell. Biol. 21, 7862–7871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffin D. (2007) in Fields Virology (Howley P., Griffin D., Lamb R., Martin M., Roizman B., Straus S. eds) pp. 1551–1585, Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 25.Garg R. K. (2002) Postgrad. Med. J. 78, 63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cattaneo R., Schmid A., Spielhofer P., Kaelin K., Baczko K., ter Meulen V., Pardowitz J., Flanagan S., Rima B. K., Udem S. A. (1989) Virology 173, 415–425 [DOI] [PubMed] [Google Scholar]
- 27.Cattaneo R. (1994) Curr. Opin. Genet. Dev. 4, 895–900 [DOI] [PubMed] [Google Scholar]
- 28.Toth A. M., Devaux P., Cattaneo R., Samuel C. E. (2009) J. Virol. 83, 961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang P., Samuel C. E. (2007) J. Virol. 81, 8192–8200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang P., Jacobs B. L., Samuel C. E. (2008) J. Virol. 82, 840–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Devaux P., von Messling V., Songsungthong W., Springfeld C., Cattaneo R. (2007) Virology 360, 72–83 [DOI] [PubMed] [Google Scholar]
- 32.Karber G. (1931) Naunyn Schmiedeberg's Arch. Pharmacol. 162, 480–483 [Google Scholar]
- 33.Navaratnarajah C. K., Leonard V. H., Cattaneo R. (2009) Curr. Top. Microbiol. Immunol. 329, 59–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bass B. L., Weintraub H. (1987) Cell 48, 607–613 [DOI] [PubMed] [Google Scholar]
- 35.Rebagliati M. R., Melton D. A. (1987) Cell 48, 599–605 [DOI] [PubMed] [Google Scholar]
- 36.Ashkenazi A., Dixit V. M. (1998) Science 281, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 37.Rüegg U. T., Burgess G. M. (1989) Trends Pharmacol. Sci. 10, 218–220 [DOI] [PubMed] [Google Scholar]
- 38.Abraham N., Stojdl D. F., Duncan P. I., Méthot N., Ishii T., Dubé M., Vanderhyden B. C., Atkins H. L., Gray D. A., McBurney M. W., Koromilas A. E., Brown E. G., Sonenberg N., Bell J. C. (1999) J. Biol. Chem. 274, 5953–5962 [DOI] [PubMed] [Google Scholar]
- 39.Panne D., Maniatis T., Harrison S. C. (2007) Cell 129, 1111–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hiscott J. (2007) J. Biol. Chem. 282, 15325–15329 [DOI] [PubMed] [Google Scholar]
- 41.Zhang P., Samuel C. E. (2008) J. Biol. Chem. 283, 34580–34587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uematsu S., Akira S. (2007) J. Biol. Chem. 282, 15319–15323 [DOI] [PubMed] [Google Scholar]
- 43.Yoneyama M., Fujita T. (2009) Immunol. Rev. 227, 54–65 [DOI] [PubMed] [Google Scholar]
- 44.Lin R., Heylbroeck C., Pitha P. M., Hiscott J. (1998) Mol. Cell. Biol. 18, 2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bankamp B., Wilson J., Bellini W. J., Rota P. A. (2005) Virology 336, 120–129 [DOI] [PubMed] [Google Scholar]
- 46.Reutter G. L., Cortese-Grogan C., Wilson J., Moyer S. A. (2001) Virology 285, 100–109 [DOI] [PubMed] [Google Scholar]
- 47.Devaux P., Cattaneo R. (2004) J. Virol. 78, 11632–11640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hartner J. C., Schmittwolf C., Kispert A., Müller A. M., Higuchi M., Seeburg P. H. (2004) J. Biol. Chem. 279, 4894–4902 [DOI] [PubMed] [Google Scholar]
- 49.Wang Q., Miyakoda M., Yang W., Khillan J., Stachura D. L., Weiss M. J., Nishikura K. (2004) J. Biol. Chem. 279, 4952–4961 [DOI] [PubMed] [Google Scholar]
- 50.Iizasa H., Nishikura K. (2009) Nat. Immunol. 10, 16–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartner J. C., Walkley C. R., Lu J., Orkin S. H. (2009) Nat. Immunol. 10, 109–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nie Y., Hammond G. L., Yang J. H. (2007) J. Virol. 81, 917–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phuphuakrat A., Kraiwong R., Boonarkart C., Lauhakirti D., Lee T. H., Auewarakul P. (2008) J. Virol. 82, 10864–10872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clerzius G., Gélinas J. F., Daher A., Bonnet M., Meurs E. F., Gatignol A. (2009) J. Virol. 83, 10119–10128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doria M., Neri F., Gallo A., Farace M. G., Michienzi A. (2009) Nucleic Acids Res., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jayan G. C., Casey J. L. (2002) J. Virol. 76, 12399–12404 [DOI] [PMC free article] [PubMed] [Google Scholar]