

Abstract
Tristetraprolin (TTP) is a CCCH zinc finger-containing protein that destabilizes mRNA by binding to an AU-rich element. Mice deficient in TTP develop a severe inflammatory syndrome mainly because of overproduction of tumor necrosis factor α. We report here that TTP also negatively regulates NF-κB signaling at the transcriptional corepressor level, by which it may repress inflammatory gene transcription. TTP expression inhibited NF-κB-dependent transcription. However, overexpression of TTP did not affect reporter mRNA stability. Instead, TTP functioned as a corepressor of p65/NF-κB. In support of this concept, we found that TTP physically interacted with the p65 subunit of NF-κB and was also associated with HDAC1, -3, and -7 in vivo. Treatment with histone deacetylase inhibitors or small interfering RNA induced HDAC1 or HDAC3 knockdown completely or partly abolished the inhibitory activity of TTP on NF-κB reporter activation. Consistently, chromatin immunoprecipitation showed decreased recruitment of HDAC1 and increased recruitment of CREB-binding protein on the Mcp-1 promoter in TTP−/− cells compared with wild-type cells. Moreover, overexpression of TTP blocked CREB-binding protein-induced acetylation of p65/NF-κB. Taken together, these data suggest that TTP may also function in vivo as a modulator in suppressing the transcriptional activity of NF-κB.
The transcription factor NF-κB mediates the major inflammatory signal pathways and regulates the most inflammatory gene expression (1). Excessive and prolonged activation of NF-κB can cause massive damage to host tissue and can result in human inflammatory diseases such as atherosclerosis and arthritis (2). Thus, the activation of NF-κB must be terminated through multiple mechanisms, including recruitment of transcriptional corepressors (3–5).
TTP2 is an RNA-binding protein required for the rapid degradation of mRNAs containing AU-rich elements (6). Targets regulated by TTP include the mRNAs encoding TNFα (7), granulocyte-macrophage colony-stimulating factor (8), and interleukin-2 (9), etc. Mice deficient in TTP develop an inflammatory syndrome characterized by cachexia, spontaneous arthritis, dermatitis, and neutrophilia (10). The inflammatory syndrome in TTP−/− mice is caused mainly by overproduction of TNFα, as neutralizing antibodies reactive with TNFα prevent most of the inflammatory symptoms in TTP−/− mice (10). Overexpression of TNFα in TTP−/− mice may be explained by its prolonged mRNA half-life, but other mechanisms may also exist. Accumulating evidence indicates that TTP may have additional functions besides influencing cytokine mRNA stability. In Schizosaccharomyces pombe, a TTP-related protein is required for effective transmission of a pheromone-induced Ras/mitogen-activated protein kinase (MAPK) signal (11). In addition, a nim/cdc25 mutant can be complemented by either the Cdc2 kinase or a TTP/TIS11 gene, suggesting a cell cycle effect (12). A TTP/TIS11-related protein in Saccharomyces cerevisiae is required for normal metabolism and retards cell growth when overexpressed (13). TTP is induced during apoptosis in response to the breast cancer susceptibility protein BRCA1 (14). Furthermore, continuous expression of TTP at physiological levels causes apoptotic cell death (15, 16). These observations indicate that TTP protein might influence regulatory pathways that regulate survival, differentiation, or proliferation. In a genome-wide analysis of TTP-affected glucocorticoid targets, the half-lives of many TTP target mRNAs were not increased in TTP−/− cells, suggesting a regulatory role for TTP not limited to mRNA turnover (17). In addition, TTP is shuttled between the cytoplasm and nucleus (18). It promotes mRNA decay in the cytoplasm. However, what it does in the nucleus is unknown.
We report here that TTP also negatively regulates NF-κB signaling at the transcriptional corepressor level. It suppresses the transcriptional activity of p65/NF-κB by recruiting HDACs on the NF-κB target gene promoters. These results suggest that TTP may control the inflammatory response through multiple mechanisms, including inhibition of transcription in the nucleus and promotion of mRNA decay in the cytoplasm.
MATERIALS AND METHODS
Cells
Littermate wild-type and TTP−/− day 14.5 embryos were used to generate MEF cell lines 67+/+ and 66−/−, respectively (provided by Dr. Perry J. Blackshear, NIEHS, NIH, Research Triangle Park, NC). Cells were grown as a monolayer in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal bovine serum, 2 mm l-glutamine, and 100 units/ml each penicillin and streptomycin. The mouse macrophage cell line RAW264.7 and HEK293 cells were cultured as described previously (19).
Plasmids
The TNFα-Luc reporter construct was kindly provided by Dr. Dmitry V. Kuprash (Russian Academy of Science) and was described previously (20). NF-κB-TK-Luc was purchased from Stratagene (La Jolla, CA). The pGL3-Control vector was from Promega. HA-tagged TTP and TTP-C124R expression plasmids were kindly provided by Dr. Perry J. Blackshear. The pGal4-p65-(270–591) plasmid was kindly provided by Dr. Brian P. Ashburner (University of Toledo). Gal4-TK-Luc and pcDNA-p65 were described previously (19). pGST-p65-(1–305), pGST-p65-(245–355), and pGST-p65-(345–551) were gifts from Dr. David R. Jones (University of Virginia). FLAG-HDAC1, FLAG-HDAC2, FLAG-HDAC3, and FLAG-HDAC7 were kindly provided by Dr. Ronald M. Evans (Salk Institute). CMX-CBP and CMX-SMRT expression plasmids were provided by the laboratory of Dr. Mangelsdorf. CMV-FLAG-KNP1 was generated in this laboratory.
Reagents
Antibodies against phospho-IKKβ (Ser180), phospho-IκBα (Ser32), acetyl-p65, HDAC1, HDAC3, CBP, and HA tag were purchased from Cell Signaling Technology Inc. Anti-p65/NF-κB and anti-TTP (G-20) antibodies and siRNAs for TTP, HDAC1, and HDAC3 were from Santa Cruz Biotechnology. Anti-FLAG tag antibody, an anti-HA immunoprecipitation kit, LPS (Escherichia coli 026:B6-derived), human recombinant interferon-γ, IL-1β, and TNFα were purchased from Sigma. Anti-SMRT antibody was purchased from Abcam. Glutathione-Sepharose 4B was purchased from GE Healthcare. Apicidin was purchased from BIOMOL.
Transfection
Transient transfection and luciferase assay were performed as described previously (19).
Protein Isolation and Western Blotting
Protein isolation and Western blotting were essentially performed as described previously (19).
Co-immunoprecipitation
Co-immunoprecipitation assays were performed by using an anti-HA immunoprecipitation kit or a protein G immunoprecipitation kit (Sigma) following the manufacturer's instruction. Briefly, subconfluent HEK293 cells on a 10-cm plate were transfected with 10 μg of the indicated combinations of expression vectors by the calcium phosphate method. After 24 h, the transfected cells were left untreated or treated with TNFα for 15 min. Cells were lysed in CelLytic M cell lysis buffer with protease inhibitors and phosphatase inhibitors. Cell lysates were incubated with anti-HA beads overnight at 4 °C. After extensive washing, immunoprecipitated proteins were resolved by 6–10% SDS-PAGE and analyzed by Western blotting with anti-HA or anti-p65 antibodies. Membranes were developed with enhanced chemiluminescence (Amersham Biosciences). For endogenous immunoprecipitations, RAW264.7 cells were treated with LPS (1 μg/ml) for 0, 2, 4, and 8 h. The cell lysates were incubated with anti-TTP polyclonal antibody or rabbit anti-IgG as a control. The immune complexes were collected by incubation (2 h, 4 °C) with protein G-agarose (Sigma). Western blotting for detection of endogenous TTP, p65, and HDAC1 was performed with anti-TTP, anti-p65, and anti-HDAC1 polyclonal antibodies, respectively.
GST Fusion Protein Pulldown Assay
Plasmids pGST, pGST-p65-(1–305), pGST-p65-(245–355), and pGST-p65-(345–551) were transformed into E. coli BL21 cells by the CaCl2 method. At A600 = 0.8, the cells were induced with isopropyl β-d-thiogalactopyranoside (0.2 mm final concentration) at 30 °C for 3 h and then lysed in buffer containing 25 mm Tris-HCl (pH 7.4), 150 mm NaCl, 10% glycerol, 1 mm dithiothreitol, protease inhibitor mixture (Complete Mini, Roche Applied Science), and 0.05% Triton X-100, followed by three 10-s pulses of sonication. Glutathione-Sepharose CL-4B beads (GE Healthcare) were incubated with the lysates for 1 h. The beads containing the bound GST-p65 fusion protein or the bound GST protein were pelleted and extensively washed with lysis buffer. Protein contents were quantified against known amounts of bovine serum albumin on Coomassie Brilliant Blue-stained SDS-polyacrylamide gels. Equal quantities of GST-p65 fusion protein or GST protein bound to the glutathione-Sepharose CL-4B beads were separately added to the precleared TTP-HA cell lysates and incubated overnight at 4 °C. The bound proteins from the TTP-HA-expressing cell lysates (pulldown assays) were pelleted with the beads at 2000 rpm in a microcentrifuge, washed, and separated on SDS-polyacrylamide gels. The separated proteins were then analyzed by Western blotting with anti-HA antibody to detect the pulled-down TTP-HA, followed by Coomassie Brilliant Blue staining to confirm the presence of GST protein or GST-p65 fusion protein in samples as appropriate.
Chromatin Immunoprecipitation Assay
ChIP assay was performed using the ChIP assay kit (Upstate Biotechnology) according to the manufacturer's instructions. The immunoprecipitation-enriched DNA was quantified by real-time PCR, and the values were normalized by the input DNA. The primers used to amplify the Mcp-1 promoter were 5′-CACCCCATTACATCTCTTCCCC-3′ and 5′-TGTTTCCCTCTCACTTCACTCTGTC-3′.
Statistics
Data are expressed as means ± S.D. For comparison between two groups, the unpaired Student's test was used. For multiple comparisons, analysis of variance followed by the unpaired Student's test was used. A p value of <0.05 was considered significant.
RESULTS
TTP Expression Inhibits TNFα Promoter Activity
TTP is the prototype member of a CCCH zinc finger protein family, which down-regulates expression of many inflammatory genes by destabilizing their mRNAs. Recently, we identified the novel CCCH zinc finger protein MCPIP1 (MCP-1-induced protein 1), which down-regulates cytokine-induced TNFα expression by repressing its mRNA transcription but does not affect its mRNA stability (19). In luciferase reporter experiments, TTP was transfected to serve as a control. Surprisingly, we found that overexpression of TTP strongly inhibited mouse TNFα promoter activity in a dose-dependent manner in both HEK293 and RAW264.7 cell lines (Fig. 1, A and B). Although TTP can mediate mRNA stability, this is unlikely to explain the results in Fig. 1 (A and B) because the mRNA transcribed from the reporter construct lacked the AU-rich element associated with TTP-mediated mRNA destabilization. Nevertheless, to exclude this possibility, we determined the half-life of luciferase mRNA under the influence of TTP. To accomplish this, HEK293 cells were transfected with the luciferase reporter driven by the TNFα promoter with or without the TTP plasmid and then stimulated with IL-1β 24 h after transfection. After 8 h of stimulation, cells were treated with actinomycin D (10 μg/ml), and RNA was harvested at serial time points as indicated. Luciferase mRNA extracted from these samples was quantified by Northern blotting. As shown in Fig. 1 (C and D), TTP coexpression did not influence the half-life of luciferase mRNA. These results indicate that TTP may regulate TNFα expression at the level of transcription.
FIGURE 1.

TTP expression inhibits TNFα promoter activity. A and B, HEK293 or RAW264.7 cells were cotransfected with a luciferase reporter plasmid under the transcriptional control of the mouse TNFα promoter and increasing amounts of TTP-HA (0, 25, 100, and 400 ng/well). After being quiescent for 24 h, cells were treated with or without IL-1β (10 ng/ml) or LPS (0.1 μg/ml) as shown and collected for analysis of reporter gene activity 24 h later. Data represent means ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01 versus IL-1β stimulation without the TTP-HA-transfected group. The gels under the graph in A are the protein levels of TTP-HA and actin in the transfected cell lysates detected by Western blotting. C, HEK293 cells were transiently transfected with the pcDNA3.1 or pTTP-HA expression construct. After 24 h, the cells were quiescent for 18 h and then treated with IL-1β (10 ng/ml) for 8 h, and actinomycin D (10 μg/ml) was added to stop transcription. RNA was harvested after different time points as indicated. The luciferase (Luc.) mRNA level was examined by Northern blotting. D, the intensity of the bands in B was quantified using an AlphaImager 2200 system (Alpha Innotech). The value of the 0 point was set as 100%, and the values of the other points were transformed according to the 0 point. The data from three independent experiments were averaged and plotted.
TTP Inhibits NF-κB-dependent Gene Activation
TNFα production is coordinately regulated by NF-κB-, AP-1 (activator protein 1)-, and STAT-mediated signal pathways (20). To further understand how TTP represses TNFα transcription, we tested whether TTP affects the activation of NF-κB-, AP-1-, and STAT-mediated signaling by luciferase assays. As shown in Fig. 2A, TTP overexpression significantly inhibited the basal and stimulus-induced NF-κB activation in a concentration-dependent manner. TTP did not affect the basal reporter activity of STAT, but a high level of TTP transfection did reduce interferon-γ- and LPS-induced STAT reporter activation as well as phorbol 12-myristate 13-acetate-induced AP-1 reporter activation (data not shown). As it was noted previously that TTP overexpression leads to nonspecific “squelching” of reporter constructs (7), we tested the effect of TTP on an SV40-driven control reporter. The same concentrations of the TTP plasmid did not affect SV40 promoter-mediated gene expression. To further confirm that the suppressive effect of TTP on the NF-κB reporter is specific rather than caused by “promoter squelching,” we tested the effect of the unrelated protein FLAG-KNP1 on the NF-κB reporter. As shown in Fig. 2C, expression of TTP significantly inhibited TNFα-induced NF-κB activation. However, expression of KNP1 did not affect NF-κB reporter activity at all. Expression of both TTP and FLAG-KNP1 was controlled by the cytomegalovirus promoter. Their expression in the transfected cells is shown in Fig. 2C (lower panels). Interestingly, TTP-C124R, the first zinc finger mutant of TTP, also significantly repressed TNFα-induced NF-κB activation but was less potent compared with the wild-type TTP protein (Fig. 2C).
FIGURE 2.

TTP represses NF-κB-dependent gene expression. A and B, HEK293 cells were cotransfected with the NF-κB-TK-Luc (A) or pGL3-Control (B) vector as indicated with increasing amounts of pTTP-HA (0, 25, 100, and 400 ng/well) by the calcium phosphate method. After being quiescent for 16 h, cells were treated with or without TNFα (10 ng/ml) (A) and collected for analysis of reporter gene activity 24 h later. *, p < 0.01 versus stimulation without the TTP-HA-transfected group. C, HEK293 cells were cotransfected with NF-κB-TK-Luc with or without expression plasmids as indicated and subjected to the same treatment as described above. The proper protein expression is shown in the lower panels. D, NF-κB-Luc was transfected into TTP+/+ or TTP−/− MEFs by FuGENE 6. 24 h later, the cells were stimulated with LPS (0.1 μg/ml) or IL-1β (10 ng/ml) for another 24 h. The luciferase (Luc.) activity was measured and normalized by β-galactosidase activity. The number on each bar represents -fold over the untreated group in TTP+/+ cells. E, scrambled control (siControl) or TTP (siTTP) siRNA was cotransfected with NF-κB-Luc into RAW264.7 cells by electroporation (Amaxa). 24 h later, the cells were quiescent for 12 h and then stimulated with LPS (0.1 μg/ml) for another 24 h. The luciferase activity was measured and normalized by β-galactosidase activity. The TTP and β-actin protein levels in the transfected cells were analyzed by Western blotting and are shown in the lower panels.
To further explore the role of endogenous TTP in NF-κB regulation, we transfected the NF-κB reporter into TTP+/+ and TTP−/− MEFs. 24 h later, the cells were stimulated with LPS (0.1 μg/ml) or IL-1β (10 ng/ml) for 24 h. The basal reporter activity was increased by 7-fold in TTP−/− MEFs compared with TTP+/+ MEFs. LPS and IL-1β induced an ∼2-fold increase in reporter activity in both TTP+/+ and TTP−/− MEFs (Fig. 2D). In addition, we observed the effect of knockdown of TTP on NF-κB-dependent gene expression by using siRNA. These experiments were performed in RAW264.7 cells transiently cotransfected with the NF-κB reporter and siRNA for TTP or the scrambled control and stimulated with LPS (0.1 μg/ml) for 24 h. As shown in Fig. 2E, LPS induced NF-κB reporter activity by 6.3-fold in RAW264.7 cells transfected with scrambled control siRNA. The induction of NF-κB reporter activity was further increased by 11.2-fold in cells transfected with TTP siRNA. Western blotting demonstrated that the siRNA effectively reduced TTP protein levels by >60% relative to the control, whereas the internal control β-actin was not affected. Taken together, these results suggest that TTP feedback controls LPS-induced NF-κB activation.
TTP Represses p65/NF-κB Transactivation
To further examine how TTP regulates NF-κB signaling, we investigated the effect of TTP on IKKβ and IκBα phosphorylation. The transfected HEK293 cells were quiescent and then stimulated with TNFα for various times as indicated. The whole cell lysates were subjected to Western blot analysis using anti-phospho-IKKβ and anti-phospho-IκBα antibodies. As shown in Fig. 3A, there were no differences between TTP-overexpressing and normal cells in the extent of TNFα-induced IKKβ and IκBα phosphorylation and degradation. In addition, the overall protein concentration of NF-κB p65 was not affected by overexpression of TTP.
FIGURE 3.
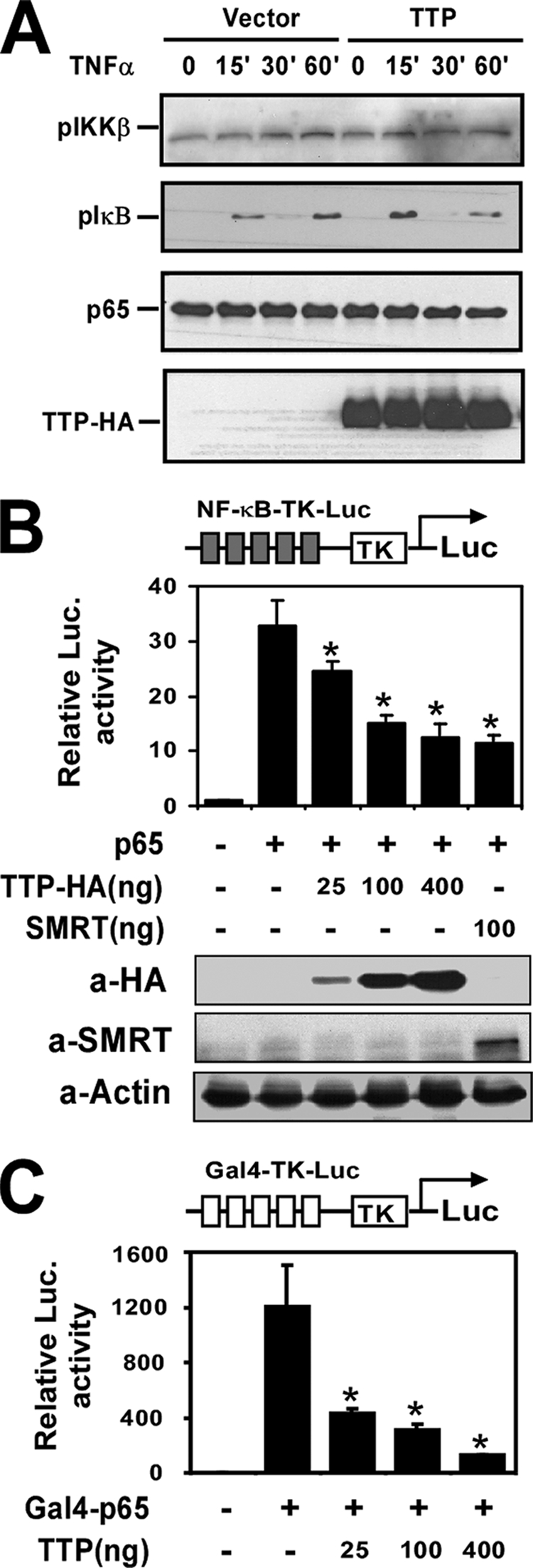
TTP represses transactivation of p65. A, HEK293 cells were transfected with pcDNA3.1 or pTTP-HA. After 24 h, the transfected cells were quiescent for 16 h and then stimulated with TNFα (10 ng/ml) for the indicated times. Whole cell extracts were isolated and subjected to Western blot analysis using anti-phospho-IKKβ, anti-phospho-IκBα, anti-p65, or anti-HA antibody as indicated. B and C, HEK293 cells were transfected with NF-κB-TK-Luc, p65, and increasing amounts of pTTP-HA as indicated or SMRT (B) or with Gal4-TK-Luc with an expression construct encoding the Gal4-p65(TAD) fusion protein and increasing amounts of pTTP-HA (C) as indicated. 36 h later, the cells were collected for analysis of reporter gene activity and protein expression. *, p < 0.01 versus stimulation without the TTP-HA-transfected group. Luc., luciferase.
To determine whether TTP represses p65/NF-κB transactivation activity, NF-κB-TK-Luc was cotransfected with the expression construct p65 and increasing amounts of HA-TTP or SMRT (a well known corepressor of NF-κB and other transcription factors) into HEK293 cells. As shown in Fig. 3B, expression of p65 activated NF-κB-dependent reporter expression by 32.8-fold. Expression of TTP repressed this activation in a concentration-dependent manner. Used as a positive control, SMRT expression also markedly repressed p65-induced NF-κB reporter activation. Overexpression of TTP and SMRT in the transfected cells is shown in Fig. 3B (lower panels).
p65/NF-κB contains a TAD and a Rel homology domain. To further confirm that TTP affects the transactivation activity but not activation and DNA binding of p65, we cotransfected the Gal4 reporter containing five copies of the Gal4-binding site (Gal4-TK-Luc) with the Gal4-p65(TAD) fusion construct and increasing amounts of TTP-HA. As shown in Fig. 3C, TTP also dramatically suppressed Gal4-p65(TAD)-activated reporter expression. These results suggest that TTP may directly repress the p65/NF-κB transactivation domain.
TTP Physically Interacts with p65
Next, we investigated whether TTP physically interacts with the p65 subunit of NF-κB. HEK293 cells were transfected with p65 and pTTP-HA expression plasmids. 24 h post-transfection, the cells were untreated or treated with TNFα for 30 min. Protein extracts were prepared and subjected to immunoprecipitation with anti-HA beads followed by immunoblotting with both anti-p65 and anti-HA antibodies. As shown in Fig. 4A, TTP physically interacted with p65 in untreated cells. TNFα treatment reduced TTP binding to p65. To further test whether endogenous TTP protein is also associated with endogenous p65, RAW264.7 cells were treated with LPS (0.1 μg/ml) for 0, 2, 4, and 8 h. The cell lysates were immunoprecipitated by anti-TTP antibody followed by protein G purification. Both whole cell extracts and immunoprecipitates were subjected to Western blot analysis with anti-TTP and anti-p65 antibodies. As shown in Fig. 4B, endogenous TTP associated with p65 peaked at 4 h after LPS stimulation but declined at 8 h after LPS stimulation. Consistent with previous reports, TTP protein was significantly induced by LPS (22). Taken together, these results suggest that TTP can physically interact with p65, which is dynamically regulated by TNFα and LPS.
FIGURE 4.

TTP physically interacts with p65. A, HEK293 cells were transiently cotransfected with p65 with or without the pTTP-HA expression plasmid. The transfected cells were stimulated with or without TNFα for 30 min. The whole cell lysates (WCE) were subjected to co-immunoprecipitation (IP) with anti-HA antibody followed by Western blotting with anti-p65 or anti-HA antibody. The same lysates were subjected to SDS-PAGE and probed with anti-p65 or anti-HA antibody for proper protein expression. B, RAW264.7 cells were treated with LPS (0.1 μg/ml) for 0, 2, 4, and 8 h. The cell extracts were subjected to immunoprecipitation with anti-TTP antibody. Both whole cell extracts and immunoprecipitates were analyzed by Western blotting with anti-TTP and p65 antibodies. C, HEK293 cells were transfected with the pTTP-HA plasmid. After 24 h of transfection, the cells were treated with TNFα for 30 min. Aliquots of cell lysates were subjected to purification with glutathione-Sepharose beads already bound to GST, GST-p65-(1–305), GST-p65-(245–355), or GST-p65-(345–551). Proteins co-precipitated with GST beads were analyzed by Western blotting with anti-HA antibody. Whole cell extracts were also analyzed by Western blot analysis with anti-HA antibody to monitor the expression of transfected DNA. Coomassie (Comas.) Brilliant Blue stain showed the expression of GST and GST fusion proteins.
To examine the TTP interaction region of p65, HEK293 lysates overexpressing TTP-HA were incubated with GST, GST-p65-(1–305), GST-p65-(245–355), or GST-p65-(345–551); washed; and separated on SDS-polyacrylamide gels. The separated proteins were then analyzed by Western blotting with anti-HA antibody to detect the pulled-down TTP-HA followed by Coomassie Brilliant Blue staining to confirm the presence of GST protein or GST-p65 fusion protein in samples as appropriate. As shown in Fig. 4C, TTP was found to interact with GST-p65-(245–355) and GST-p65-(345–551) but not GST-p65-(1–305). Thus, the C-terminal region (positions 345–355) of p65 may be responsible for binding to TTP.
Involvement of HDACs in the TTP-mediated Inhibitory Effect on NF-κB
As documented, the transactivation function of NF-κB is regulated through interaction with HDAC corepressor proteins (23). By analogy, TTP may also associate with HDACs. To test this possibility, FLAG-tagged HDAC1, HDAC2, HDAC3, and HDAC7 were cotransfected with HA-tagged TTP into HEK293 cells. Cell lysates of the transfected cells were immunoprecipitated with HA-specific antibody and subjected to Western blot analysis for HDACs and TTP. As shown in Fig. 5A, HDAC1, HDAC3, and HDAC7 were readily co-immunoprecipitated with TTP. HDAC2 was not found to be immunoprecipitated with TTP, which may be due to the low expression of HDAC2 in whole cell extracts (Fig. 1A). The interaction of endogenous HDACs with endogenous TTP was also determined by co-immunoprecipitation. As shown in Fig. 5B, HDAC1 was specifically precipitated by anti-TTP antibody but not by IgG or anti-SRA antibody in LPS-treated RAW264.7 cell extracts.
FIGURE 5.

Involvement of HDACs in the TTP-mediated inhibitory effect on NF-κB. A, HEK293 cells were cotransfected with pTTP-HA and the FLAG-HDAC1, FLAG-HDAC2, FLAG-HDAC3, or FLAG-HDAC7 expression plasmid. After 24 h of transfection, the whole cell lysates (WCE) were subjected to co-immunoprecipitation (IP) with anti-HA antibody followed by Western blotting (immunoblotting (IB)) with anti-FLAG or anti-HA antibody as indicated. The same lysates were subjected to SDS-PAGE and probed with anti-FLAG or anti-HA antibody for proper protein expression. B, RAW264.7 cells were treated with LPS for 8 h. The cell extracts were subjected to immunoprecipitation by anti-TTP antibody, no specific IgG, or an unrelated antibody followed by Western blot analysis with anti-TTP or HDAC1 antibody. The HDAC1 protein level in whole cell extracts was also probed by anti-HDAC1 antibody. C, HEK293 cells were cotransfected with NF-κB-TK-Luc and TTP-HA or the empty vector as indicated. The transfected cells were treated with or without HDAC inhibitor (apicidin at 0.1, 0.2, and 0.5 μm) for 2 h and then stimulated with TNFα (10 ng/ml) for 24 h. Cells were collected for analysis of reporter gene activity. The data are from three independent experiments (means ± S.D.). *, p < 0.01 versus TTP-HA-transfected without the apicidin treatment group. D, HEK293 cells were transfected with scrambled control siRNA (siControl), HDAC1 siRNA (siHDAC1), or HDAC3 siRNA (siHDAC3) together with NF-κB-Luc and the TTP-HA plasmid as indicated. 24 h later, the cells were quiescent for 12 h and then stimulated with TNFα for 24 h. The reporter activity was analyzed. The HDAC1 and HDAC3 protein levels in the cell lysates were detected by Western blot analysis. *, p < 0.01 versus the scrambled control siRNA- and TTP-HA-transfected group. E, HEK293 cells were cotransfected with p65, CBP, and pTTP-HA as indicated. Cell lysates were extracted and analyzed by Western blotting with anti-acetyl-p65, anti-p65, and anti-HA antibodies.
To determine whether TTP represses the transcriptional activity of NF-κB through HDACs, apicidin, a potent tetrapeptide HDAC inhibitor, was used to treat the transfected HEK293 cells. As shown in Fig. 5C, TNFα induced NF-κB promoter activation by 23-fold. Overexpression of TTP repressed its activity to 7.5-fold. Interestingly, the inhibitory activity of TTP on NF-κB-mediated gene activation was completely abolished in the presence of 500 nm apicidin. Thus, the inhibitory activity of TTP on NF-κB is mediated by HDAC activity. To further test whether endogenous HDAC1 and HDAC3 mediate the inhibitory effect of TTP on NF-κB, HEK293 cells were cotransfected with control, HDAC1, or HDAC3 siRNA and TTP-HA and the NF-κB reporter. 24 h after transfection, the cells were treated with TNFα for another 24 h. As shown in Fig. 5D, siRNA-mediated knockdown of HDAC1 or HDAC3 partly rescued the effect of TTP on NF-κB reporter activation. However, knockdown of both HDAC1 and HDAC3 completely rescued the effect of TTP on NF-κB activation, suggesting that both HDAC1 and HDAC3 are involved in the TTP-mediated inhibitory effect on NF-κB. Western blotting demonstrated that the specific siRNAs effectively reduced the protein expression of HDAC1 and HDAC3 by >80% relative to control siRNA (Fig. 5D, lower panel).
It is well known that acetylation of p65 by CBP/p300 is associated with increased transcription without altering its binding to DNA (24). To further explore how TTP represses p65 transactivation activity, we observed the effect of TTP expression on CBP-induced acetylation of p65. As shown in Fig. 5E, cotransfection CBP induced p65 acetylation. Overexpression of TTP completely abolished CBP-induced acetylation of p65. The results further support that TTP may function as a transcriptional corepressor of p65 by recruiting HDACs and diminishing p65 acetylation.
Absence of TTP Decreases HDAC1 but Increases CBP Recruitment to the Mcp-1 Promoter
It has been reported that HDACs and CBP are dynamically recruited to the NF-κB target promoter to precisely control gene expression. To further examine the influence of endogenous TTP on the recruitment of HDAC1 and CBP to the Mcp-1 promoter, we performed ChIP assay in TNFα-treated TTP+/+ and TTP−/− MEFs with anti-HDAC1 and anti-CBP antibodies (Fig. 6). Consistent with the results above, HDAC1 recruitment was significantly decreased in TTP−/− MEFs compared with TTP+/+ MEFs. In contrast, CBP recruitment was increased in TTP−/− MEFs. As noted, the absence of TTP did not affect the binding of p65 to the Mcp-1 promoter. TTP recruitment on the Mcp-1 promoter was induced by TNFα in TTP+/+ cells. As expected, no TTP recruitment was detected in TTP−/− cells.
FIGURE 6.
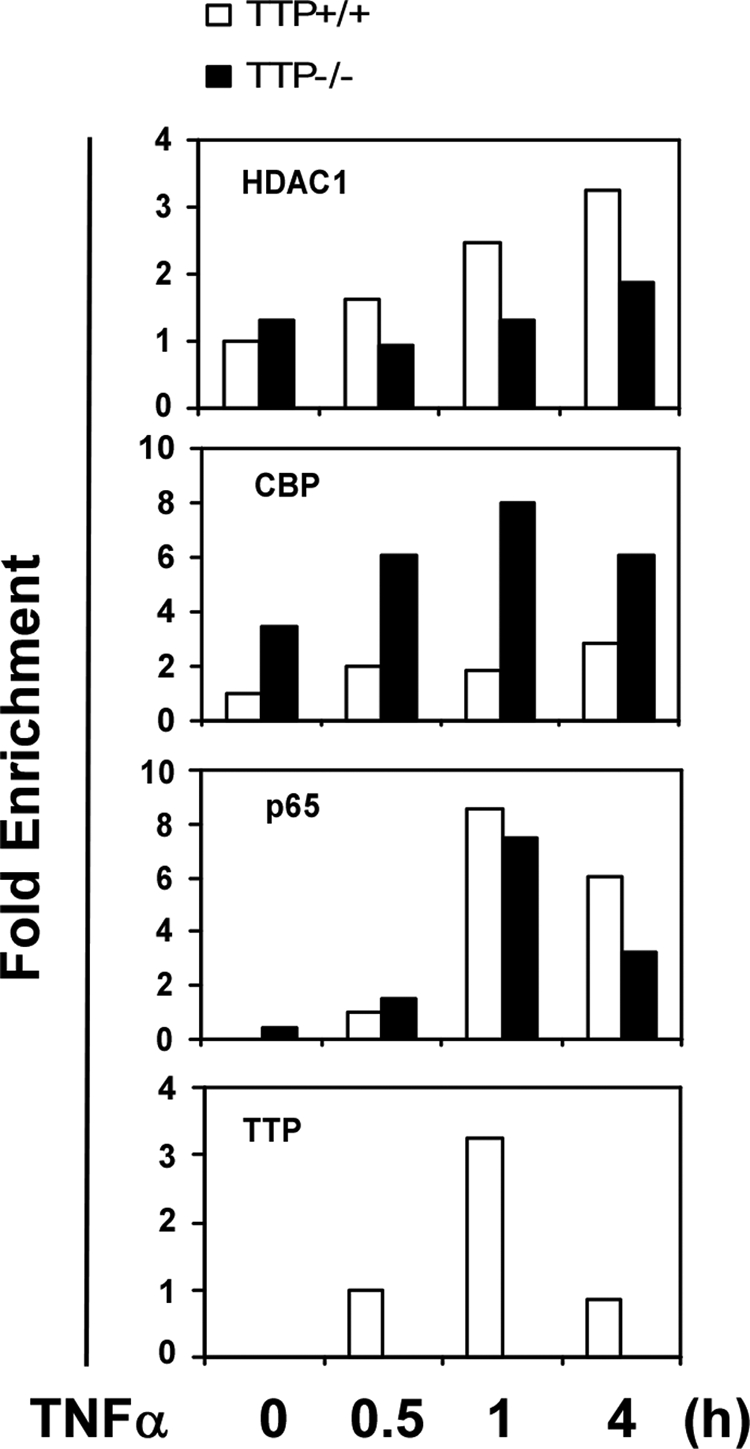
ChIP analysis of the Mcp-1 promoter in TTP−/− MEFs. ChIP assays were performed with MEFs from wild-type (+/+) or TTP null (−/−) littermates untreated or treated with TNFα (10 ng/ml). Protein-DNA complexes were immunoprecipitated with the indicated antibodies, and immunoprecipitated DNA was analyzed by quantitative PCR with specific primers against the IκB site in the murine Mcp-1 promoter. The values were normalized by input DNA. The experiment was independently repeated twice with similar results.
DISCUSSION
TTP is a tandem CCCH zinc finger protein that was characterized as a destabilizing factor of inflammatory mRNAs (6). In this work, we have provided evidence that TTP also negatively regulates NF-κB signaling in an HDAC-dependent manner. Using reporter assays, we have shown that TTP inhibits NF-κB-mediated gene activation and that such an inhibitory activity of TTP can be overcome by suppressing HDAC activity or expression. By in vivo co-immunoprecipitation and GST pulldown, TTP was found to physically interact with the p65 subunit of NF-κB and to associate with HDAC1, HDAC3, and HDAC7. Consistently, TTP did not interfere with NF-κB signaling activation. Instead, TTP diminished p65 acetylation. Furthermore, knockdown of TTP enhanced LPS-induced NF-κB reporter activation, which is consistent with increased CBP recruitment and decreased HDAC1 recruitment on the NF-κB target promoter in TTP−/− cells. Combined with many other reports, these results suggest that TTP may critically regulate inflammatory gene expression through multiple mechanisms, including mRNA transcription and decay. However, the precise mechanisms by which TTP represses mRNA transcription are not completely understood. Our studies suggest that TTP exerts this effect at least partly through recruitment of HDACs to the target promoter and diminishment of p65 acetylation. However, other mechanisms may also exist.
Consistent with previously reports, there is low level of TTP protein in resting RAW264.7 cells. LPS treatment caused a marked increase in TTP protein. However, the pattern of p65 co-immunoprecipitation with TTP did not follow the pattern of TTP accumulation (Fig. 4B). It has been reported that LPS regulates TTP at both the transcriptional and post-transcriptional levels. LPS-activated signaling pathways can dramatically induce TTP expression, which could also induce phosphorylation of TTP (25). It was observed previously that p38-mediated phosphorylation of TTP can shuttle TTP from the nucleus to the cytoplasm and inhibit TTP binding to mRNA (26, 27). Our results suggest that LPS stimulates the binding of TTP to p65 at an early time point (4 h) by induction of TTP expression and prevents TTP interaction with p65 at a late time point (8 h) by phosphorylation. This may also explain why TNFα treatment reduced TTP interaction with p65 in the overexpressing cell line (Fig. 4A). Interestingly, Zhu et al. (28) also observed that p38-mediated TTP phosphorylation can release the suppressive effect of TTP on TNFα transcription.
Our results suggest that TTP may also negatively regulate AP-1- and STAT-mediated signaling pathways, as overexpression of TTP also repressed AP-1 and STAT reporter activity (data not shown). However, there is no direct association of TTP with c-Fos/c-Jun or STAT1, suggesting that TTP may repress AP-1- and STAT-mediated signaling pathways through different mechanisms.
One may ask whether the inhibitory effect of TTP on NF-κB signaling is dependent on its RNA-destabilizing activity. We observed that the first zinc finger mutant of TTP also significantly suppressed NF-κB activation with less potency compared with the wild-type TTP protein. Because any zinc finger mutation of TTP protein completely loses its RNA-binding activity (29), our data suggest that the effect of TTP on NF-κB signaling may be independent of its RNA-destabilizing activity but that its two zinc fingers may be required for transcription repression.
As reported previously, expression of TTP not only causes apoptotic cell death but also sensitizes cells to induction of apoptosis by TNFα (15, 16). However, the molecular mechanisms underlying these events are not clear. It was well established that maturing activated monocytes face a choice between NF-κB-dependent survival and differentiation and death by TNFα-induced apoptosis. Our results suggest that overexpression of TTP may promote TNFα-induced apoptosis through repression of NF-κB signaling.
At least 55 CCCH zinc finger genes exist in the human genome (30). Although most of the CCCH proteins have been less characterized, most of the characterized CCCH proteins are RNA-binding proteins and regulate RNA metabolism, including mRNA maturation, export, modification, and turnover (30). However, there are also some CCCH proteins that regulate mRNA transcription. For instance, PIE-1 is a Caenorhabditis elegans protein that contains two central CCCH zinc fingers. PIE-1 can function as a transcription repressor by targeting to the basic transcriptional machinery (31). Recently, we identified the novel CCCH protein MCPIP1, which also represses TNFα transcription but does not affect its mRNA stability (19). Actually, TTP was also reported to interfere with the transcriptional activity of Tax by directly interacting with Tax protein (32). Zhu et al. (28) also observed that TTP can inhibit TNFα and IL-8 promoter activity. Considering that TTP is a nucleocytoplasmic shuttling protein, these results strongly indicate that TTP is a candidate to couple transcription in the nucleus and mRNA decay in the cytoplasm. In yeast, the RNA-binding protein Rpb4/7 couples the two processes of transcription in the nucleus and mRNA decay in the cytoplasm (21).
In summary, we report that TTP, an RNA-destabilizing protein, also functions as a transcriptional corepressor of NF-κB. These results suggest that TTP may critically control the inflammatory response by multiple mechanisms and is a potential therapeutic target for human inflammatory diseases.
Acknowledgments
We thank Drs. Perry J. Blackshear and Wi S. Lai for providing the wild-type and TTP knock-out MEFs and the HA-tagged TTP and TTP-C124R plasmids and for critical reading of this manuscript and helpful comments. We also thank Drs. Brian P. Ashburner, David R. Jones, Dmitry V. Kuprash, and Ronald M. Evans for providing plasmids used in this work.
This work was supported by a James and Esther King New Investigator research grant and an American Heart Association beginning grant-in-aid (to M. F.).
- TTP
- tristetraprolin
- TNFα
- tumor necrosis factor α
- HDAC
- histone deacetylase
- MEF
- mouse embryonic fibroblast
- HA
- hemagglutinin
- CBP
- cAMP-responsive element-binding protein-binding protein
- IKKβ
- IκB kinase β
- siRNA
- small interfering RNA
- LPS
- lipopolysaccharide
- IL-1β
- interleukin-1β
- GST
- glutathione S-transferase
- ChIP
- chromatin immunoprecipitation
- STAT
- signal transducer and activator of transcription
- TAD
- transcriptional activation domain.
REFERENCES
- 1.O'Neill L. A. (2006) Nat. Rev. Drug Discov. 5, 549–563 [DOI] [PubMed] [Google Scholar]
- 2.Pfitzner E., Kliem S., Baus D., Litterst C. M. (2004) Curr. Pharm. Des. 10, 2839–2850 [DOI] [PubMed] [Google Scholar]
- 3.Natoli G., Chiocca S. (2008) Sci. Signal. 1, pe1. [DOI] [PubMed] [Google Scholar]
- 4.Ogawa S., Lozach J., Jepsen K., Sawka-Verhelle D., Perissi V., Sasik R., Rose D. W., Johnson R. S., Rosenfeld M. G., Glass C. K. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 14461–14466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wormald S., Hilton D. J. (2004) J. Biol. Chem. 279, 821–824 [DOI] [PubMed] [Google Scholar]
- 6.Carrick D. M., Lai W. S., Blackshear P. J. (2004) Arthritis Res. Ther. 6, 248–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carballo E., Lai W. S., Blackshear P. J. (1998) Science 281, 1001–1005 [DOI] [PubMed] [Google Scholar]
- 8.Carballo E., Lai W. S., Blackshear P. J. (2000) Blood 95, 1891–1899 [PubMed] [Google Scholar]
- 9.Ogilvie R. L., Abelson M., Hau H. H., Vlasova I., Blackshear P. J., Bohjanen P. R. (2005) J. Immunol. 174, 953–961 [DOI] [PubMed] [Google Scholar]
- 10.Taylor G. A., Carballo E., Lee D. M., Lai W. S., Thompson M. J., Patel D. D., Schenkman D. I., Gilkeson G. S., Broxmeyer H. E., Haynes B. F., Blackshear P. J. (1996) Immunity 4, 445–454 [DOI] [PubMed] [Google Scholar]
- 11.Kanoh J., Sugimoto A., Yamamoto M. (1995) Mol. Biol. Cell 6, 1185–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warbrick E., Glover D. (1994) Gene 151, 243–246 [DOI] [PubMed] [Google Scholar]
- 13.Thompson M. J., Lai W. S., Taylor G. A., Blackshear P. J. (1996) Gene 174, 225–233 [DOI] [PubMed] [Google Scholar]
- 14.Harkin D. P., Bean J. M., Miklos D., Song Y. H., Truong V. B., Englert C., Christians F. C., Ellisen L. W., Maheswaran S., Oliner J. D., Haber D. A. (1999) Cell 97, 575–586 [DOI] [PubMed] [Google Scholar]
- 15.Johnson B. A., Geha M., Blackwell T. K. (2000) Oncogene 19, 1657–1664 [DOI] [PubMed] [Google Scholar]
- 16.Johnson B. A., Blackwell T. K. (2002) Oncogene 21, 4237–4246 [DOI] [PubMed] [Google Scholar]
- 17.Ishmael F. T., Fang X., Galdiero M. R., Atasoy U., Rigby W. F., Gorospe M., Cheadle C., Stellato C. (2008) J. Immunol. 180, 8342–8353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips R. S., Ramos S. B., Blackshear P. J. (2002) J. Biol. Chem. 277, 11606–11613 [DOI] [PubMed] [Google Scholar]
- 19.Liang J., Wang J., Azfer A., Song W., Tromp G., Kolattukudy P. E., Fu M. (2008) J. Biol. Chem. 283, 6337–6346 [DOI] [PubMed] [Google Scholar]
- 20.Kuprash D. V., Udalova I. A., Turetskaya R. L., Kwiatkowski D., Rice N. R., Nedospasov S. A. (1999) J. Immunol. 162, 4045–4052 [PubMed] [Google Scholar]
- 21.Goler-Baron V., Selitrennik M., Barkai O., Haimovich G., Lotan R., Choder M. (2008) Genes Dev. 22, 2022–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao H., Tuttle J. S., Blackshear P. J. (2004) J. Biol. Chem. 279, 21489–21499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner B. P., Westerheide S. D., Baldwin A. S., Jr. (2001) Mol. Cell. Biol. 21, 7065–7777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L. F., Greene W. C. (2004) Nat. Rev. Mol. Cell Biol. 5, 392–401 [DOI] [PubMed] [Google Scholar]
- 25.Fairhurst A. M., Connolly J. E., Hintz K. A., Goulding N. J., Rassias A. J., Yeager M. P., Rigby W., Wallace P. K. (2003) Arthritis Res. Ther. 5, R214–R225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson B. A., Stehn J. R., Yaffe M. B., Blackwell T. K. (2002) J. Biol. Chem. 277, 18029–18036 [DOI] [PubMed] [Google Scholar]
- 27.Carballo E., Cao H., Lai W. S., Kennington E. A., Campbell D., Blackshear P. J. (2001) J. Biol. Chem. 276, 42580–42587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu W., Brauchle M. A., Di Padova F., Gram H., New L., Ono K., Downey J. S., Han J. (2001) Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L499–L508 [DOI] [PubMed] [Google Scholar]
- 29.Lai W. S., Kennington E. A., Blackshear P. J. (2002) J. Biol. Chem. 277, 9606–9613 [DOI] [PubMed] [Google Scholar]
- 30.Liang J., Song W., Tromp G., Kolattukudy P. E., Fu M. (2008) PLoS ONE 3, e2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Batchelder C., Dunn M. A., Choy B., Suh Y., Cassie C., Shim E. Y., Shin T. H., Mello C., Seydoux G., Blackwell T. K. (1999) Genes Dev. 13, 202–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Twizere J. C., Kruys V., Lefèbvre L., Vanderplasschen A., Collete D., Debacq C., Lai W. S., Jauniaux J. C., Bernstein L. R., Semmes O. J., Burny A., Blackshear P. J., Kettmann R., Willems L. (2003) J. Natl. Cancer Inst. 95, 1846–1859 [DOI] [PubMed] [Google Scholar]