

Abstract
Aminoacylphosphatidylglycerol synthases (aaPGSs) are multiple peptide resistance factors that transfer amino acids from aminoacyl-tRNAs to phosphatidylglycerol (PG) in the cytoplasmic membrane. Aminoacylation of PG is used by bacteria to decrease the net negative charge of the cell envelope, diminishing affinity for charged molecules and allowing for adaptation to environmental changes. Lys-PGS, which transfers lysine to PG, is essential for the virulence of certain pathogens, providing resistance to both host cationic antimicrobial peptides and therapeutic antibiotics. Ala-PGS was also recently described, but little is known about the possible activities of other members of the highly diverse aaPGS family of proteins. Systematic deletion of the predicted membrane-inserted domains of several aaPGSs revealed that the carboxyl-terminal hydrophilic domain alone is sufficient for aminoacylphosphatidylglycerol transferase catalytic activity. In contrast to previously characterized aaPGSs, the Enterococcus faecium enzyme used an expanded repertoire of amino acids to modify PG with Ala, Arg, or Lys. Reexamination of previously characterized aaPGSs also revealed broader than anticipated substrate specificity, for example Bacillus subtilis Lys-PGS was shown to also catalyze Ala-PG synthesis. The relaxed substrate specificities of these aaPGSs allows for more elaborate remodeling of membrane lipids than previously thought, potentially providing bacteria that harbor these enzymes resistance to a broad spectrum of antibiotics and environmental stresses.
To adapt to changing environmental conditions, such as those encountered during host infection, bacteria must alter the properties of their cellular envelope by adjusting the composition and abundance of their membrane lipids. Bacteria choose from a variety of lipid components that vary in fatty acid chain length and saturation levels and that bear a repertoire of different polar head groups. Aminoacylphosphatidylglycerol synthases (aaPGSs)2 are enzymes embedded within the bacterial membrane that are responsible for the transfer of amino acids from aminoacyl-tRNAs (aa-tRNAs) to the polar head groups of phosphatidylglycerol (PG). Addition of amino acids to membrane PG is a critical mechanism evolved by bacteria to decrease the net negative charge of the cell membrane (1). This alteration of charge diminishes the membrane affinity for cationic antimicrobial peptides used by the host immune system (i.e. defensins), and for other bactericidal agents (2). During infection, aaPGSs have been shown to be essential for the virulence of several pathogenic microorganisms by facilitating the evasion of antibiotic activity. Their role in virulence and their broad distribution in bacterial species make aaPGSs attractive targets for new therapeutic strategies to combat pathogenic microbes.
Lys-PG in the membrane allows for the evasion of neutrophils and enhances the virulence of Staphylococcus aureus both in mice (3) and in endovascular infection of rabbits (4). Similar observations were made for Listeria monocytogenes, in which Lys-PG enhances infectivity of epithelial cells and macrophages in mice (5, 6). Moreover, Lys-PG provides S. aureus with resistance to other classes of cationic bactericidal agents such as vancomycin (glycopeptide) and daptomycin (lipopeptide) (7, 8), which are often used as a last resort to treat infections. A new aaPGS with altered specificity was recently discovered in Clostridium perfringens and Pseudomonas aeruginosa (9, 10). This enzyme, Ala-PGS, is responsible for the formation of Ala-PG in the membrane. Despite the net neutral charge of Ala-PG, this modification has been shown to enhance bacterial resistance to certain cationic antimicrobial peptides (10). Both Lys-PG and Ala-PG have also been shown to enhance the resistance of S. aureus and P. aeruginosa to several negatively charged β-lactams (e.g. oxacillin, methicillin, cefsulodin) (7, 10, 11). These latter findings suggest that aaPGs confer antibiotic resistance both by diminishing the net negative charge of the membrane and by modulating more general biophysical properties such as membrane fluidity and permeability (12). In addition, aaPGs not only decrease membrane permeability to cationic antimicrobial peptides, but also to protons and osmolytes (e.g. lactate), providing bacteria with resistance to challenging osmotic or acidic growth conditions such as those encountered during fermentative growth (10, 13, 14).
Despite recent progress, many questions remain unanswered concerning PG aminoacylation in bacteria. Since the identification of Lys-PGS and Ala-PGS and the discovery of resistances associated with these enzymes the amino acid specificities of only seven aaPGSs have been directly characterized (2, 5, 9, 10, 13, 15–18). Predicted aaPGS sequences from different organisms display a remarkable level of secondary structural diversity. To date, 348 putative aaPGS genes in 213 distinct species have been identified in 96 different genera of microorganisms, covering almost all known groups of bacteria. Also, 49 of the available genome sequences encode two or more aaPGS paralogs. This dual occurrence is most often encountered in Gram-positive bacteria, particularly in members of the Actinobacteria class (e.g. Mycobacterium, Streptomyces, etc.). Here, we describe structure/function-based assignment of aaPGS catalytic domains that subsequently allowed us to identify new amino acid specificities of PG aminoacylation. The coexistence of several such activities in the same organism expands the set of unique aaPGSs that are available for tuning of membrane properties and may provide these bacteria with resistance to a far broader spectrum of antimicrobial agents and environmental conditions than previously appreciated. In addition, our work shows that although some aaPGSs display strict substrate specificity (e.g. Lys-PGS from C. perfringens), some aaPGSs display relaxed substrate specificity in vitro. For example, Enterococcus faecium aaPGS exhibits a triple specificity for Ala-, Arg-, and Lys-tRNA, and Bacillus subtilis displays a dual specificity for Lys- and, Ala-tRNA.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, and General Methods
The open reading frames encoding the aaPGSs from E. faecium DO (gi: 69245409 and 69249189) and Agrobacterium tumefaciens (gi: 15889786) were amplified by PCR and cloned under the control of the T7 promoter at the NcoI/XhoI restriction sites of the vector pet33b (kanR; Novagen). The same vector was previously used for the cloning of the aaPGSs from B. subtilis and C. perfringens (9, 15). Recombinant vectors were used to transform the Escherichia coli strain C41 DE3 containing the plasmid pRARE2 (camR; Novagen) expressing tRNAs for translation of rare codons. Protein production was performed according to procedures described before (15) in autoinduction medium incubated overnight at 30 °C under agitation. To remove the membrane domain of aaPGSs partially or totally, site-directed mutagenesis was performed with the QuikChange kit (Stratagene) according to the manufacturer's instructions. Affinity-tagged lysyl-tRNA synthetase (Lys-RS) and alanyl-tRNA synthetase (Ala-RS) from E. coli and arginyl-tRNA synthetase (Arg-RS) from jack bean were produced and purified according to procedures described previously (19–21).
Membrane Extracts and Lipid Analysis
Membrane suspensions from aaPGS-expressing strains were prepared by successive centrifugation steps. Briefly, the cells were disrupted by sonication in a buffer containing 50 mm Tris-HCl, pH 8.0, 1 mm diisopropylfluorophosphate, 1 mm phenylmethylsulfonyl fluoride, 3 mm 2-mercaptoethanol. Cell debris were removed by centrifugation for 15 min at 10,000 × g. Membranes were sedimented from the supernatant by ultracentrifugation for 45 min at 250,000 × g. Membrane suspension was obtained after gentle sonication of the pellet and was then stored at −80 °C in the lysis buffer containing 20% glycerol. The concentration of protein in the membrane suspension was 15–20 mg/ml, as measured by the method of Bradford (Bio-Rad). Total lipids were extracted from cells using the procedure of Bligh and Dyer (22) with 120 mm potassium acetate, pH 4.5, in the aqueous phase. Lipids were analyzed by TLC on Silica Gel HL plates (Analtech) developed in one dimension or two dimensions by using, successively, the solvents chloroform:methanol:water (14:6:1, vol/vol/vol) in the first dimension and chloroform:methanol:acetic acid (13:5:2, vol/vol/vol) in the second dimension. Lipids were visualized by fluorescence after staining with primuline.
tRNA-dependent AaPG Synthesis
aaPGS activity within the membrane suspensions obtained from aaPGS-producing strains was assayed in a reaction medium containing 100 mm Hepes-NaOH, pH 7.2, 30 mm KCl, and 15 mm MgCl2, 8 mm ATP, 2 mg/ml total tRNA from E. coli MRE 600 (Roche Applied Science), 20 μm 14C-labeled amino acids, 1 μm appropriate aminoacyl-tRNA synthetase, 3.25 mg/ml membrane extract, and a 2 mg/ml suspension of egg-PG (Avanti) prepared as described. After 30 min of incubation at 37 °C, lipids were extracted by the Bligh and Dyer procedure (22), dried, and resuspended in 20 μl of a solution of chloroform:methanol (2:1). 2 μl of the extract was then subjected to analysis by TLC (see above). Transcripts of tRNAArg from jack bean and tRNALys UUU from E. faecium were synthesized as previously described, and used in the aaPG biosynthesis assay in the presence of E. coli Lys-RS or jack bean Arg-RS, respectively.
Alignment and Phylogenetic Analysis
Sequences were retrieved by Blast (23) using the Lys-PGS from B. subtilis as a template. 437 aaPGS sequences displaying an E value < 10E-6 were aligned using ClustalX 2.0 (24). The alignment was split according to the three different domains present in aaPGSs (membrane, hydrophilic, and Lys-RS domains), and the sequences were independently realigned. Phylogenetic analyses were conducted with the Phylip package 3.68 with a representative subset of 230 sequences displaying <95% identity. The tree was constructed from a bootstrap of 100 replicates of a degapped alignment of 218 residues using the neighbor-joining method. A consensus tree was edited with iTOL (25). Prediction of transmembrane helices in aaPGSs was performed using TMHMM (26) and TOPCONS (27).
RESULTS
E. faecium Possesses a Dual-specificity Arg/Lys-PGS
Previous reports describe the presence of several amino acids associated with phosphatidylglycerol in the membrane of E. faecium (formerly Streptococcus faecalis) (28). These studies showed that Lys-PG is present, along with significant amounts of Arg-PG, in the membrane of this organism, suggesting the possible presence of two distinct aaPGSs responsible for the biosynthesis of these two aaPGs. The presence of two aaPGS paralogs responsible for the biosynthesis of two distinct aaPGs (Lys-PG and Ala-PG) has recently been reported in C. perfringens (9). Using B. subtilis Lys-PGS as a guide, BLAST search analyses of the publicly available genome sequences of E. faecium (E. faecium DO, TX1330, 1141733, 1231501, Com12, and Com15) revealed the presence of two aaPGS paralogs encoded in all six organisms. Both open reading frames (aaPGS1 and aaPGS2, accession numbers ZP_00603404 and ZP_00604896, respectively) from E. faecium DO were cloned in pet33b and expressed in E. coli. The aaPGS activities of membrane extracts from the producing strains were tested with Lys-tRNALys and Arg-tRNAArg as aminoacyl group donors. This analysis failed to reconstitute the activity of aaPGS1 but showed unambiguously the dual aa-tRNA specificity (Lys/Arg) of aaPGS2 (Fig. 1). These results suggest that the presence of Arg-PG and Lys-PG in the membrane of E. faecium may be due to a single aaPGS that bears two aa-tRNA specificities. Further in vitro characterization of these activities with tRNA transcripts (i.e. tRNALys UUU from E. faecium and tRNAArg from jack bean) revealed that aaPGS2 displays a specific activity for lysylation of PG only 6.5-fold higher than that for arginylation. This result prompted us to reinvestigate the specificities of known aaPGSs using an expanded range of aa-tRNAs and to determine the specificity of a previously uncharacterized aaPGS.
FIGURE 1.
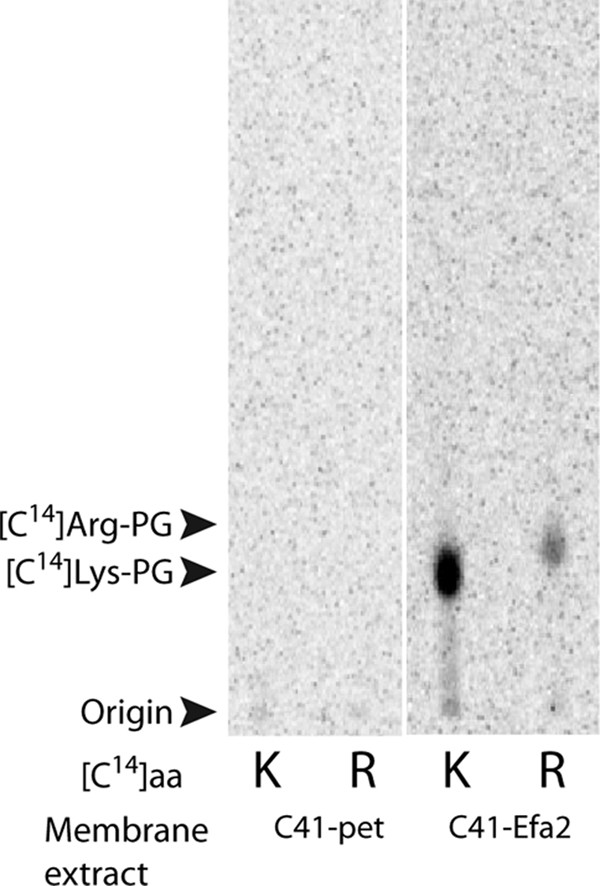
Activity of aaPGS2 from E. faecium. AaPGS activities in the membrane fraction of the E. coli strain C41 transformed with the vector pet33b empty (C41-pet) or producing aaPGS2 from E. faecium (C41-Efa2) were assayed as described under Experimental Procedures in the presence of [14C]Lys (K) or [14C]Arg (R).
AaPGSs Display Divergent Specificity for Aa-tRNAs in Vitro
The specificities of the Lys-PGS from B. subtilis and of the Ala-PGS from C. perfringens have been determined previously (9, 18) and were now reinvestigated using Lys-, Arg- and Ala-tRNA as substrates. An aaPGS homologous to Lys-PGS has also been reported in the Rhizobiales (i.e. Sinorhizobium medicae) for which it conferred resistance to lethal acidic conditions (14). However, the function of aaPGS in Rhizobiales remained unclear because no Lys-PG was detected (14). These previous results prompted us also to characterize the substrate specificity of a Rhizobiales aaPGS, in this case from the plant pathogen A. tumefaciens. All aaPGSs tested displayed activity in vivo (Fig. 2) and a moderate specificity toward their aa-tRNAs. For example, the B. subtilis enzyme, previously reported as a Lys-PGS, was also able to synthesize Ala-PG, but Arg-PG was not detected in this case (Fig. 2B). A. tumefaciens aaPGS synthesized Lys-PG, but no trace amounts of either Arg-PG or Ala-PG could be detected when this enzyme was assayed with Ala-tRNAAla and Arg-tRNAArg (Fig. 2C). Although the B. subtilis aaPGS displayed relaxed substrate specificity, Ala-PGS from C. perfringens exhibited a more stringent specificity for Ala-tRNAAla because neither Lys-PG nor Arg-PG was synthesized by this enzyme (Fig. 2D). Reexamination of E. faecalis aaPGS2 revealed that it was able to make Ala-PG in addition to Arg-PG and Lys-PG (Fig. 2E). Our findings show that known aaPGSs fall into three categories according to their specificities: (i) aaPGSs that display a single, strict, specificity (e.g. Ala-PGS from C. perfringens and Lys-PGS from A. tumefaciens) and produce no detectable traces of other aaPGs; (ii) aaPGSs with a “primary” specificity (i.e. Lys-PGS from B. subtilis) that are also capable of producing several other aaPGs less efficiently; and (iii) aaPGS with relaxed specificity (Ala/Arg/Lys-PGS from E. faecium) able to synthesize several aaPGs at comparable levels.
FIGURE 2.

Specificity of aaPGSs from various origins. Membrane extracts from the E. coli strain C41 containing the empty vector pet33b (C41-pet; A) or producing the aaPGS from B. subtilis (C41-bsu; B), A. tumefaciens (C41-Atu; C); C. perfringens (C41-Cpe; D); or E. faecium (C41-Efa2; E). Activities were assayed as described under Experimental Procedures in the presence of [14C]Lys (K), [14C]Arg (R), or [14C]Ala (A).
AaPGS Structural Heterogeneity
To correlate the divergent specificity of aaPGSs with structural features, 230 representative sequences from 204 organisms were aligned and compared (Fig. 3). AaPGSs are predicted to be composed of a membrane-inserted amino terminus appended to a hydrophilic carboxyl terminus, but the precise role of each of these domains is unknown. The membrane domain varies in size and in sequence and is even absent from aaPGS homologs in a few cases (e.g. Streptomyces, Burkholderia). Based on the predicted secondary structure, the membrane-inserted domain is thought to consist of a variable number of α-helices (4 to 14) that orient the amino-terminal extremity and direct the hydrophilic carboxyl-terminal moiety toward the cytoplasm where aa-tRNAs, the substrate of aaPGSs, are synthesized. Biochemically characterized aaPGSs possess a membrane domain containing 14 predicted α-helices, except for the Ala-PGS from C. perfringens that contains only six helices. More distant aaPGS, such as the paralogs found in actinomycetes, are fused with a Lys-RS domain that displays 40% identity with E. coli Lys-RS. This domain exhibits all of the features associated with tRNA lysylation such as a tRNA anticodon binding domain (OB-fold) along with all the conserved positions in Lys-RSs necessary for substrate binding (Lys, ATP, and tRNA) and enzyme dimerization (data not shown). The presence of this domain in aaPGS is probably involved in the channeling of the Lys-tRNA toward the membrane modification site. However, these enzymes possess a notably shorter membrane domain, containing six transmembrane α-helices. These secondary structural comparisons suggest that the aaPGS active site is likely located within the hydrophilic carboxyl-terminal moiety of the polypeptide.
FIGURE 3.

Phylogeny and modular organization of aaPGSs. Aa-tRNA specificity of the aaPGSs that have been characterized in vitro is indicated (black arrow). The modular organization and number of predicted helices within the integral membrane domain of each sequence, respectively, are reported beside each named organism and by a colored background. Branches displaying a bootstrap support >80% are indicated by gray circles. The number of aaPGS paralogs within a given organism is indicated in brackets.
AaPGSs Contain a Catalytic Carboxyl-terminal Domain
To determine whether the membrane-inserted domain of aaPGS plays a role in catalysis, the amino termini of the C. perfringens Ala-PGS and of the B. subtilis Lys-PGS were truncated to leave either one or none of the hydrophobic helices intact. It is worth noting that these predicted membrane domains are quite divergent: B. subtilis Lys-PGS contains14 helices whereas C. perfringens Ala-PGS contains only 6. The activity of the different constructs was then investigated in vivo and in vitro. Although the presence of a single helix resulted in little or no activity in vitro, removal of the entire membrane domain led to variants with significant activity, indicating that aaPG synthesis is catalyzed by the hydrophilic amino terminus alone (Fig. 4). In contrast, only the full-length aaPGSs, but none of the truncated variants, displayed aaPG synthetic activity in vivo (Fig. 5).
FIGURE 4.

Activity of truncated aaPGSs. A, schematic representation of the B. subtilis (Bsu) and C. perfringens (Cpe) aaPGSs. Truncation within the amino-terminal membrane domain of both proteins is indicated. The membrane domains of B. subtilis and C. perfringens aaPGSs contain 14 and 6 α-helices, respectively (as predicted by TOPCONS). B, aaPGS activity of full-length and truncated proteins. Membrane extracts were tested as described under Experimental Procedures with the corresponding 14C-labeled aa-tRNA in a medium containing 14C-labeled amino acids, ATP, total tRNA from E. coli, and the cognate aa-RS.
FIGURE 5.

Biosynthesis of aaPG- in aaPGS-expressing strains. Full-length aaPGS2 from E. faecium (C41-Efa2), A. tumefaciens (C41-Atu), B. subtilis (C41-Bsu), and the truncated aaPGS from B. subtilis encompassing the residues 488–861 or 525–861 were expressed in the E. coli strain C41. Total lipids were extracted, separated on TLC, and revealed by primuline staining. Amino group-containing lipids were visualized by ninhydrine staining (*).
DISCUSSION
Distinct Roles of the AaPGS Amino- and Carboxyl-terminal Domains
Recent studies of aaPGSs, particularly in the context of drug-resistant mprF alleles, provided preliminary indications of the possible roles of different domains of the protein. For example, loss of daptomycin susceptibility allowed the tentative assignment of lysyl-PG translocase (flippase) activity to the N-terminal eight transmembrane helices of S. aureus Lys-PGS (29, 30). It was also proposed that the synthase domain consisted of six transmembrane helices appended to the carboxyl terminus. Our data now show that the carboxyl terminus alone is sufficient for full synthase activity in vitro. The absence of activity in vivo for the carboxyl-terminal domain alone suggests that transmembrane helices position the catalytic domain in the cytosol during aaPG synthesis. The dual role of the membrane-inserted amino terminus, both as a translocase and for anchoring the catalytic moiety, is consistent with the high degree of heterogeneity observed in this region compared with the more conserved carboxyl terminus (Fig. 3). AaPGSs are encoded in many phylogenetically distant bacteria, and divergence in the membrane-inserted domain may reflect adaptation to a wide range of cell wall morphologies and lipid compositions. The carboxyl-terminal catalytic domain instead recognizes molecules that show relatively little phylogenetic variation, aa-tRNAs, consistent with the higher degree of conservation seen within this domain across different species.
Aa-tRNA Selection by AaPGS
Pioneering studies in S. aureus first identified lysyl-tRNA-dependent PG modification by Lys-PGS as a means to increase the net positive surface charge of bacteria (2, 31). More recently, tRNA-dependent Ala-PG synthesis was also demonstrated (9), thereby establishing that the aaPGS family does not exclusively catalyze lipid lysylation. Both Arg and ornithine would be expected to have an impact on the charge of PG comparable with Lys, and both Arg-PG and ornithyl-PG have previously been identified in bacteria (for review, see 32). Although tRNA-dependent ornithyl-PG synthesis could not be detected,3 Arg-PG and Ala-PG synthase activities were readily observed for some Lys-PGs tested. The triple specific Ala/Arg/Lys-PGS from E. faecium displayed the broadest specificity because this enzyme was also able to synthesize Ala-PG at an efficiency comparable with Arg- or Lys-PG. These data suggest that aaPGSs capable of transferring bulkier amino acids such as Lys or Arg may display a moderate specificity compared with those that utilize smaller amino acids such as Ala. These observations imply that aaPGSs adapted to bind smaller amino acids (i.e. Ala) cannot accommodate bulkier substrates such as Lys or Arg. Conversely, those aaPGSs that preferentially utilize Lys or Arg as substrates can sometimes accommodate smaller amino acids such as Ala. A refined model for the molecular basis of amino acid recognition and discrimination by aaPGSs now requires detailed structural studies, but it is noteworthy that our initial findings are reminiscent of size-based amino acid discrimination strategies described for some aa-RS proteins (33, 34).
Functional Implications of Relaxed Substrate Specificity
It was shown recently that Ala-PG and Lys-PG impact cellular physiology in many similar ways, suggesting that the primary role of aaPGS is to neutralize the net negative charge of PG rather than place a particular aminoacyl group in the membrane (1, 10). In this respect, aaPGSs differ from other tRNA-dependent cell wall synthesis and remodeling enzymes that supply specific amino acids for processes such as peptidoglycan biosynthesis (35, 36). This functional difference between aaPGSs and related enzymes is consistent with the expanded range of aaPG synthesis described here. The relaxed specificity of aaPGSs allows cells to modulate their net charge using a wider range of substrates than previously anticipated. This, in turn, provides a means by which a single pathway can be used to adapt to environmental conditions under which different amino acids may become limiting at various points in the life cycle of the cell. The ability to adapt to particular environments may be facilitated further by regulation of aaPGSs of differing specificities in a single organism, such as in C. perfringens that encodes both an Ala-PGS and a Lys-PGS (9). Future studies will focus on understanding in more detail how the full range of aaPGS activities alters cellular physiology, in particular with respect to determining the roles of aaPGs in defining a wide spectrum of antibiotic resistances.
Acknowledgments
We thank Barbara E. Murray (University of Texas, Houston) for the E. faecium DO strain used in this study, Gabor L. Igloi (University of Freiburg, Germany) for the strain expressing the jack bean Arg-RS, and Kiley Dare for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM 65183.
H. Roy and M. Ibba, unpublished results.
- aaPGS
- aminoacylphosphatidylglycerol synthase
- aa-tRNA
- aminoacyl-tRNA
- Ala-RS
- alanyl-tRNA synthetase
- Arg-RS
- arginyl-tRNA synthetase
- Lys-RS
- lysyl-tRNA synthetase
- PG
- phosphatidylglycerol.
REFERENCES
- 1.Roy H., Dare K., Ibba M. (2009) Mol. Microbiol. 71, 547–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peschel A., Jack R. W., Otto M., Collins L. V., Staubitz P., Nicholson G., Kalbacher H., Nieuwenhuizen W. F., Jung G., Tarkowski A., van Kessel K. P., Van Strijp J. A. (2001) J. Exp. Med. 193, 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Staubitz P., Neumann H., Schneider T., Wiedemann I., Peschel A. (2004) FEMS Microbiol. Lett. 231, 67–71 [DOI] [PubMed] [Google Scholar]
- 4.Weidenmaier C., Peschel A., Kempf V. A., Lucindo N., Yeaman M. R., Bayer A. S. (2005) Infect. Immun. 73, 8033–8038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thedieck K., Hain T., Mohamed W., Tindall B. J., Nimtz M., Chakraborty T., Wehland J., Jänsch L. (2006) Mol. Microbiol. 62, 1325–1339 [DOI] [PubMed] [Google Scholar]
- 6.Zemansky J., Kline B. C., Woodward J. J., Leber J. H., Marquis H., Portnoy D. A. (2009) J. Bacteriol. 191, 3950–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishi H., Komatsuzawa H., Fujiwara T., McCallum N., Sugai M. (2004) Antimicrob. Agents Chemother. 48, 4800–4807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui L., Tominaga E., Neoh H. M., Hiramatsu K. (2006) Antimicrob. Agents Chemother. 50, 1079–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy H., Ibba M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 4667–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein S., Lorenzo C., Hoffmann S., Walther J. M., Storbeck S., Piekarski T., Tindall B. J., Wray V., Nimtz M., Moser J. (2009) Mol. Microbiol. 71, 551–565 [DOI] [PubMed] [Google Scholar]
- 11.Komatsuzawa H., Ohta K., Fujiwara T., Choi G. H., Labischinski H., Sugai M. (2001) FEMS Microbiol. Lett. 203, 49–54 [DOI] [PubMed] [Google Scholar]
- 12.Mishra N. N., Yang S. J., Sawa A., Rubio A., Nast C. C., Yeaman M. R., Bayer A. S. (2009) Antimicrob. Agents Chemother. 53, 2312–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sohlenkamp C., Galindo-Lagunas K. A., Guan Z., Vinuesa P., Robinson S., Thomas-Oates J., Raetz C. R., Geiger O. (2007) Mol. Plant Microbe Interact. 20, 1421–1430 [DOI] [PubMed] [Google Scholar]
- 14.Reeve W. G., Bräu L., Castelli J., Garau G., Sohlenkamp C., Geiger O., Dilworth M. J., Glenn A. R., Howieson J. G., Tiwari R. P. (2006) Microbiology 152, 3049–3059 [DOI] [PubMed] [Google Scholar]
- 15.Hachmann A. B., Angert E. R., Helmann J. D. (2009) Antimicrob. Agents Chemother. 53, 1598–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samant S., Hsu F. F., Neyfakh A. A., Lee H. (2009) J. Bacteriol. 191, 1311–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salzberg L. I., Helmann J. D. (2008) J. Bacteriol. 190, 7797–7807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy H., Ibba M. (2008) Methods 44, 164–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer A. E., Beuning P. J., Musier-Forsyth K. (1999) J. Biol. Chem. 274, 37093–37096 [DOI] [PubMed] [Google Scholar]
- 20.Ataide S. F., Ibba M. (2004) Biochemistry 43, 11836–11841 [DOI] [PubMed] [Google Scholar]
- 21.Hogg J., Schiefermayr E., Schiltz E., Igloi G. L. (2008) Protein Expr. Purif. 61, 163–167 [DOI] [PubMed] [Google Scholar]
- 22.Bligh E. G., Dyer W. J. (1959) Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 23.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson J. D., Gibson T. J., Higgins D. G. (2003) Current Protocols in Bioinformatics (Baxevanis A. D. ed) pp. 2.3.1–2.3.22 [Google Scholar]
- 25.Letunic I., Bork P. (2007) Bioinformatics 23, 127–128 [DOI] [PubMed] [Google Scholar]
- 26.Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. (2001) J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 27.Bernsel A., Viklund H., Hennerdal A., Elofsson A. (2009) Nucleic Acids Res. 37, W465–W468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.dos Santos Mota J. M., den Kamp J. A., Verheij H. M., van Deenen L. L. (1970) J. Bacteriol. 104, 611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang S. J., Xiong Y. Q., Dunman P. M., Schrenzel J., François P., Peschel A., Bayer A. S. (2009) Antimicrob. Agents Chemother. 53, 2636–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones T., Yeaman M. R., Sakoulas G., Yang S. J., Proctor R. A., Sahl H. G., Schrenzel J., Xiong Y. Q., Bayer A. S. (2008) Antimicrob. Agents Chemother. 52, 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lennarz W. J., Nesbitt J. A., 3rd, Reiss J. (1966) Proc. Natl. Acad. Sci. U.S.A. 55, 934–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roy H. (2009) IUBMB Life, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hendrickson T. L., Schimmel P. (2003) Transfer RNA-dependent amino acid discrimination by aminoacyl-tRNA synthetases, Translation Mechanisms (Lapointe J., Brakier-Gingras L. eds) pp. 34–64, Kluwer Academic Publishers, Norwell, MA [Google Scholar]
- 34.Ibba M., Soll D. (2000) Annu. Rev. Biochem. 69, 617–650 [DOI] [PubMed] [Google Scholar]
- 35.Villet R., Fonvielle M., Busca P., Chemama M., Maillard A. P., Hugonnet J. E., Dubost L., Marie A., Josseaume N., Mesnage S., Mayer C., Valéry J. M., Ethève-Quelquejeu M., Arthur M. (2007) Nucleic Acids Res. 35, 6870–6883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lloyd A. J., Gilbey A. M., Blewett A. M., De Pascale G., El Zoeiby A., Levesque R. C., Catherwood A. C., Tomasz A., Bugg T. D., Roper D. I., Dowson C. G. (2008) J. Biol. Chem. 283, 6402–6417 [DOI] [PubMed] [Google Scholar]