

Abstract
The Ras/B-Raf/C-Raf/MEK/ERK signaling cascade is critical for the control of many fundamental cellular processes, including proliferation, survival, and differentiation. This study demonstrated that small interfering RNA-dependent knockdown of diacylglycerol kinase η (DGKη) impaired the Ras/B-Raf/C-Raf/MEK/ERK pathway activated by epidermal growth factor (EGF) in HeLa cells. Conversely, the overexpression of DGKη1 could activate the Ras/B-Raf/C-Raf/MEK/ERK pathway in a DGK activity-independent manner, suggesting that DGKη serves as a scaffold/adaptor protein. By determining the activity of all the components of the pathway in DGKη-silenced HeLa cells, this study revealed that DGKη activated C-Raf but not B-Raf. Moreover, this study demonstrated that DGKη enhanced EGF-induced heterodimerization of C-Raf with B-Raf, which transmits the signal to C-Raf. DGKη physically interacted with B-Raf and C-Raf, regulating EGF-induced recruitment of B-Raf and C-Raf from the cytosol to membranes. The DGKη-dependent activation of C-Raf occurred downstream or independently of the already known C-Raf modifications, such as dephosphorylation at Ser-259, phosphorylation at Ser-338, and interaction with 14-3-3 protein. Taken together, the results obtained strongly support that DGKη acts as a novel critical regulatory component of the Ras/B-Raf/C-Raf/MEK/ERK signaling cascade via a previously unidentified mechanism.
The Ras/Raf/MEK3/ERK signaling pathway is critical for the transduction of the extracellular signals to the nucleus, regulating diverse physiological processes such as cell proliferation, differentiation, and survival (1, 2). The binding of extracellular ligands, such as growth factors and cytokines, to cell surface receptors activates Ras. The Raf serine/threonine kinase transmits signals from activated Ras to the downstream protein kinases, MEK1 and MEK2, subsequently leading to activation of ERK1 and ERK2.
In mammals, the Raf kinase consists of three isoforms, A-Raf, B-Raf, and C-Raf (Raf-1). It is clinically known that both B-Raf and C-Raf mutations are associated with human cancers (3–5). Knock-out mouse studies demonstrated that each individual Raf isoform has distinct functions, although the three Raf isoforms have high homology in the amino acid sequence (6). The mechanisms underlying C-Raf activation are complicated and thus are not completely understood (3). In response to extracellular signals, C-Raf is initially recruited from cytosol to the plasma membrane and undergo conformational changes by binding directly to the active Ras (7). In addition, other modifications and factors are required for the sufficient activation of C-Raf. For example, dephosphorylation of Ser-259 and phosphorylation of Ser-338, Tyr-341, Thr-491, and Ser-494 are critical for the activation of C-Raf (8–11). Feedback phosphorylation of C-Raf by ERK was also reported to be important for the modulation of C-Raf activity (12, 13). C-Raf activity is regulated by the interaction with 14-3-3 protein (14). Moreover, the heterodimerization of C-Raf with B-Raf, which transmits the signal to C-Raf, has been reported to play an essential role in the activation of the MEK-ERK signaling pathway (15–17). Although B-Raf and C-Raf are the central regulatory components in the Ras/B-Raf/C-Raf/MEK/ERK signaling cascade involved in a variety of pathophysiological events, the activation mechanisms of C-Raf by B-Raf are still unclear.
Diacylglycerol kinase (DGK) catalyzes the phosphorylation of diacylglycerol to generate phosphatidic acid. DGK has been recently recognized as an emerging key regulator in a wide range of cell signaling systems (18–20). To date, 10 mammalian DGK isozymes have been identified. They characteristically contain two or three protein kinase C-like C1 domains and a catalytic region and are subdivided into five subtypes according to their structural features (18–20). Their structural variety and distinct expression patterns in tissues allow us to presume that each DGK isozyme has its own biological functions. Indeed, recent studies have revealed that individual DGK isozymes play distinct roles in cell functions through interactions with unique partner proteins such as protein kinase C (21, 22), Ras guanyl nucleotide-releasing protein (23, 24), phosphatidylinositol-4-phosphate 5-kinase (25), chimerins (26, 27), AP-2 (28), and PSD-95 (29).
DGKη belongs to the type II DGKs containing a pleckstrin homology domain at the N terminus and the separated catalytic region (19, 30). Two alternative splicing products of DGKη have been identified as DGKη1 and -η2 (31). DGKη2 possesses a sterile α-motif (SAM) domain at the C terminus, whereas DGKη1 does not. This study demonstrated that the expression levels of DGKη1 and -η2 were regulated differently by glucocorticoid, and that they were translocated from the cytoplasm to endosomes in response to stress stimuli as osmotic shock and oxidative stress (31). However, the physiological roles of DGKη remain unknown.
This study showed that siRNA-dependent knockdown of DGKη inhibits cell proliferation of the HeLa cells. In addition, DGKη is required for the Ras/B-Raf/C-Raf/MEK/ERK signaling cascade activated by epidermal growth factor (EGF). Intriguingly, DGKη regulates recruitment of B-Raf and C-Raf from cytosol to membranes and their heterodimerization. Moreover, this study demonstrated that DGKη activates C-Raf but not B-Raf in an EGF-dependent manner. The data show DGKη as a novel key regulator of the Ras/B-Raf/C-Raf/MEK/ERK signaling pathway.
EXPERIMENTAL PROCEDURES
Antibodies
To raise anti-DGKη polyclonal antibody, rabbits were immunized by intramuscular multiple injections of 200 μg of glutathione S-transferase (GST) fusion protein containing amino acids 580–736 of human DGKη mixed with adjuvant. The serum obtained after the fourth injection was used. This antibody did not react with human DGKδ (data not shown). Anti-DGKδ polyclonal antibody was generated as described previously (32). Other antibodies were obtained from commercial sources as follows: anti-FLAG M2 and anti-β-actin (AC-15) antibodies (Sigma); anti-actin and anti-B-Raf antibodies (Santa Cruz Biotechnology); anti-C-Raf (clone 53) antibody (BD Biosciences); anti-Ras (RAS10) antibody (Upstate Biotechnology, Inc.); anti-ERK1/2, anti-phospho-ERK1/2 (Thr-202/Tyr-204), anti-MEK1/2, anti-phospho-MEK1/2 (Ser-217/221), anti-phospho-C-Raf (Ser-259), and anti-phospho-C-Raf (Ser-338) (56A6) antibodies (Cell Signaling Technology); and anti-14-3-3 antibody (Zymed Laboratories Inc.).
Cell Culture and Transfections
HeLa, COS7, and HEK293 cells were maintained in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal bovine serum and penicillin (100 units/ml)/streptomycin (100 μg/ml) (Invitrogen) at 37 °C in an atmosphere containing 5% CO2. HeLa and HEK293 cells were seeded in 60-mm dishes at a density of 2 × 105 and 4 × 105, respectively. The next day, the cells were transfected with 10 nm of siRNA using LipofectamineTM RNAiMAX (Invitrogen), according to the instructions from the manufacturer. After 72 h of siRNA transfection, the cells were serum-starved for 5 h in Dulbecco's modified Eagle's medium containing 0.1% bovine serum albumin and stimulated with 100 ng/ml human recombinant EGF (Wako Pure Chemical Industries) for the indicated times. HeLa and COS7 cells (∼8 × 105 cells per 60-mm dish) were transiently transfected with 1 μg of plasmid using Effectene (Qiagen), according to instructions from the manufacturer.
siRNAs
To silence the expression of human DGKη (31), DGKδ (33), B-Raf, and C-Raf, the following siRNAs were used: DGKη siRNA 1, 5′-CAAGGGAAAUCAUGUUGCGGGCAAA-3′ and 5′-UUUGCCCGCAACAUGAUUUCCCUUG-3′ (Invitrogen); DGKη siRNA 2, 5′-CCAAGACGCUAUGUGAAACUGUAAA-3′ and 5′-UUUACAGUUUCACAUAGCGUCUUGG-3′ (Invitrogen); DGKη siRNA 3, 5′-GGAUCUAGAUUCCGUAGAUdTdT-3′ and 5′-AUCUACGGAAUCUAGAUCCdGdG-3′ (Qiagen); DGKδ, 5′-UUAACAGGCGACUGAUGACUGAGCC-3′ and 5′-GGCUCAGUCAUCAGUCGCCUGUUAA-3′ (Invitrogen); B-Raf, 5′-GGACAAAGAAUUGGAUCUGGAUCAU-3′ and 5′-AUGAUCCAGAUCCAAUUCUUUGUCC-3′ (Invitrogen); and C-Raf, 5′-GGUCAAUGUGCGAAAUGGAAUGAGC-3′ and 5′-GCUCAUUCCAUUUCGCACAUUGACC-3′ (Invitrogen). These siRNA target sequences of B-Raf and C-Raf have been reported previously (34). Stealth RNA interference negative control duplexes (Invitrogen) or negative control siRNA (Qiagen) was used as control.
Plasmid Constructs
p3xFLAG-DGKη1 and -DGKη2 were generated as described previously (31). Mutations within p3xFLAG-DGKη1 were introduced using the QuikChange site-directed mutagenesis kit (Stratagene). An siRNA-resistant DGKη1 mutant was constructed by substituting the target sequence of DGKη siRNA 1 to 5′-CAAGGGAGATTATGTTGAGGGCAAA-3′ (mutated nucleotides are underlined) without changing the coding amino acids using a PCR-based site-directed mutagenesis method. p3xFLAG-DGKη-(-1–328) and p3xFLAG-DGKη-(-329–1164) were constructed by inserting PCR fragments encoding amino acids 1–328 and 329–1164, respectively, of human DGKη1 into the SalI-BamHI site of p3xFLAG-CMV-7.1 (Sigma). The full-length cDNAs of human DGKη1 and DGKη2 were amplified by PCR using p3xFLAG-DGKη1 and -DGKη2, respectively, and subcloned into pcDNA3.1 (Invitrogen) at the site of BamHI-XhoI. pAcGFP1-DGKη1 was constructed by inserting a PCR fragment of DGKη1 cDNA into the SalI-BamHI site of pAcGFP1-C1 (Clontech). pCMV-C-Raf was obtained from Clontech. pCMV-C-Raf-S338D/Y341D/T491E/S494D was generated by PCR-based site-directed mutagenesis. pDs-Red-monomer-C-Raf was constructed by inserting a PCR fragment of C-Raf cDNA into the EcoRI-BamHI site of pDsRed-monomer-C1 (Clontech). pCMV-H-RasV12 and pCMV-H-RasN17 were obtained from Clontech. The full-length cDNAs of H-RasV12 and H-RasN17 were amplified by PCR and inserted into the EcoRI-SalI sites of pECFP-C1 (Clontech) to generate pECFP-H-RasV12 and pECFP-H-RasN17, respectively. Authenticity of the constructs was confirmed by DNA sequencing.
Cell Proliferation Assay
HeLa cells were seeded in 12-well plates at a density of 2 × 104. The next day, the cells were transfected with 10 nm of siRNA. After 0, 48, and 96 h of transfections, the cells were trypsinized. Cells excluding trypan blue were counted using a hemocytometer.
Immunoprecipitation and Western Blot Analysis
The cells were lysed in 500 μl of ice-cold buffer A (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, and protease inhibitor mixture (Roche Applied Science)) containing 1 mm β-glycerophosphate, 2.5 mm sodium pyrophosphate, and 1 mm sodium vanadate. The mixture was centrifuged at 12,000 × g for 10 min at 4 °C to obtain the cell lysates. The lysates were precleared by mixing 10 μl of protein G-Sepharose 4 Fast Flow beads (GE Healthcare) for 30 min at 4 °C. The precleared lysates were incubated for 1 h at 4 °C with 1.5 μg of anti-B-Raf antibody, anti-C-Raf antibody, or normal IgG (Santa Cruz Biotechnology), followed by incubation with 10 μl of protein G-Sepharose 4 Fast Flow beads for 1 h at 4 °C. For immunoprecipitation of FLAG-tagged proteins, the cell lysates were incubated for 1 h at 4 °C with 10 μl of anti-FLAG M2 affinity gel (Sigma). For immunoprecipitation of endogenous DGKη, precleared lysates prepared from HEK293 cells were incubated for 2 h at 4 °C with 5 μl of anti-DGKη antibody or preimmune serum, followed by incubation with 10 μl of protein G-Sepharose 4 Fast Flow beads for 1 h at 4 °C. The beads were washed three times with 500 μl of buffer B (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 0.1% Nonidet P-40) and then boiled in SDS sample buffer. The cell lysates and immunoprecipitants were separated using SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Bio-Rad). The membranes were blocked with 5% low fat milk in Tris-buffered saline (TBS: 20 mm Tris-HCl, pH 7.4, and 150 mm NaCl) containing 0.1% Tween 20 for 1 h and incubated with primary antibodies. The immunoreactive bands were visualized with horseradish peroxidase-conjugated anti-mouse or -rabbit IgG antibody (Jackson ImmunoResearch) and ECL Western blotting detection system (GE Healthcare). The detected bands were quantified by densitometric analysis using ImageJ 1.34s.
Affinity Precipitation of Activated Ras
The cells were rinsed with TBS and lysed in 500 μl of ice-cold buffer A containing 5 mm MgCl2, 1 mm dithiothreitol, and 5% glycerol. After centrifugation at 12,000 × g for 10 min at 4 °C, the supernatant was incubated with 10 μl of Raf-RBD (Ras-binding domain)-GST beads (Cytoskeleton), which selectively interacted with active GTP-bound Ras, for 1 h at 4 °C. The beads were washed three times with 500 μl of buffer B containing 5 mm MgCl2 and then boiled in SDS sample buffer. Ras associated with Raf-RBD-GST and total Ras in cell lysates were detected with anti-Ras antibody using Western blot analysis.
In Vitro Raf Kinase Assay
The cells were lysed in 500 μl of ice-cold buffer A containing 1 mm β-glycerophosphate, 2.5 mm sodium pyrophosphate, and 1 mm sodium vanadate, and centrifuged at 12,000 × g for 10 min at 4 °C. The supernatant was incubated for 1 h at 4 °C with 1.5 μg of anti-C-Raf or anti-B-Raf antibodies, followed by incubation with 5 μl of protein G-Sepharose 4 Fast Flow beads for an additional hour. The beads were washed three times with 500 μl of buffer B and once with 500 μl of TBS. The immunoprecipitated Raf kinases were incubated in 20 μl of buffer containing 20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm MgCl2, 50 μm ATP, and 500 ng of His6-tagged MEK1-K97R (Upstate Biotechnology) at 30 °C for 20 min. Phosphorylated MEK1-K97R was detected with anti-phospho-MEK1/2 (Ser-217/221) antibody by Western blot.
Cell Fractionation
The cytosol and membrane fractions were isolated using ProteoExtractTM subcellular proteome extraction kit (Calbiochem), according to the manufacturer's instructions.
Fluorescence Microscopy
HeLa cells grown on type I collagen-coated glass coverslips were transiently transfected with pAcGFP-DGKη1, pDsRed-monomer-C-Raf, and either pECFP, pECFP-H-RasV12, or pECFP-H-RasN17. After 24 h of transfection, the cells were serum-starved for 5 h. The cells were fixed with 4% paraformaldehyde in phosphate-buffered saline for 10 min. The coverslips were mounted using Vectashield HardSet (Vector Laboratories). The cells were examined using a confocal laser scanning microscope (LSM510META; Carl Zeiss).
Assay of DGK Activity
The octyl glucoside-mixed micellar assay of DGK activity was performed as described previously (35).
RESULTS
Knockdown of DGKη Inhibits HeLa Cell Proliferation
To determine the roles of DGKη in cell function, initially an attempt was made to knock down the expression of DGKη by transfecting HeLa cells, which are derived from cervical cancer, with siRNA. DGKη-specific siRNA successfully silenced the expression of DGKη protein 48 and 96 h after transfection (Fig. 1A). Of the two DGKη isoforms, DGKη1 was predominantly expressed in HeLa cells, consistent with the mRNA expression patterns of most tissues and tumor-derived cells (31). Intriguingly, knockdown of DGKη in HeLa cells showed a significant proliferation defect 96 h after transfection with the siRNA (Fig. 1B). These results suggest that DGKη plays a key role in promoting cell growth.
FIGURE 1.

Silencing of DGKη expression inhibits HeLa cell proliferation. HeLa cells were transfected with control or DGKη-specific siRNA 1. After 48 and 96 h, the cells were harvested. A, cell lysates prepared from HeLa cells transfected with siRNAs and COS7 cells transfected with pcDNA3.1-DGKη1 or -η2 were separated by SDS-PAGE. The protein levels of DGKη and actin in cell lysates were analyzed using Western blot (WB). B, cells were counted with a hemocytometer. The cell numbers are shown as means ± S.D. (n = 3). Statistical significance was determined using Student's t test (*, p < 0.05). Results are representative of three independent experiments.
DGKη Activates the Ras/B-Raf/C-Raf/MEK/ERK Signaling Pathway Induced by EGF
To elucidate the mechanism by which DGKη enhances cell proliferation, EGF-induced ERK1/2 activation in DGKη-silenced HeLa cells was subsequently examined. EGF-induced ERK1/2 activation is known to be critical for cell growth. The ERK1/2 activation was assessed by its phosphorylation; EGF stimulation maximally induced phosphorylation of ERK1/2 within 5 min (Fig. 2A). As expected, DGKη depletion significantly inhibited the increase in ERK1/2 phosphorylation by ∼60 and ∼40% after 2 and 5 min of EGF stimulation, respectively. To confirm specific silencing of DGKη, another set of DGKη siRNA was used. As siRNA 2, which recognizes a DGKη sequence different from that of siRNA 1, efficiently inhibited the expression of DGKη, EGF-dependent phosphorylation of ERK1/2 was impaired (supplemental Fig. S1A). In addition, DGKη depletion by siRNA 3, which also recognizes a DGKη sequence different from those of siRNAs 1 and 2, also inhibited ERK1/2 phosphorylation induced by EGF (supplemental Fig. S1B). These results confirm that the down-regulation of ERK1/2 activation is because of the specific silencing of DGKη expression. In addition to HeLa cells, DGKη knockdown by siRNA also inhibited EGF-induced ERK1/2 activation in HEK293 cells derived from human embryonic kidney cells (supplemental Fig. S1C), indicating that the ERK activation by DGKη is not an event restricted to one cell line.
FIGURE 2.

Activation of ERK1/2 by EGF requires DGKη but not its catalytic activity. A, HeLa cells were transfected with control siRNA or DGKη siRNA 1. After 72 h, the cells were serum-starved for 5 h and stimulated with 100 ng/ml EGF for the indicated times. ERK1/2 phosphorylation and total ERK1/2 in the cell lysates were analyzed by Western blot (WB). Upper panels, representative results of Western blot analysis are shown. Bottom panel, the phospho-ERK1/2 levels were quantified by densitometry. Phospho-ERK1/2 levels in cells, which were treated with control siRNA and stimulated with EGF for 2 min, were set to 100. The data are shown as means ± S.D. of three independent experiments. B, HeLa cells were transfected with control siRNA or DGKη siRNA 1. After 24 h, the cells were further transfected with p3xFLAG vector or p3xFLAG-DGKη1-WT (wild-type) or -G392D (a kinase-dead mutant), which were introduced with siRNA-resistant mutations. After 48 h of transfection with the expression plasmids, the cells were serum-starved for 5 h and stimulated with EGF for 2 min. Phospho-ERK1/2, ERK1/2, 3xFLAG-tagged DGKη1, and DGKη in the cell lysates were detected by Western blot. Upper panels, representative results of Western blot analysis are shown. Bottom panel, visualized bands of phospho-ERK1/2 were quantified by densitometry. Phospho-ERK1/2 levels in the cells that were transfected with control siRNA and p3xFLAG vector were set to 100. The data are shown as means ± S.D. of three independent experiments.
To verify the positive effect of DGKη on ERK activity, complementation experiments were performed. DGKη-silenced HeLa cells were transiently transfected with a plasmid encoding wild-type DGKη1 (DGKη siRNA-resistant cDNA), followed by stimulation with EGF. As shown in Fig. 2B, wild-type DGKη1 successfully compensated for a defect in ERK1/2 activation induced by DGKη depletion, further supporting a critical role of DGKη in the regulation of the Ras/B-Raf/C-Raf/MEK/ERK signaling cascade.
To examine whether the catalytic activity of DGKη is required for EGF-induced activation of ERK1/2, DGKη-knocked down HeLa cells were transiently transfected with a plasmid encoding DGKη1-G392D (DGKη siRNA-resistant cDNA), a kinase-dead mutant where Gly-392 is replaced with Asp. It was confirmed that DGK activity of the G392D mutant was less than 1% of the wild-type control, measured by in vitro assay using their immunoprecipitants (data not shown). Unexpectedly, the G392D mutant exhibited almost the same effect as the wild-type enzyme (Fig. 2B). Although the possibility that subtle phosphatidic acid produced by DGKη is involved in ERK1/2 activation by EGF cannot be excluded, this study supports the model that DGKη activates the ERK1/2 signaling pathway by acting as a scaffold/adaptor protein.
To identify a target molecule(s) of DGKη in the Ras/B-Raf/C-Raf/MEK/ERK pathway, the effects of DGKη silencing on EGF-induced phosphorylation of upstream kinases of ERK1/2, MEK1/2 was subsequently examined. Phosphorylation of MEK1/2 reached a maximum by 2 min after EGF stimulation and returned to almost a basal level within 10 min (Fig. 3). Depletion of DGKη also inhibited MEK1/2 phosphorylation by ∼60% after 2 and 5 min of EGF stimulation, respectively, indicating that a target component of DGKη is at least upstream of MEK1/2.
FIGURE 3.

Activation of MEK1/2 by EGF requires DGKη. The HeLa cells were transfected with control siRNA or DGKη siRNA 1. After 72 h, the cells were serum-starved for 5 h and stimulated with 100 ng/ml EGF for the indicated times. Phospho-MEK1/2 at Ser-217/221, total MEK1/2, and DGKη in the cell lysates were detected using Western blot (WB). Upper panels, representative results of Western blot analysis are shown. Bottom panel, phospho-MEK1/2 levels were quantified by densitometry. Phospho-MEK1/2 levels in the cells, which were treated with control siRNA and stimulated with EGF for 2 min, were set to 100. The data are shown as means ± S.D. of three independent experiments.
Some DGK isozymes (α, ζ, and ι) have been reported to influence the signaling of Ras, an upstream molecule of the B-Raf/C-Raf/MEK/ERK pathway, by modulating activities of Ras guanyl nucleotide-releasing protein (23, 24, 36). Therefore, the effects of DGKη on Ras activation induced by EGF were investigated. After knockdown of DGKη expression by siRNA, endogenous levels of Ras-GTP were measured by the GST-Raf-RBD pulldown assay. The level of Ras-GTP maximally increased 2 min after EGF stimulation (Fig. 4A). However, DGKη depletion had no significant effect on EGF-induced Ras activation. It was examined whether the inhibition of MEK1/2 activities by DGKη depletion occurs when RasV12, a constitutively active form of Ras, is expressed in the cells. Transfection with DGKη siRNA into cells markedly inhibited the MEK1/2 activation induced by RasV12 expression as observed in EGF-stimulated cells (Fig. 4B). These results exclude the possibility that DGKη directly affects the activities of Ras-GEFs or Ras-GAPs in the EGF-stimulated cells. Thus, the data also indicate that a target component of DGKη is at least downstream of Ras.
FIGURE 4.

DGKη does not affect the EGF-induced activation of Ras but regulates the MEK1/2 activities in a Ras-dependent manner. A, HeLa cells were transfected with control siRNA or DGKη siRNA 1. After 72 h, the cells were serum-starved for 5 h and stimulated with 100 ng/ml EGF for the indicated times. Ras-GTP precipitated with GST-Raf-RBD and total Ras in the cell lysates were detected using Western blot (WB). Upper panels, representative results of Western blot analysis are shown. Bottom panel, visualized bands of Ras-GTP were quantified by densitometry. Ras-GTP levels in the cells that were treated with control siRNA and stimulated with EGF for 2 min were set to 100. The data are shown as means ± S.D. of three independent experiments. B, HeLa cells were transfected with control siRNA or DGKη siRNA 1. After 24 h, the cells were transfected with pcDNA3.1 or pCMV-H-RasV12. After 48 h of transfection with the expression plasmids, the cells were serum-starved for 5 h. The cells transfected with pcDNA3.1 were stimulated with 100 ng/ml EGF as controls (EGF) for 2 min. Phospho-MEK1/2, MEK1/2, and Ras were detected by Western blot (WB). Upper panels, representative results of Western blot analysis are shown. Bottom panel, visualized bands of phospho-MEK1/2 were quantified by densitometry. Phospho-MEK1/2 levels in the cells that were transfected with control siRNA and pcDNA3.1 and stimulated with EGF for 2 min were set to 100. The data are shown as means ± S.D. of three independent experiments.
DGKδ belongs to type II DGKs and is closely related to DGKη (30). To evaluate the specificity of the effect of DGKη on MEK1/2 phosphorylation induced by active Ras (Fig. 4B), expression of DGKδ was knocked down by transfection with siRNA into HeLa cells. However, depletion of DGKδ did not affect MEK1/2 activation by RasV12 (supplemental Fig. S2). Collectively, these results strongly suggest that DGKη specifically regulates the MEK/ERK pathway downstream of Ras.
DGKη Is Required for EGF-dependent Activation of C-Raf but Not B-Raf
B-Raf and C-Raf fulfill the criteria that a molecule, the target component of DGKη obtained earlier, acts downstream of Ras as well as upstream of MEK in the Ras/B-Raf/C-Raf/MEK/ERK cascade. To observe the effects of DGKη on Raf kinase activities, endogenous C-Raf and B-Raf in the lysates from DGKη-silenced HeLa cells were immunoprecipitated with anti-C-Raf and B-Raf antibodies, respectively, and used for in vitro Raf kinase assay. EGF clearly induced the activation of C-Raf within 2 min after stimulation (Fig. 5A). Interestingly, DGKη silencing remarkably inhibited C-Raf kinase activities prepared from EGF-stimulated cells. In contrast, the knockdown of DGKη did not affect B-Raf activities (Fig. 5B). MEK phosphorylation by the Raf kinases in vitro, but not the endogenous MEK phosphorylation that was carried out, was verified by abolishing the phosphorylation caused by withdrawal of ATP from the assay mixtures. These experiments demonstrate that DGKη is indispensable for the activation of C-Raf, but not for B-Raf, in response to EGF.
FIGURE 5.

DGKη is required for kinase activation of C-Raf in response to EGF but not B-Raf. HeLa cells were transfected with control siRNA or DGKη siRNA 1. After 72 h, the cells were serum-starved for 5 h and stimulated with 100 ng/ml EGF for 2 min. Endogenous C-Raf and B-Raf in the cell lysates were immunoprecipitated with anti-C-Raf antibody (A) and anti-B-Raf antibody (B), respectively. A, C-Raf kinase activities of the immunoprecipitants were measured in vitro using His6-tagged MEK1-K97R as a substrate in the presence or absence of ATP. Phosphorylation of His6-tagged MEK1-K97R was analyzed by Western blot (WB) using anti-phospho-MEK1/2 antibody. Upper panels, representative results of Western blot analysis are shown. Bottom panel, C-Raf activities were measured in the presence of ATP, and visualized bands of phospho-MEK1 were quantified by densitometry. C-Raf activities of the cells that were transfected with control siRNA and stimulated with EGF for 2 min were set to 100. The data are shown as means ± S.D. of three independent experiments. B, B-Raf kinase activities of immunoprecipitants were measured and quantified using the same methods as the C-Raf activity assay described in A. Upper panels, representative results of Western blot analysis are shown. Bottom panel, B-Raf activities of the cells that were transfected with control siRNA and stimulated with EGF for 2 min were set to 100. The data are shown as means ± S.D. of three independent experiments.
DGKη Activates C-Raf Downstream or Independently of Ras-C-Raf Interaction, C-Raf Phosphorylation, Feedback C-Raf Phosphorylation by ERK, and C-Raf Interaction with 14-3-3 Protein
C-Raf activity is known to be regulated by five pathways as follows: (a) interaction of C-Raf with activated Ras (7, 37); (b) phosphorylation at Ser-338, Tyr-341, Thr-491, and Ser-494, and dephosphorylation at Ser-259 in C-Raf (8–11); (c) interaction of C-Raf with 14-3-3 protein (14, 38); (d) feedback phosphorylation of C-Raf by ERK (12, 13); and (e) heterodimerization of C-Raf with B-Raf (15–17). Thus, the next step was to determine the pathways that are involved in the DGKη-dependent C-Raf activation.
First, this study examined if the DGKη-dependent C-Raf activation is regulated by Ras-C-Raf interaction (7, 37). Immunoprecipitation experiments showed that DGKη depletion did not affect the binding of RasV12 to C-Raf (supplemental Fig. S3), indicating that DGKη regulates the C-Raf activity downstream of the active Ras-dependent activation.
Activity of C-Raf is known to be regulated by phosphorylation. Dephosphorylation at Ser-259 and phosphorylation at Ser-338, Tyr-341, Thr-491, and Ser-494 play critical roles in C-Raf activation in response to growth factors (3). A test was conducted to determine whether DGKη influences the C-Raf phosphorylation by EGF. However, DGKη depletion by siRNA did not significantly affect the C-Raf phosphorylation of Ser-259 (data not shown) or Ser-338 (supplemental Fig. S4A). C-Raf phosphorylation at Tyr-341, Thr-491, or Ser-494 could not be detected with available phosphoantibodies even in cells stimulated with EGF. This could probably be due to their limited levels of phosphorylation. As mutations of Ser-338 to Asp/Tyr-341 to Asp/Thr-491 to Glu/Ser-494 to Asp (DDED) in C-Raf were reported to mimic their phosphorylation and confer constitutive activity (10), we alternatively examined the effects of DGKη depletion on MEK1/2 phosphorylation induced by transient expression of the C-Raf-DDED mutant in HeLa cells. The C-Raf-DDED mutant remarkably elevated MEK1/2 phosphorylation without any stimulation (supplemental Fig. S4B). However, the effects of DGKη depletion on MEK1/2 activation were clearly detected even when the C-Raf-DDED mutant was expressed. These results strongly suggest that DGKη activates C-Raf downstream or independently of the phosphorylation of these residues.
The 14-3-3 proteins were repeatedly reported to interact with and regulate C-Raf (14, 38). Although the interaction of C-Raf with 14-3-3 proteins in DGKη-depleted cells was examined, no significant difference in the interaction was observed with or without EGF stimulation (supplemental Fig. S5). Thus, it has been suggested that DGKη modulates C-Raf activity downstream or independently of the interaction between C-Raf and the 14-3-3 protein.
Recent studies have shown that feedback of C-Raf phosphorylation by ERK modulates C-Raf activity and that treatment with U0126, a MEK inhibitor, inhibited hyperphosphorylation of C-Raf (12, 13). The effect of U0126 on the in vitro activity of C-Raf isolated from EGF-stimulated HeLa cells was tested. U0126 did not affect the in vitro C-Raf activity 2 min after EGF stimulation despite a strong inhibition of ERK1/2 phosphorylation (supplemental Fig. S6), indicating that feedback C-Raf phosphorylation by ERK is not involved in DGKη-dependent activation of C-Raf under the conditions employed.
DGKη Enhances EGF-induced Heterodimerization of B-Raf and C-Raf
Recent studies have reported that active Ras and growth factor stimulation induce heterodimerization of B-Raf and C-Raf (15, 17). Heterodimerization of B-Raf and C-Raf exhibits a potent Raf kinase activity in vitro (17). B-Raf, which is biochemically purified as an activator of C-Raf (39), behaves as an upstream activator of C-Raf (16, 40). Finally, it was examined whether DGKη influences a complex formation between B-Raf and C-Raf. To this end, endogenous C-Raf in cell lysates was immunoprecipitated with anti-C-Raf antibody, and co-immunoprecipitated B-Raf was detected using Western blot. Upon stimulation with EGF, endogenous B-Raf was clearly co-precipitated (Fig. 6A). Interestingly, the depletion of DGKη by siRNA significantly inhibited the complex formation between C-Raf and B-Raf. Consistent with these findings, DGKη knockdown was also observed to inhibit heterodimerization of B-Raf and C-Raf in cells that transiently expressed RasV12 (supplemental Fig. S7).
FIGURE 6.

Heterodimerization of B-Raf with C-Raf induced by EGF requires DGKη. A, HeLa cells were transfected with control siRNA or DGKη siRNA 1. After 72 h, the cells were serum-starved for 5 h and stimulated with 100 ng/ml EGF for 2 min. Endogenous C-Raf in the cell lysates was immunoprecipitated with anti-C-Raf antibody. B-Raf associated with C-Raf was detected by Western blot (WB). Normal rabbit IgG was used as a negative control. Upper panels, representative results of Western blot analysis are shown. Bottom panel, visualized bands of co-precipitated B-Raf were quantified by densitometry. Relative intensities of the interaction are shown as means ± S.D. of three independent experiments. B, HeLa cells were transfected with control and B-Raf, C-Raf, and DGKη-specific siRNAs. After 72 h, the cells were serum-starved for 5 h and stimulated with 100 ng/ml EGF for 2 min. Phospho-MEK1/2, MEK1/2, B-Raf, C-Raf, and DGKη were detected by Western blot using their specific antibodies. Upper panels, representative results of Western blot analysis are shown. Bottom panel, phospho-MEK1/2 levels were quantified by densitometry. Phospho-MEK1/2 levels in the cells, which were treated with control siRNA and stimulated with EGF for 2 min, were set to 100. The data are shown as means ± S.D. of three independent experiments. TCL, total cell lysates; IP, immunoprecipitate.
The next attempt was to confirm whether B-Raf, in addition to C-Raf, plays an essential role in MEK1/2 phosphorylation in HeLa cells by silencing its expression using siRNA. A B-Raf-specific siRNA successfully knocked down the expression of B-Raf in HeLa cells (Fig. 6B). Depletion of B-Raf remarkably inhibited MEK1/2 phosphorylation induced by EGF, as observed in DGKη-knocked down cells, supporting that B-Raf is required for DGKη-dependent activation of C-Raf. It was verified that the silencing of C-Raf also significantly inhibited EGF-dependent MEK1/2 phosphorylation. Taken together, these results strongly suggest that DGKη regulates C-Raf activity, at least partly, through controlling the heterodimerization between C-Raf and B-Raf.
DGKη Interacts with B-Raf and C-Raf
Earlier experiments showed that DGKη regulates heterodimerization between B-Raf and C-Raf (Fig. 6 and supplemental Fig. S7). Moreover, it was previously suggested that DGKη activates the ERK1/2 signaling pathway by acting as a scaffold/adaptor protein (Fig. 2B). Therefore, we determined whether DGKη physically interacts with B-Raf and C-Raf. To this end, 3xFLAG-tagged DGKη1 was transiently expressed in HeLa cells and immunoprecipitated with anti-FLAG antibody. When DGKη1 was immunoprecipitated, endogenous B-Raf and C-Raf were apparently co-precipitated (Fig. 7A). As their interaction did not show any dependence on EGF stimulation or RasV12 expression (data not shown), DGKη1 may stably interact with B-Raf and C-Raf in cells. To examine the region of DGKη that is responsible for the interaction with C-Raf, HeLa cells were transfected with the plasmids encoding the 3xFLAG-tagged N- or C-terminal half of DGKη1 (Fig. 7B). After immunoprecipitation, the C-terminal half of DGKη1 (DGKη1-(329–1164)) interacted with C-Raf as strongly as the full-length of DGKη1, whereas the N-terminal half of DGKη (DGKη1-(1–328)) almost failed to interact with C-Raf (Fig. 7C). These immunoprecipitation experiments revealed that the C-terminal half of the DGKη1 containing catalytic domains are responsible for the interaction between DGKη1 and C-Raf. Further deletion analysis showed that DGKη interacts with C-Raf via multiple sites in the C-terminal half (data not shown). The regions of DGKη1 responsible for their weak interaction with B-Raf could not be accurately determined. To confirm that DGKη is physiologically associated with C-Raf, interaction of endogenous DGKη with C-Raf in HEK293 cells was examined. Consistent with the results obtained from transfected cells, endogenous C-Raf was slightly but clearly co-immunoprecipitated with DGKη (Fig. 7D).
FIGURE 7.

DGKη1 interacts with B-Raf and C-Raf. A, HeLa cells were transiently transfected with p3xFLAG vector or p3xFLAG-DGKη1. After 48 h, the cells were lysed. 3xFLAG-tagged DGKη1 in the cell lysates was immunoprecipitated using anti-FLAG M2 affinity gel. The cell lysates (Input) were also subjected to Western blot (WB) analysis. B-Raf, C-Raf, and FLAG-tagged protein in the cell lysates and immunoprecipitants were detected using their specific antibodies as indicated. Similar results were obtained in three independent experiments. B, schematic diagrams of the DGKη1 truncated mutants used are shown. C, HeLa cells were transfected with plasmids encoding 3xFLAG-tagged DGKη1 mutants. After 48 h, the cells were lysed. 3xFLAG-tagged DGKη1 in the cell lysates (Input) was immunoprecipitated using anti-FLAG M2 affinity gel. Co-immunoprecipitation of C-Raf with DGKη1 mutants was detected by Western blot (WB). The cell lysates (Input) were also subjected to Western blot analysis. Similar results were obtained in three independent experiments. D, HEK293 cell extracts were immunoprecipitated with anti-DGKη antibody or pre-immune serum. Endogenous C-Raf and DGKη were detected by Western blot using anti-C-Raf and anti-DGKη antibodies, respectively. With longer exposures, DGKη was detectable in the Input lane. Input represents 2% of the starting materials. IP, immunoprecipitate; PH, pleckstrin homology.
DGKη Enhances Recruitment of B-Raf and C-Raf to Membranes Induced by EGF
As the translocation of B-Raf and C-Raf to membranes where their upstream factor, Ras, is activated is thought to be an important step to associate with each other and activate C-Raf (16), it was necessary to determine whether DGKη knockdown affects the subcellular localizations of B-Raf and C-Raf. A cell fractionation assay showed that EGF stimulation apparently recruited both B-Raf and C-Raf to membrane fractions (Fig. 8A). DGKη silencing by siRNA significantly inhibited translocation of both B-Raf and C-Raf from cytosol to membranes after EGF stimulation. The recruitment of both B-Raf and C-Raf was also seen in cells transiently expressing RasV12 (Fig. 8B). Consistent with EGF stimulation, DGKη depletion by siRNA significantly inhibited recruitment of both B-Raf and C-Raf to membranes induced by RasV12 expression. Activated Ras is known to recruit B-Raf and C-Raf to the plasma membrane, leading to activation of the Raf kinases (41). However, as shown in supplemental Fig. S3, immunoprecipitation experiments showed that DGKη depletion did not affect the binding of RasV12 to C-Raf. The binding of B-Raf to RasV12 could not be detected, because the levels of RasV12 interacting with endogenous B-Raf were too low to detect in Western blot. These results suggest that DGKη enhances, at least, the recruitment of C-Raf to membranes in a Ras-C-Raf interaction-independent manner.
FIGURE 8.

DGKη is necessary for recruitment of B-Raf and C-Raf to membranes induced by EGF and active Ras. HeLa cells were transfected with control siRNA or DGKη siRNA 1. A, after 72 h, the cells were serum-starved for 5 h and stimulated with 100 ng/ml EGF for 2 min. B, after 24 h of transfection with siRNAs, the cells were transiently transfected with either pcDNA3.1 or pCMV-H-RasV12. After 48 h of transfection with the expression plasmids, the cells were serum-starved for 5 h. The cytosol and membrane fractions were isolated using ProteoExtract subcellular proteome extraction kit. Each fraction (10 μl) was analyzed by Western blot (WB) using anti-B-Raf and anti-C-Raf antibodies. Upper panels, representative results of Western blot analysis are shown. Bottom panel, intensity of the each band was quantified by densitometry, and the percentages of B-Raf and C-Raf in the membrane fractions are shown as the means ± S.D. of three independent experiments. Statistical significance was determined using Student's t test (*, p < 0.05; **, p < 0.01). C, co-localization of DGKη1 with RasV12 and C-Raf. HeLa cells grown on type I collagen-coated glass coverslips were co-transfected with plasmids encoding AcGFP1-tagged DGKη1, DsRed-monomer-tagged C-Raf, and either ECFP, ECFP-tagged H-RasV12, or ECFP-tagged H-RasN17. After 24 h of transfection, the cells were serum-starved for 5 h. The cells were fixed with 4% paraformaldehyde and observed by confocal laser scanning microscopy. ECFP-, AcGFP1-, and DsRed-monomer fusion proteins are shown in blue, green, and red, respectively. Scale bar, 10 μm.
Subcellular localization of DGKη was analyzed. In the absence of dominant-active Ras (RasV12), DGKη was located in the cytoplasm (Fig. 8C). On the other hand, RasV12 enhanced translocation of DGKη from the cytoplasm to the plasma membrane and concomitant co-localization of DGKη with the active Ras and C-Raf. However, dominant-negative Ras (RasN17) did not show such effects. Taken together, these results imply that, in addition to an interaction between C-Raf and active Ras, the translocation of DGKη carries at least DGKη-associated C-Raf to the plasma membrane where active Ras and B-Raf are located. Reciprocally, DGKη can also convey B-Raf to the membrane in which active Ras and C-Raf exist.
DISCUSSION
The Ras/B-Raf/C-Raf/MEK/ERK signaling cascade is essential for the transduction of extracellular signals to the nucleus, regulating a wide variety of pathophysiological processes such as cell proliferation, differentiation, and survival and carcinogenesis (1, 2). This study demonstrated for the first time that DGKη is a critical component in the Ras/B-Raf/C-Raf/MEK/ERK signaling pathway activated by EGF in HeLa cells (Fig. 9). In addition to HeLa cells derived from cervical cancer, DGKη critically controlled the EGF-induced ERK1/2 activation in HEK293 cells derived from human embryonic kidney cells also (supplemental Fig. S1C), indicating that the ERK pathway regulation by DGKη is not an event restricted to one cell line. DGKη transcripts were clearly detectable in most of the normal tissues (31). Therefore, these results may lend support to the view that DGKη regulates a wide variety of physiological events including cell growth (e.g. see Fig. 1) through controlling the Ras/B-Raf/C-Raf/MEK/ERK signaling pathway. Moreover, we had previously reported that DGKη was also expressed in all of the tumor-derived cells examined. Intriguingly, high DGKη expression was detected in 8 of 10 stomach cancers compared with the adjacent noncancerous stomach.4 Thus, it can be speculated that, in addition to proliferation of normal cells, DGKη may play an important role in pathological processes, development, and/or progression of human cancers, including stomach cancer, via regulating the Ras/B-Raf/C-Raf/MEK/ERK signaling pathway.
FIGURE 9.
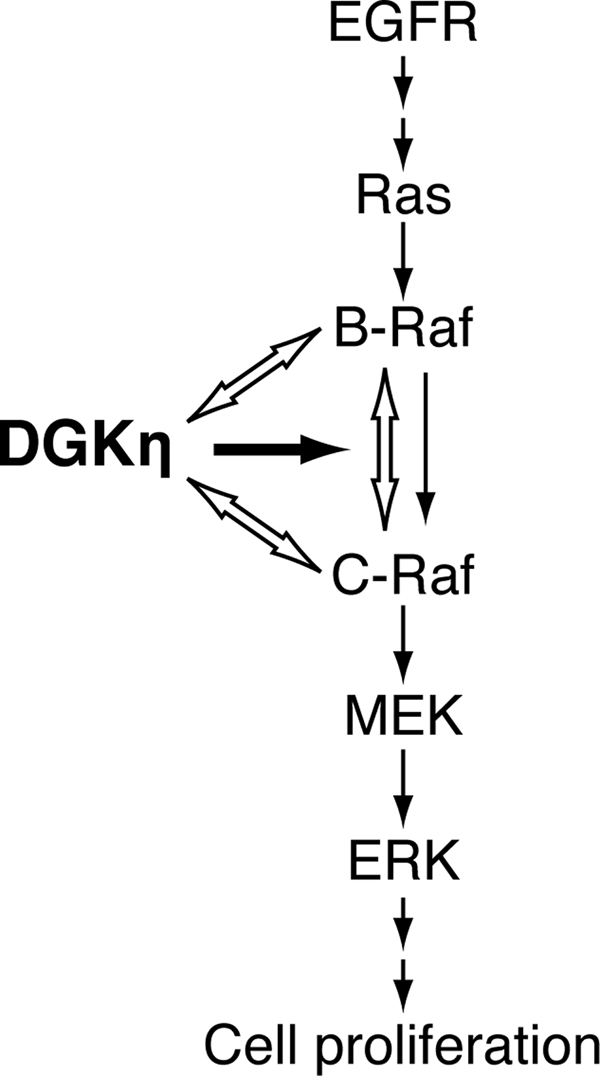
A working model of DGKη-mediated C-Raf activation. The data are consistent with a model in which DGKη physically interacts with B-Raf and C-Raf and enhances EGF-dependent heterodimerization between B-Raf and C-Raf. As B-Raf activates C-Raf via the heterodimerization with C-Raf, DGKη augments C-Raf activity. Black and white arrows indicate “activation” and “interaction,” respectively.
B-Raf and C-Raf are the pivotal regulatory components in the Ras/B-Raf/C-Raf/MEK/ERK signaling cascade (3). Recent studies have reported that heterodimerization of B-Raf and C-Raf is required to exhibit a potent Raf kinase activity (17). Interaction of C-Raf with wild-type B-Raf occurs at the plasma membranes, which is dependent on active Ras (16). In addition, B-Raf activates C-Raf, but C-Raf does not activate B-Raf. However, the molecular mechanism of their heterodimerization is poorly understood. In this study, it was demonstrated that DGKη is a novel critical activator of C-Raf but not B-Raf (Fig. 5). Moreover, DGKη enhanced B-Raf-C-Raf heterodimer formation (Fig. 6A), which is essential for C-Raf activation. Moreover, DGKη physically interacted with B-Raf and C-Raf (Fig. 7). Thus, the data reveal DGKη to be a novel key regulator of B-Raf-C-Raf heterodimer formation and subsequent C-Raf activation. Moreover, because B-Raf is essential for the DGKη-dependent C-Raf activation (Fig. 6B), it is strongly suggested that DGKη at least in part regulates C-Raf activity through controlling the heterodimerization between C-Raf and B-Raf. The B-Raf-C-Raf heterodimer formation was reported to be regulated by interaction of C-Raf with 14-3-3 protein (17). However, knockdown of DGKη failed to affect the interaction of C-Raf with 14-3-3 (supplemental Fig. S5), indicating that the DGKη-dependent augmentation of B-Raf-C-Raf heterodimerization occurs independently or downstream of the 14-3-3 protein binding to C-Raf. Rheb, a member of the Ras/Rap/Ral subfamily of Ras proteins, was recently reported to inhibit C-Raf activity and heterodimerization of B-Raf and C-Raf (42). In contrast, DGKη is required for the activation of C-Raf and heterodimerization. Thus, it is possible that DGKη and Rheb reversely regulate each other. However, it is unlikely that DGKη is involved in the Rheb-dependent C-Raf activation mechanism, for the following reasons. (a) Rheb interacts with only B-Raf, but not with C-Raf to which DGKη is bound (Fig. 7). (b) Rheb regulates C-Raf phosphorylation at Ser-338, which is not affected by DGKη knockdown (supplemental Fig. S4). (c) Rheb inhibits kinase activity of B-Raf but not C-Raf, which is regulated by DGKη (Fig. 5).
Expression of DGKη could compensate for ERK phosphorylation impaired by DGKη-siRNA treatment, independent of the catalytic activity of DGKη (Fig. 2B). It is difficult to rule out the possibility that subtle phosphatidic acid produced by DGKη influences the activation of the ERK pathway, because the catalytic activity that remained slightly in the DGKη-G392D mutant may introduce effective levels of phosphatidic acid by its overexpression. It is likely that DGKη could serve as a scaffold/adaptor protein. Indeed, DGKη is physically bound to B-Raf and C-Raf (Fig. 7). These observations support a paradigm that regulation of C-Raf activation is controlled within the assembled signaling complex composed of B-Raf, C-Raf, and DGKη. Hence, it appears that DGKη does not simply perform the mandated task of converting diacylglycerol to phosphatidic acid, both signaling lipids, but unexpectedly function as a scaffold/adaptor protein bridging the integral members of the MEK/ERK pathway, B-Raf, and C-Raf. As DGKη obviously exhibits a catalytic activity (31), we speculate that, in addition to the function of scaffold/adaptor protein, DGKη has an unknown primary function via its catalytic action. If the catalytic activity-dependent function is explored, then DGKη may become a new nexus/crossroad linking the Ras/B-Raf/C-Raf/MEK/ERK cascade and an unidentified signaling lipid (diacylglycerol and/or phosphatidic acid)-regulated pathway.
DGKη positively regulates the EGF-induced translocation of B-Raf and C-Raf from cytosol to membranes (Fig. 8). The DGKη-dependent translocation is likely to contribute to the heterodimer formation processes, because heterodimerization has been reported to occur at the plasma membrane (16), where their upstream regulator/interaction partner, Ras, is activated. DGKη was associated with B-Raf and C-Raf, and the interactions were not affected by EGF stimulation. Thus, translocation of DGKη to the plasma membrane and its co-localization with active Ras (Fig. 8C) are likely to enhance translocation of at least DGKη-associated C-Raf. Because active Ras and B-Raf are located on the plasma membrane, the concomitant translocation of DGKη-associated C-Raf to the plasma membrane may facilitate heterodimerization of C-Raf with B-Raf. However, the possibility that DGKη has other effects on B-Raf and C-Raf, such as induction of their conformational changes and enhancements of interactions with their regulatory proteins, cannot be denied.
DGKη is known to consist of two splice variants, DGKη1 and -η2. DGKη2 possesses a SAM domain at its C terminus but not DGKη1. The SAM domain is responsible for homo- and hetero-oligomerization of DGKη and DGKδ (31, 43). Unfortunately, as the expression levels of DGKη2 were considerably low in complementation experiments, it was not possible to evaluate whether the SAM domain is essential for DGKη-dependent activation of the ERK pathway. However, the SAM domain does not seem to contribute to the interaction of DGKη with C-Raf because both the DGKs, η1 and η2, interacted with C-Raf to almost the same extent in the immunoprecipitation assays (data not shown). DGKη1 was detected in most of the normal tissues and all the examined tumor-derived cells, including HeLa cells (Fig. 1A) (31). On the other hand, DGKη2 was expressed in limited tissues. As the Ras/B-Raf/C-Raf/MEK/ERK pathway is a central signaling cascade in most of the tissues and cell lines (1–3), the results imply that DGKη1 is mainly responsible for the regulation of the Ras/B-Raf/C-Raf/MEK/ERK signaling pathway.
DGKδ possessing pleckstrin homology and SAM domains also belongs to type II DGKs containing DGKη. Analysis using DGKδ knock-out mice revealed that knock-out of DGKδ attenuates EGF receptor (EGFR) phosphorylation by protein kinase C, resulting in degradation and impaired signaling of EGFR (22). In this study, DGKη is also observed to regulate EGFR signaling by activating C-Raf but not EGFR itself (Fig. 6A). Thus, δ- and η-isoforms belonging to the same type of DGK (type II) control the same signaling pathway, the EGFR/Ras/B-Raf/C-Raf/MEK/ERK cascade. However, they modulate different sites, EGFR and C-Raf, in the pathway. DGKs, δ and η, utilize different control mechanisms by consuming a protein kinase C activator, diacylglycerol, in a catalytic activity-dependent manner and by acting as a scaffold/adaptor protein in a catalytic activity-independent manner, respectively. It is therefore evident that DGK isoforms, even in the same subgroup, regulate important cell signal transductions via a variety of mechanisms.
The results of this study reveal a previously unappreciated role for DGKη and expanded the repertoire of DGK function to the scaffold/adaptor protein. The identification of DGKη as a novel key regulator of heterodimerization of C-Raf and B-Raf (Fig. 9) may help to elucidate the activation mechanism of C-Raf by B-Raf, which is poorly understood at present, despite its importance in a wide variety of pathophysiological processes, such as cell proliferation and tumorigenesis. Further study on the mechanism underlying the Raf signaling regulated by DGKη is needed to better understand the complex regulation of an important signaling pathway, the Ras/B-Raf/C-Raf/MEK/ERK cascade.
Supplementary Material
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Northern Advancement Center for Science and Technology of Hokkaido, Japan, the Japan Diabetes Foundation, The Suhara Memorial Foundation, Novo Nordisk Pharma Ltd. (Japan), The Takeda Science Foundation, The Suzuken Memorial Foundation, and The Akiyama Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7.
S. Yasuda, M. Takeishi, A. Takemoto, and F. Sakane, unpublished data.
- MEK
- mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- DGK
- diacylglycerol kinase
- EGF
- epidermal growth factor
- ERK
- extracellular signal-regulated kinase
- GST
- glutathione S-transferase
- RBD
- Ras-binding domain
- SAM
- sterile α-motif
- TBS
- Tris-buffered saline
- siRNA
- small interfering RNA
- EGFR
- EGF receptor.
REFERENCES
- 1.Marshall C. J. (1995) Cell 80, 179–185 [DOI] [PubMed] [Google Scholar]
- 2.Kolch W. (2000) Biochem. J. 351, 289–305 [PMC free article] [PubMed] [Google Scholar]
- 3.Wellbrock C., Karasarides M., Marais R. (2004) Nat. Rev. Mol. Cell Biol. 5, 875–885 [DOI] [PubMed] [Google Scholar]
- 4.Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M. J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., Hall S., Hawes R., Hughes J., Kosmidou V., Menzies A., Mould C., Parker A., Stevens C., Watt S., Hooper S., Wilson R., Jayatilake H., Gusterson B. A., Cooper C., Shipley J., Hargrave D., Pritchard-Jones K., Maitland N., Chenevix-Trench G., Riggins G. J., Bigner D. D., Palmieri G., Cossu A., Flanagan A., Nicholson A., Ho J. W., Leung S. Y., Yuen S. T., Weber B. L., Seigler H. F., Darrow T. L., Paterson H., Marais R., Marshall C. J., Wooster R., Stratton M. R., Futreal P. A. (2002) Nature 417, 949–954 [DOI] [PubMed] [Google Scholar]
- 5.Zebisch A., Staber P. B., Delavar A., Bodner C., Hiden K., Fischereder K., Janakiraman M., Linkesch W., Auner H. W., Emberger W., Windpassinger C., Schimek M. G., Hoefler G., Troppmair J., Sill H. (2006) Cancer Res. 66, 3401–3408 [DOI] [PubMed] [Google Scholar]
- 6.Hagemann C., Rapp U. R. (1999) Exp. Cell Res. 253, 34–46 [DOI] [PubMed] [Google Scholar]
- 7.Terai K., Matsuda M. (2005) EMBO Rep. 6, 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mason C. S., Springer C. J., Cooper R. G., Superti-Furga G., Marshall C. J., Marais R. (1999) EMBO J. 18, 2137–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhillon A. S., Meikle S., Yazici Z., Eulitz M., Kolch W. (2002) EMBO J. 21, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong H., Lee J., Guan K. L. (2001) EMBO J. 20, 3716–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubicek M., Pacher M., Abraham D., Podar K., Eulitz M., Baccarini M. (2002) J. Biol. Chem. 277, 7913–7919 [DOI] [PubMed] [Google Scholar]
- 12.Balan V., Leicht D. T., Zhu J., Balan K., Kaplun A., Singh-Gupta V., Qin J., Ruan H., Comb M. J., Tzivion G. (2006) Mol. Biol. Cell 17, 1141–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dougherty M. K., Müller J., Ritt D. A., Zhou M., Zhou X. Z., Copeland T. D., Conrads T. P., Veenstra T. D., Lu K. P., Morrison D. K. (2005) Mol. Cell 17, 215–224 [DOI] [PubMed] [Google Scholar]
- 14.Fischer A., Baljuls A., Reinders J., Nekhoroshkova E., Sibilski C., Metz R., Albert S., Rajalingam K., Hekman M., Rapp U. R. (2009) J. Biol. Chem. 284, 3183–3194 [DOI] [PubMed] [Google Scholar]
- 15.Weber C. K., Slupsky J. R., Kalmes H. A., Rapp U. R. (2001) Cancer Res. 61, 3595–3598 [PubMed] [Google Scholar]
- 16.Garnett M. J., Rana S., Paterson H., Barford D., Marais R. (2005) Mol. Cell 20, 963–969 [DOI] [PubMed] [Google Scholar]
- 17.Rushworth L. K., Hindley A. D., O'Neill E., Kolch W. (2006) Mol. Cell. Biol. 26, 2262–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mérida I., Avila-Flores A., Merino E. (2008) Biochem. J. 409, 1–18 [DOI] [PubMed] [Google Scholar]
- 19.Sakane F., Imai S., Kai M., Yasuda S., Kanoh H. (2007) Biochim. Biophys. Acta 1771, 793–806 [DOI] [PubMed] [Google Scholar]
- 20.Topham M. K., Epand R. M. (2009) Biochim. Biophys. Acta 1790, 416–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo B., Prescott S. M., Topham M. K. (2003) J. Cell Biol. 160, 929–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crotty T., Cai J., Sakane F., Taketomi A., Prescott S. M., Topham M. K. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 15485–15490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Regier D. S., Higbee J., Lund K. M., Sakane F., Prescott S. M., Topham M. K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 7595–7600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Topham M. K., Prescott S. M. (2001) J. Cell Biol. 152, 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo B., Prescott S. M., Topham M. K. (2004) Cell. Signal. 16, 891–897 [DOI] [PubMed] [Google Scholar]
- 26.Yasuda S., Kai M., Imai S., Kanoh H., Sakane F. (2007) FEBS Lett. 581, 551–557 [DOI] [PubMed] [Google Scholar]
- 27.Yasuda S., Kai M., Imai S., Kanoh H., Sakane F. (2008) Biochem. J. 409, 95–106 [DOI] [PubMed] [Google Scholar]
- 28.Kawasaki T., Kobayashi T., Ueyama T., Shirai Y., Saito N. (2008) Biochem. J. 409, 471–479 [DOI] [PubMed] [Google Scholar]
- 29.Kim K., Yang J., Zhong X. P., Kim M. H., Kim Y. S., Lee H. W., Han S., Choi J., Han K., Seo J., Prescott S. M., Topham M. K., Bae Y. C., Koretzky G., Choi S. Y., Kim E. (2009) EMBO J. 28, 1170–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klauck T. M., Xu X., Mousseau B., Jaken S. (1996) J. Biol. Chem. 271, 19781–19788 [DOI] [PubMed] [Google Scholar]
- 31.Murakami T., Sakane F., Imai S., Houkin K., Kanoh H. (2003) J. Biol. Chem. 278, 34364–34372 [DOI] [PubMed] [Google Scholar]
- 32.Sakane F., Imai S., Kai M., Wada I., Kanoh H. (1996) J. Biol. Chem. 271, 8394–8401 [DOI] [PubMed] [Google Scholar]
- 33.Sakane F., Imai S., Yamada K., Murakami T., Tsushima S., Kanoh H. (2002) J. Biol. Chem. 277, 43519–43526 [DOI] [PubMed] [Google Scholar]
- 34.Sharma A., Trivedi N. R., Zimmerman M. A., Tuveson D. A., Smith C. D., Robertson G. P. (2005) Cancer Res. 65, 2412–2421 [DOI] [PubMed] [Google Scholar]
- 35.Imai S., Kai M., Yasuda S., Kanoh H., Sakane F. (2005) J. Biol. Chem. 280, 39870–39881 [DOI] [PubMed] [Google Scholar]
- 36.Sanjuán M. A., Pradet-Balade B., Jones D. R., Martínez-A C., Stone J. C., Garcia-Sanz J. A., Mérida I. (2003) J. Immunol. 170, 2877–2883 [DOI] [PubMed] [Google Scholar]
- 37.Marais R., Light Y., Paterson H. F., Marshall C. J. (1995) EMBO J. 14, 3136–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajalingam K., Wunder C., Brinkmann V., Churin Y., Hekman M., Sievers C., Rapp U. R., Rudel T. (2005) Nat. Cell Biol. 7, 837–843 [DOI] [PubMed] [Google Scholar]
- 39.Mizutani S., Inouye K., Koide H., Kaziro Y. (2001) FEBS Lett. 507, 295–298 [DOI] [PubMed] [Google Scholar]
- 40.Wan P. T., Garnett M. J., Roe S. M., Lee S., Niculescu-Duvaz D., Good V. M., Jones C. M., Marshall C. J., Springer C. J., Barford D., Marais R. (2004) Cell 116, 855–867 [DOI] [PubMed] [Google Scholar]
- 41.Marais R., Light Y., Paterson H. F., Mason C. S., Marshall C. J. (1997) J. Biol. Chem. 272, 4378–4383 [DOI] [PubMed] [Google Scholar]
- 42.Karbowniczek M., Robertson G. P., Henske E. P. (2006) J. Biol. Chem. 281, 25447–25456 [DOI] [PubMed] [Google Scholar]
- 43.Imai S., Sakane F., Kanoh H. (2002) J. Biol. Chem. 277, 35323–35332 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.