

Abstract
A major mechanism of bacterial resistance to β-lactam antibiotics (penicillins, cephalosporins, carbapenems, etc.) is the production of β-lactamases. A handful of class A β-lactamases have been discovered that have acquired the ability to turn over carbapenem antibiotics. This is a disconcerting development, as carbapenems are often considered last resort antibiotics in the treatment of difficult infections. The GES family of β-lactamases constitutes a group of extended spectrum resistance enzymes that hydrolyze penicillins and cephalosporins avidly. A single amino acid substitution at position 170 has expanded the breadth of activity to include carbapenems. The basis for this expansion of activity is investigated in this first report of detailed steady-state and pre-steady-state kinetics of carbapenem hydrolysis, performed with a class A carbapenemase. Monitoring the turnover of imipenem (a carbapenem) by GES-1 (Gly-170) revealed the acylation step as rate-limiting. GES-2 (Asn-170) has an enhanced rate of acylation, compared with GES-1, and no longer has a single rate-determining step. Both the acylation and deacylation steps are of equal magnitude. GES-5 (Ser-170) exhibits an enhancement of the rate constant for acylation by a remarkable 5000-fold, whereby the enzyme acylation event is no longer rate-limiting. This carbapenemase exhibits kcat/Km of 3 × 105 m−1s−1, which is sufficient for manifestation of resistance against imipenem.
Bacterial production of β-lactamases is a primary mechanism of resistance to β-lactam antibiotics (1). These enzymes hydrolytically process the β-lactam bond of the antibiotic, and by so doing, inactivate them. Four classes of β-lactamases, A, B, C, and D, are known, of which the class A enzymes are most prevalent (1, 2). Enzymes belonging to classes A, C, and D are serine-dependent. A critical active site serine in these enzymes experiences acylation by the antibiotic followed by deacylation. The mechanistic details of the deacylation steps vary for each class (1). Class B enzymes are zinc-dependent, and their mechanistic details are distinct. Random mutations in the genes for these enzymes have allowed for selection of novel variants within the clinic having an increased breadth for substrate preference (3). Variants of β-lactamases with the ability to turn over both penicillins and cephalosporins are referred to as extended spectrum β-lactamases (4). Extended spectrum β-lactamases typically do not have the ability to hydrolyze carbapenems; however, exceptions to this rule are emerging among members of classes A, B, and D and are called carbapenemases (5, 6). Within class A, members of the KPC and GES families are becoming increasingly problematic within the clinic (3, 5, 7). The GES type class A β-lactamases were identified for the first time only 10 years ago, but they have been increasingly detected worldwide among Gram-negative bacteria (3). The first variants detected were plasmid borne and isolated from Klebsiella pneumoniae, Pseudomonas aeruginosa, Escherichia coli, and Enterobacter cloacae (8–14); however, newer variants have been identified in the chromosome of P. aeruginosa (15, 16). A distinguishing characteristic of this family of enzymes is its ability to develop resistance to all classes of β-lactam antibiotics, including third-generation cephalosporins, cephamycins, monobactams, and/or carbapenems. This is in stark contrast to “classical” β-lactamases, such as the TEM family of enzymes, which also rapidly evolved to produce numerous extended spectrum and inhibitor-resistant variants, yet have failed to evolve activity against carbapenem antibiotics. This combination of resistance to both extended spectrum β-lactams and carbapenem antibiotics makes the organisms that harbor the genes for these enzymes dangerous in a clinical setting as treatment options dwindle.
GES-1, the first member of the GES family to be identified, has no significant ability to hydrolyze carbapenems, leading to its classification as only an extended spectrum β-lactamase (10). Other GES-type enzymes were later discovered with increased resistance to carbapenems. Two of these variants, GES-2 and -5, contain only a single amino acid substitution compared with the sequence of GES-1, both at position 170 (Ambler numbering is used) (17). This position is located within an Ω-loop forming one of the walls of the active site (1). The canonical residue at this position of class A β-lactamases is asparagine, which is found in GES-2. However, GES-1 contains a glycine, and GES-5 contains a serine (17). We recently solved the x-ray crystal structure of the GES-1 enzyme; however, it was not clear from the structural data why such a substitution would convey resistance in GES-2 and -5, in contrast to the case of GES-1, which does not (18). The mechanistic basis for the extended profile for resistance is not known, and it is the subject of study in this report. Previous kinetic analyses of class A carbapenemases, including GES enzymes, have been limited to steady-state kinetic parameters (8, 10, 11). In this report, we have performed an in-depth analysis of the GES family, including both steady-state and the first pre-steady-state analyses (for any class A carbapenemase) to elucidate the nature of the microscopic steps (binding, acylation, deacylation, and product release) in the turnover process by these clinically important enzymes.
EXPERIMENTAL PROCEDURES
Minimal Inhibitory Concentration Determinations
The minimal inhibitory concentrations (MICs)2 of β-lactam antibiotics were determined using the broth microdilution method, as recommended by the Clinical and Laboratory Standards Institute (19). GES-1, -2, and -5 were expressed in E. coli JM83 using the plasmids pHF:GES-1, pHF:GES-2, and pHF:GES-5, respectively. E. coli JM83 harboring pHF016 was used as a control. MICs were determined in Mueller-Hinton II broth (Difco) using a bacterial inoculum of 5 × 105 colony-forming units/ml. All plates were incubated at 37 °C for 16–20 h before results were interpreted.
Data Collection and Analysis
All spectrophotometric data were collected on a Cary 50 spectrophotometer (Varian) at 22 °C. Analyses were performed using the nonlinear regression program Prism 5 (GraphPad Software, Inc.) using data obtained from at least three independent experiments.
Determination of Steady-state Kinetic Parameters
Reactions containing 50 mm NaPi (pH 7.0), 100 mm NaCl, and varying concentrations of the β-lactam substrate were initiated by the addition of enzyme. The absorbance was monitored at the following wavelengths: imipenem (λ = 297 nm and Δϵ = −10,930 cm−1 m−1) and nitrocefin (λ = 500 nm and Δϵ = +15,900 cm−1 m−1). The steady-state velocities (v) were determined from the linear phase of the reaction time courses. The observed rate constants (kobs = v/[E]) were plotted as a function of the β-lactam concentration and fit non-linearly with the Michaelis-Menten equation to obtain the steady-state kinetic parameters kcat and Km. The parameters for nitrocefin hydrolysis by GES-1, -2, and -5 are given in supplemental Table 1.
Determination of Dissociation Constants
The substrate dissociation constant (Ks) for imipenem, with GES-1, -2, and -5, was determined by treating it as an inhibitor of nitrocefin hydrolysis as described previously (20). Reactions containing 50 mm NaPi (pH 7.0), 100 mm NaCl, 160 μm (GES-1), 50 μm (GES-2), or 120 μm (GES-5) of nitrocefin, and varying concentrations of imipenem were initiated by the addition of the enzyme (200 pm final for GES-1 and GES-5 or 10 nm final for GES-2). The absorbance was monitored at 500 nm, and the steady-state velocities were determined from the linear phase of the reaction time courses. The steady-state velocity data were plotted as a function of imipenem concentration and fit with Equation 1.
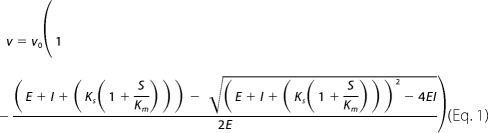 |
where v is the steady-state velocity, v0 is the steady-state velocity in the absence of imipenem, E is the enzyme concentration, I is the imipenem concentration, Ks is the dissociation constant for imipenem, S is the nitrocefin concentration, and Km is the Michaelis constant for nitrocefin hydrolysis.
The dissociation constants for the product of imipenem hydrolysis (Ki,p) were determined as follows. Reactions containing 50 mm NaPi (pH 7.0), 100 mm NaCl, 160 μm (GES-1), 5 μm (GES-2), or 200 μm (GES-5) nitrocefin, and varying concentrations of hydrolyzed imipenem (up to 4 mm) were initiated by the addition of the enzyme (200 pm GES-1 and -5 or 10 nm GES-2 final). The absorbance was monitored at 500 nm, and the steady-state velocities were determined from the linear phase of the reaction time courses.
Determination of the Microscopic Rate Constants for Acylation and Deacylation
The first-order rate constants describing acylation (k2) of GES-1, -2, and -5 by imipenem were determined under single turnover conditions. Reaction mixtures contained 50 mm NaPi (pH 7.0), 100 mm NaCl, 75 μm enzyme, and 15 μm imipenem. Acylation was detected by monitoring the opening of the β-lactam ring of imipenem at 297 nm (Δϵ297 = −10,930 cm−1 m−1) with the aid of an SFA-20 stopped-flow apparatus (Hi-Tech Scientific, Salisbury, UK), and the time courses were fit with Equation 2.
where At is the absorbance at time t, A0 is the initial absorbance, A∞ is the final absorbance, and k2 is the observed first-order rate constant for enzyme acylation. Acylation of GES-5 by imipenem under single turnover conditions resulted in two distinct exponential phases, which was fit with Equation 3.
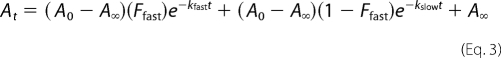 |
where At, A0, and A∞ are as defined in Equation 2, Ffast is the fraction of the reaction corresponding to the fast phase, kfast is the observed first-order rate constant for the fast phase, and kslow is the observed first-order rate constant for the slow phase.
The rate at which imipenem was displaced from the active site of GES-1, -2, and -5 was determined as described previously (21). Briefly, a reaction containing 50 mm NaPi (pH 7.0), 100 mm NaCl, and 1 μm GES-1 and -2 or 750 nm GES-5 in the absence and presence of 20 μm (in the case of GES-1), 5 μm (in the case of GES-2), or 15 μm (in the case of GES-5) imipenem (10 × Km) was incubated at room temperature for 2 min. This reaction was diluted 1:100 and/or 1:1,000 into a reaction mixture containing 50 mm NaPi (pH 7.0), 100 mm NaCl, and 800 μm (in the case of GES-1), 50 μm (in the case of GES-2), or 600 μm (in the case of GES-5) of nitrocefin, and nitrocefin hydrolysis was monitored at 500 nm. The rate constant describing recovery of enzymatic activity (k3) was determined by fitting the absorbance versus time data with Equation 4.
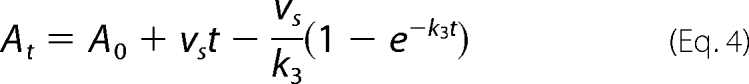 |
where At is the absorbance at time t, A0 is the initial absorbance, vs is the steady-state velocity, and k3 is the rate constant for the recovery of enzymatic activity (i.e. deacylation).
Calculation of the Microscopic Rate Constants Describing Imipenem Hydrolysis
Hydrolysis of imipenem by GES-1, -2, and -5 can be described by Scheme 1. The following kinetic parameters can be derived for Scheme 1.
 |
 |
 |
 |
The values for k1 and k−1 for GES-1, -2, and -5 and the value for k3 for GES-5 were calculated using Equations 5–8 and the experimentally determined values for kcat, Km, Ks, k2, and/or k3.
SCHEME 1.
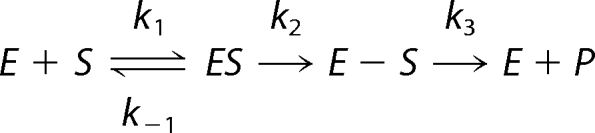
RESULTS AND DISCUSSION
β-Lactam Resistance Profile
We constructed a constitutive expression vector to directly compare the MICs of β-lactam antibiotics conferred by individual GES enzymes under an identical promoter in E. coli JM83. As the results of Table 1 (and supplemental Table 2) reveal, the breadth of activity for all three enzymes is broad. All are resistant to penicillins, including the penicillinase-stable oxacillin, and first- and second-generation cephalosporins. The expression of GES-1 and -2 confers higher resistance to the third-generation cephalosporins than is the case with GES-5, but the organism that expresses the latter enzyme has instead gained resistance to cephamycins. Most alarming is the increase in resistance toward carbapenems (imipenem, meropenem, ertapenem, and doripenem) by GES-5 (Table 1). We note that the elevated MICs for carbapenems within a laboratory E. coli strain (1 μg/ml for imipenem with GES-5) are considered acceptable clinically. However, the emergence of variants such as GES-5 in combination with other mechanisms of carbapenem resistance, such as the loss of certain porins, results in full blown resistance to carbapenems in clinical isolates (6, 22). These differences in MIC values highlight the importance of the residue at position 170 in determining the substrate specificity of these enzymes.
TABLE 1.
MICs of carbapenem antibiotics for E. coli JM83 producing various GES enzymes
All MIC data are reported in μg/ml.
| Antibiotic | Controla | GES-1 | GES-2 | GES-5 |
|---|---|---|---|---|
| imipenem | 0.125 | 0.25 | 0.25 | 1 |
| meropenem | 0.031 | 0.031 | 0.031 | 0.5 |
| ertapenem | 0.004 | 0.031 | 0.031 | 0.25 |
| doripenem | 0.031 | 0.031 | 0.031 | 0.25 |
a Parental E. coli JM83 strain harboring the vector pHF016 containing no β-lactamase gene.
Steady-state Kinetic Parameters and Dissociation Constants
The GES-1, -2, and -5 enzymes were purified to homogeneity for kinetic analyses. Class A β-lactamases (including GES enzymes) are acylated by β-lactam antibiotics at Ser-70, a step that is facilitated by Lys-73 (23). Glu-166 and Asn-170 (the residue that varies in GES-1, -2, and -5) anchor the hydrolytic water molecule within the active site. This water promotes deacylation of the acyl-enzyme intermediate to complete substrate turnover. We evaluated the kinetics of imipenem turnover in detail (Table 2). Turnover of imipenem by GES-1 and -2 was poor compared with the case of GES-5. The significant effect on steady-state parameters is manifested on kcat, resulting in a roughly two orders of magnitude increase in kcat/Km for hydrolysis of imipenem going from GES-1 to -5. Enzyme saturation for substrate turnover (as judged by Km) is achieved at low micromolar concentrations, but enzyme acylation was sufficiently sluggish to allow evaluation of the dissociation constants for the non-covalent enzyme-imipenem complexes (Ks). The Ks value for imipenem was 10 nm for GES-1 but significantly higher for GES-2 and -5, although it was still in the high nanomolar range. The value for Km is greater than the Ks for each GES variant. This reveals that acylation (k2) is more rapid than dissociation of the substrate from the non-covalent ES complex (k−1), contrary to what is generally assumed for reactions following Michaelis-Menten kinetics (24).
TABLE 2.
Kinetic parameters for hydrolysis of imipenem by GES enzymes
| Parameter | GES-1 | GES-2 | GES-5 |
|---|---|---|---|
| kcat (s−1) | 0.0059 ± 0.0003 | 0.012 ± 0.001 | 0.44 ± 0.01 |
| Km (μm) | 1.9 ± 0.7 | 0.7 ± 0.5 | 1.5 ± 0.3 |
| kcat/Km (m−1 s−1) | (3 ± 1) × 103 | (2 ± 1) × 104 | (2.9 ± 0.6) × 105 |
| Ks (nm) | 11 ± 2 | 224 ± 15 | 491 ± 45 |
| k1 (m−1 s−1)a | 3 × 103 | 2 × 104 | 3 × 105 |
| k−1 (s−1)a | 3 × 10−5 | 5 × 10−3 | 1.4 × 10−1 |
| k2 (s−1) | 0.007 ± 0.001 | 0.021 ± 0.001 | 34 ± 2b |
| k3 (s−1) | 0.03 ± 0.01 | 0.028 ± 0.001 | 0.45a |
a Not measured experimentally.
b Acylation under single turnover conditions resulted in biphasic kinetics. The rapid phase likely represents acylation under multiple turnover conditions.
Microscopic Rate Constants for Acylation and Deacylation
We also determined the microscopic rate constant (k2) for acylation of GES-1, -2, and -5 by imipenem (Table 2 and supplemental Fig. 1). The acylation rate constant (k2) was small for GES-1 and -2 and was comparable to kcat in the case of GES-1, indicating that acylation is the rate-determining step in imipenem hydrolysis by this enzyme. This observation was unexpected as the deacylation step (k3) has been shown to be rate-limiting in the turnover of carbapenems by class A enzymes for which the determination has been made (25). The time course for acylation of GES-5 did not follow a single exponential time course. Data fitting revealed that there existed an initial rapid phase accounting for one third of the total turnover (k = 34 ± 2 s−1), followed by a slower phase (k = 0.15 ± 0.01 s−1). We note that imipenem exists in solution as a distinct mixture of two side chain rotational isomers (Z:E ratio of 2) that interconvert (Fig. 1) (26). It is conceivable that GES-5 discriminates between the two rotational isomers, presumably hydrolyzing the E isomer more rapidly than the Z isomer. Under steady-state turnover, GES-5 would bind both isomers. However, the equilibrium would favor hydrolysis of the E isomer, from a kinetic standpoint, due to the 225-fold difference in the acylation rate constants for the two isomers. This assertion is also supported by the fact that the measured kcat is greater than the rate constant describing the slower phase. Alternatively, two distinct enzyme populations might exist that experience acylation by imipenem with different rate constants. We propose that the rapid phase represents acylation of GES-5 under steady-state conditions and therefore that the acylation rate constant is 75-fold larger than kcat and hence is not rate-limiting for this enzyme variant. This represents a remarkable 5000- and 1500-fold enhancement of the rate of acylation of the GES-5 enzyme over that of GES-1 and -2, respectively, by imipenem.
FIGURE 1.

The chemical structures of imipenem and the product of imipenem hydrolysis.
We evaluated the deacylation rate constant (k3) for the turnover of imipenem by monitoring recovery of enzyme activity (Table 2 and supplemental Fig. 2) (21). We note that we prepared the product of imipenem hydrolysis (an inseparable and interconvertible mixture of Δ1 and Δ2 tautomers, Fig. 1) by chemical synthesis. Analysis of this product with the GES variants revealed the dissociation of the non-covalent enzyme-product complex is readily achieved (Ki, p > 4 mm) and would not affect the recovery of enzymatic activity. The GES-1 acyl-enzyme intermediate experiences deacylation with a rate constant of 0.03 ± 0.01 s−1, confirming that acylation by imipenem is indeed rate-limiting for this enzyme. The GES-2 acyl-enzyme intermediate undergoes deacylation with a rate constant of 0.028 ± 0.001 s−1, very close to the value for the acylation event, which reveals that neither acylation nor deacylation is entirely rate-limiting for this variant. The upper limit of detection by this method is 0.03 s−1, which is smaller than the value of kcat for hydrolysis of imipenem by GES-5. Thus, we were unable to evaluate k3 for GES-5 by this method, although the value for this rate constant can be estimated using Equation 5 and our experimental values for kcat and k2 (Table 2). The deacylation rate constant for GES-5 is virtually identical to kcat, and k2 is larger than k3, indicating that this step is now rate-limiting, in contrast to the cases of GES-1 and -2. The GES-5 variant has experienced a 15-fold enhancement in the deacylation rate constant, over that of GES-1 and -2, which is necessary for the manifestation of imipenem resistance. Despite the fact that the magnitude of the enhancement for the acylation step is significantly larger than it is for deacylation, it would appear that the enhanced rate of acylation in conjunction with the more modest augmentation in k3 are the critical events for the emergence of carbapenemase resistance in GES-5.
Microscopic Rate Constants for Substrate Binding and Release
Although direct evaluation of microscopic rate constants for substrate binding (k1) and substrate release (k−1) are not possible, these rate constants were estimated using our experimental data and Equation 5–8 (Table 2). The GES-1 variant, containing Gly at position 170, has the highest affinity for imipenem (KS), primarily due to a very small rate constant for substrate release (k−1). It is interesting to note that each substitution at position 170, from Gly to Asn and finally Ser (GES-1, -2, and -5, respectively), progressively results in 10-fold increases in the rate constant for imipenem binding (k1) and a progressive 100-fold increase in the rate constants for imipenem release. This leads to a decreased affinity for imipenem from GES-1 to GES-2 to GES-5. This is desired for an enzyme that becomes a more competent catalyst as the loss of affinity for the substrate (larger Ks) avoids the undesired prospect of substrate inhibition.
Structural Comparison and Molecular Dynamics Simulations
Comparison of the structures of GES-1 (Fig. 2A) and other carbapenemases to class A β-lactamases, which do not hydrolyze carbapenems, reveal subtle differences in their active sites. The x-ray structure provides only a snap shot of the enzyme, whereas in reality the protein structure should experience dynamic motion in solution, which may explain its ability to hydrolyze carbapenem antibiotics. The Ω-loop motif, which includes both residues 166 (critical for deacylation) and 170, assumes different conformations in various class A enzymes (Fig. 2) and, at least in the case of a subset of the TEM mutants, it has been argued to be a mobile element (27–29). Both TEM-1 (Fig. 2B) and NMC-A β-lactamases (Fig. 2C) are able to bind carbapenems; however, only NMC-A is able to efficiently turn them over. We have previously shown that movement of the Ω-loop in NMC-A facilitates hydrolysis of carbapenems (25). We wondered whether the Ω-loop within the active site of GES-1 is a mobile element. If so, this could explain how at least GES-1 could bind and slowly hydrolyze carbapenems. If indeed mobile, this factor might be critical for the GES-5 enzyme, as it has an enhanced rate of deacylation for imipenem. A 6-ns dynamics simulation revealed mobility in the Ω-loop (Glu-166 Cα moved into the active site at least by ∼1 Å, Fig. 2D). The dynamic simulations also revealed the existence of a water molecule between residues Glu-166 and Gly-170 within the active site of GES-1, presumably the deacylation water molecule. This suggests that as in the case of NMC-A, movement of the Ω-loop may facilitate enzyme deacylation.
FIGURE 2.

Crystal and molecular dynamics structures of class A carbapenemases. A stereoview representation of the active-site conformations for the crystal structure of the GES-1 β-lactamase (A), the acyl-enzyme species of imipenem bound to the class A TEM-1 β-lactamase from E. coli (B) (30), 6α-(hydroxypropyl)penicillanate bound to the NMC-A β-lactamase from E. cloacae (C) (25), and molecular motions of two extreme conformations of GES-1 from dynamics simulations (D). The active sites (A–C) are depicted as Connolly solvent-accessible surfaces, with residues at positions 70, 73, 166, 169, and 170 shown as capped sticks. (Carbon atoms are colored in gray, nitrogen in blue, oxygen in red, and hydrogen in cyan, with a loop shown as a yellow tube for visual access to the active site.) A conserved water molecule is shown between residues 170 and 166 (A) as a sphere (in magenta). The role of the 6α-(hydroxypropyl) moiety in the structure of C is the same as that of the 6α-(hydroxyethyl) group of imipenem shown in B; namely, an impediment to the travel of the hydrolytic water molecule to the acyl-enzyme carbonyl. The deacylation step is rate-limiting in these two examples. Two extreme conformations are shown in D with motion of residue 166 Cα by ∼1 Å. The protein and Ω-loop of one conformer are colored in green and the Ω-loop of another conformer in orange, with residues shown in capped sticks. (Atoms are colored the same as in A–C, except the carbon atoms from the second conformer that are colored in orange.) The water molecule shown in capped stick is from the second conformer.
Conclusion
A single amino acid substitution at position 170 of GES enzymes has a dramatic impact on the turnover of the carbapenem antibiotic imipenem. The GES-1 enzyme with Gly-170 has a high affinity for imipenem but is unable to efficiently turn it over due to the low rate constants for acylation and deacylation. Substitution to Asn-170, in GES-2, results in the canonical residue at this position for typical class A β-lactamases. This variant shows no improvement in the rate constant for deacylation, but the rate constant for acylation is enhanced such that neither is rate-determining. The GES-5 enzyme, with Ser-170, shows an enhancement of both microscopic rate constants; however, the defining alteration in the kinetic profile of GES-5 is the 5000- and 1500-fold increase in the rate constant for acylation over that of GES-1 and GES-2, respectively. This results in the alteration of the rate-determining step of the reaction from acylation to deacylation and reveals that the enhanced rate of deacylation is responsible for the increased resistance to imipenem. It is not obvious at the present, from a structural perspective, why the residue at position 170 should have an effect on the rate of acylation and/or deacylation. Elucidation of these questions awaits the future availability of x-ray information for the complexes of these enzymes with carbapenem antibiotics.
Supplementary Material

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures,” Tables 1 and 2, Figs. 1 and 2, and additional references.
- MIC
- minimal inhibitory concentration.
REFERENCES
- 1.Fisher J. F., Meroueh S. O., Mobashery S. (2005) Chem. Rev. 105, 395–424 [DOI] [PubMed] [Google Scholar]
- 2.Babic M., Hujer A. M., Bonomo R. A. (2006) Drug Resist. Updat. 9, 142–156 [DOI] [PubMed] [Google Scholar]
- 3.Walther-Rasmussen J., Høiby N. (2007) J. Antimicrob. Chemother. 60, 470–482 [DOI] [PubMed] [Google Scholar]
- 4.Page M. G. (2008) Clin. Microbiol. Infect. 14, Suppl. 1, 63–74 [DOI] [PubMed] [Google Scholar]
- 5.Queenan A. M., Bush K. (2007) Clin. Microbiol. Rev. 20, 440–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nordmann P., Poirel L. (2002) Clin. Microbiol. Infect. 8, 321–331 [DOI] [PubMed] [Google Scholar]
- 7.Nordmann P., Cuzon G., Naas T. (2009) Lancet Infect. Dis. 9, 228–236 [DOI] [PubMed] [Google Scholar]
- 8.Bae I. K., Lee Y. N., Jeong S. H., Hong S. G., Lee J. H., Lee S. H., Kim H. J., Youn H. (2007) Diagn. Microbiol. Infect. Dis. 58, 465–468 [DOI] [PubMed] [Google Scholar]
- 9.Vourli S., Giakkoupi P., Miriagou V., Tzelepi E., Vatopoulos A. C., Tzouvelekis L. S. (2004) FEMS Microbiol. Lett. 234, 209–213 [DOI] [PubMed] [Google Scholar]
- 10.Poirel L., Le Thomas I., Naas T., Karim A., Nordmann P. (2000) Antimicrob. Agents Chemother. 44, 622–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poirel L., Weldhagen G. F., Naas T., De Champs C., Dove M. G., Nordmann P. (2001) Antimicrob. Agents Chemother. 45, 2598–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giakkoupi P., Tzouvelekis L. S., Tsakris A., Loukova V., Sofianou D., Tzelepi E. (2000) Antimicrob. Agents Chemother. 44, 2247–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wachino J., Doi Y., Yamane K., Shibata N., Yagi T., Kubota T., Arakawa Y. (2004) Antimicrob. Agents Chemother. 48, 2905–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wachino J., Doi Y., Yamane K., Shibata N., Yagi T., Kubota T., Ito H., Arakawa Y. (2004) Antimicrob. Agents Chemother. 48, 1960–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poirel L., Brinas L., Fortineau N., Nordmann P. (2005) Antimicrob. Agents Chemother. 49, 3593–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mavroidi A., Tzelepi E., Tsakris A., Miriagou V., Sofianou D., Tzouvelekis L. S. (2001) J. Antimicrob. Chemother. 48, 627–630 [DOI] [PubMed] [Google Scholar]
- 17.Ambler R. P., Coulson A. F., Frère J. M., Ghuysen J. M., Joris B., Forsman M., Levesque R. C., Tiraby G., Waley S. G. (1991) Biochem. J. 276, Pt 1, 269–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith C. A., Caccamo M., Kantardjieff K. A., Vakulenko S. (2007) Acta Crystallogr. D Biol. Crystallogr. 63, 982–992 [DOI] [PubMed] [Google Scholar]
- 19.Clinical Institute Laboratory Standards (2006) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard, 7th Ed., Clinical and Laboratory Standards Institute, Wayne, Pennsylvania [Google Scholar]
- 20.Zafaralla G., Manavathu E. K., Lerner S. A., Mobashery S. (1992) Biochemistry 31, 3847–3852 [DOI] [PubMed] [Google Scholar]
- 21.Taibi P., Mobashery S. (1995) J. Am. Chem. Soc. 117, 7600–7605 [Google Scholar]
- 22.Kim S. Y., Park Y. J., Yu J. K., Kim H. S., Park Y. S., Yoon J. B., Yoo J. Y., Lee K. (2007) Diagn. Microbiol. Infect. Dis. 57, 85–91 [DOI] [PubMed] [Google Scholar]
- 23.Meroueh S. O., Fisher J. F., Schlegel H. B., Mobashery S. (2005) J. Am. Chem. Soc. 127, 15397–15407 [DOI] [PubMed] [Google Scholar]
- 24.Fersht A. (1999) Structure and Mechanism in Protein Science : A Guide to Enzyme Catalysis and Protein Folding, pp. 103–131, Freeman W.H., New York [Google Scholar]
- 25.Mourey L., Miyashita K., Swaren P., Bulychev A., Samama J. P., Mobashery S. (1998) J. Am. Chem. Soc. 120, 9382–9383 [Google Scholar]
- 26.Ratcliffe R. W., Wildonger K. J., Dimichele L., Douglas A. W., Hajdu R., Goegelman R. T., Springer J. P., Hirshfield J. (1989) J. Org. Chem. 54, 653–660 [Google Scholar]
- 27.Vakulenko S. B., Taibi-Tronche P., Tóth M., Massova I., Lerner S. A., Mobashery S. (1999) J. Biol. Chem. 274, 23052–23060 [DOI] [PubMed] [Google Scholar]
- 28.Taibi-Tronche P., Massova I., Vakulenko S. B., Lerner S. A., Mobashery S. (1996) J. Am. Chem. Soc. 118, 7441–7448 [Google Scholar]
- 29.Palzkill T., Le Q. Q., Venkatachalam K. V., LaRocco M., Ocera H. (1994) Mol. Microbiol. 12, 217–229 [DOI] [PubMed] [Google Scholar]
- 30.Maveyraud L., Mourey L., Kotra L. P., Pedelacq J. D., Guillet V., Mobashery S., Samama J. P. (1998) J. Am. Chem. Soc. 120, 9748–9752 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.