

Abstract
Polycyclic aromatic hydrocarbon (PAH) o-quinones produced by aldo-keto reductases are ligands for the aryl hydrocarbon receptor (AhR) (Burczynski, M. E., and Penning, T. M. (2000) Cancer Res. 60, 908–915). They induce oxidative DNA lesions (reactive oxygen species-mediated DNA strand breaks and 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dGuo) formation) in human lung cells. We tested whether the AhR enhances PAH o-quinone-mediated oxidative DNA damage by translocating these ligands to the nucleus. Using the single cell gel electrophoresis (comet) assay to detect DNA strand breaks in murine hepatoma Hepa1c1c7 cells and its AhR- and aryl hydrocarbon receptor nuclear translocator-deficient variants, benzo[a]pyrene-7,8-dione (B[a]P-7,8-dione) produced fewer DNA strand breaks in AhR-deficient cells compared with aryl hydrocarbon receptor nuclear translocator-deficient and wild type Hepa1c1c7 cells. Decreased DNA strand breaks were also observed in human bronchoalveolar H358 cells in which the AhR was silenced by siRNA. The antioxidant α-tocopherol and the iron chelator/antioxidant desferal decreased the formation of B[a]P-7,8-dione-mediated DNA strand breaks indicating that they were reactive oxygen species-dependent. By coupling the comet assay to 8-oxoguanine glycosylase (hOGG1), which excises 8-oxo-Gua, strand breaks dependent upon this lesion were measured. hOGG1 treatment produced more DNA single strand breaks in B[a]P-7,8-dione-treated Hepa cells and H358 cells than in its absence. The levels of hOGG1-dependent DNA strand breaks mediated by B[a]P-7,8-dione were lower in AhR-deficient Hepa and AhR knockdown H358 cells. The AhR antagonist α-naphthoflavone also attenuated B[a]P-7,8-dione-mediated DNA strand breaks. The decrease in 8-oxo-dGuo levels in AhR-deficient Hepa cells and AhR knockdown H358 cells was validated by immunoaffinity capture stable isotope dilution ([15N5]8-oxo-dGuo) liquid chromatography-electrospray ionization/multiple reaction monitoring/mass spectrometry. We conclude that the AhR shuttles PAH o-quinone genotoxins to the nucleus and enhances oxidative DNA damage.
Polycyclic aromatic hydrocarbons (PAHs)2 are ubiquitous environmental pollutants that include over 200 compounds with two or more fused benzene rings. PAHs are formed as a result of incomplete combustion of fossil fuels (e.g. coal and oil) and are present in car and diesel exhaust and smoked or charbroiled food (1–3). They are also found in cigarette smoke condensate and tobacco products and are suspect agents in the causation of human lung cancer (4, 5). PAHs must be metabolically activated to reactive genotoxins to cause their mutagenic and carcinogenic effects.
Two major metabolic activation pathways are possible starting from the proximate PAH carcinogen (−)B[a]P-7,8-trans-dihydrodiol (Fig. 1). The P4501A1/1B1 pathway converts (−)B[a]P-7,8-trans-dihydrodiol to yield (+)-anti-7,8-dihydroxy-9α,10β-epoxy-7,8,9,10-tetrahydroB[a]P (6–8). This diol epoxide forms stable N2-2′-deoxyguanosine (dGuo) adducts in vitro and in vivo (9, 10) and leads to mutation in H-ras (11) and may account for mutations in “hot spots” in p53 observed in lung cancer (12). The G to T transversions most often observed in these genes might arise because of the action of one or more trans-lesional by-pass DNA polymerases that read through stable diol-epoxide DNA adducts with low processivity and fidelity (13, 14).
FIGURE 1.

PAH activation by AKRs to cause oxidative DNA damage.
As an alternative, human aldo-keto reductases (AKR1A1 and AKR1C1-AKR1C4) catalyze the NADP+-dependent oxidation of (±)B[a]P-7,8-trans-dihydrodiol to produce the electrophilic and redox-active B[a]P-7,8-dione (15, 16). In this pathway, AKRs convert B[a]P-7,8-trans-dihydrodiol to form a ketol that rearranges to a catechol. The catechol then undergoes two subsequent one-electron oxidations to yield the fully oxidized o-quinone. Once formed, B[a]P-7,8-dione amplifies reactive oxygen species (ROS) by entering futile redox cycles that deplete cellular reducing equivalents (e.g. NADPH) (17). PAH o-quinones can undergo 1,4- or 1,6-Michael addition with guanine and adenine bases to form stable N2-dGuo and N6-dAdo adducts in vitro (18–20). They can also react with the N7 position of guanine to yield depurinating adducts (21). It is possible that these covalent PAH o-quinone adducts could give to G to T transversion mutations.
PAH o-quinones also cause oxidative DNA damage in vitro and in vivo (22–25). Nanomolar concentrations of PAH o-quinones under redox cycling conditions (NADPH and Cu(II)) lead to significant 8-oxo-dGuo formation in bulk DNA, and the responsible oxidant was found to be singlet oxygen (1O2) (24, 26). Under these conditions, PAH o-quinones produced 8-oxo-dGuo as the most dominant lesion among the three types of oxidative lesions measured (abasic sites, 8-oxo-dGuo, and oxidized pyrimidines) (26). In a yeast reporter gene assay, which scored loss-of-function mutations in p53, PAH o-quinones were found to be highly mutagenic but only under redox cycling conditions. The dominant mutation observed was a G to T transversion that was suppressed by ROS scavengers (27). Subsequent HPLC analysis coupled with electrochemical detection showed that there was a linear correlation between 8-oxo-dGuo formation in p53 and mutation frequency, indicating that 8-oxo-dGuo was the likely adduct responsible for the G to T transversions observed (28). These data suggest that oxidative DNA lesions caused by PAH o-quinones are more relevant in causing mutation than covalent PAH o-quinone-DNA adducts. In the latter case even if these adducts form, they do not appear to be mutagenic on p53.
Recently, using either a hOGG1-coupled comet assay or an immunoaffinity capture-stable isotope dilution liquid chromatography/electrospray ionization/multiple reaction monitoring/mass spectrometry (LC/ESI/MRM/MS) assay, it was shown that both the AKR substrate (B[a]P-7,8-trans-dihydrodiol) and the AKR product (B[a]P-7,8-dione) caused significant DNA strand breaks and 8-oxo-dGuo formation in human lung adenocarcinoma A549 cells (25). Similar results were not observed with (+)-anti-7,8-dihydroxy-9α,10β-epoxy-7,8,9,10-tetrahydroB[a]P or the regioisomer B[a]P-4,5-trans- dihydrodiol in these AKR-expressing cells. Subsequent use of the fluorescent dye dichlorofluorescein diacetate revealed that B[a]P-7,8-dione generated ROS in the nuclear compartment of the cells, suggesting that the PAH o-quinone was transported into the nucleus to increase the ROS-mediated DNA strand breaks and 8-oxo-dGuo (25). In addition, earlier disposition studies detected significant amounts of [3H]B[a]P-7,8-dione in the cell pellets of primary rat hepatocytes within 0.5 h, which caused extensive strand scission of the genomic DNA (29), suggesting that B[a]P-7,8-dione reached the nucleus. However, how PAH o-quinones gain entry into the nucleus and induce oxidative DNA damage is currently unknown.
PAH o-quinones are ligands for the aryl hydrocarbon receptor (AhR) (30). These quinones can promote translocation of AhR to nucleus to induce P4501A1 expression. Upon binding with PAH o-quinones, the AhR dissociates from heat shock protein 90 and is rapidly translocated into nucleus where it dimerizes with the aryl hydrocarbon receptor nuclear translocator (ARNT) (31, 32). The quinone-bound AhR·ARNT complex then binds to the xenobiotic response element (XRE) and robustly activates the expression of AhR-regulated genes (30). These data raise the possibility that oxidative DNA damage caused by PAH o-quinones occurs because of their transportation and concentration in the nucleus mediated by the AhR. However, this hypothesis has not been formally tested.
We now show that B[a]P-7,8-dione produces AhR-dependent DNA strand breaks and 8-oxo-dGuo formation using murine Hepa1c1c7 cells but not in its AhR-deficient variant. Similar results were obtained in human bronchoalveolar carcinoma H358 cells, but these effects were attenuated when the AhR was knocked down with siRNA. DNA lesions were measured by using the comet assay, which was coupled with hOGG1. These results were also confirmed by LC-ESI/MRM/MS assay for 8-oxo-dGuo. Our finding shows that PAH o-quinones produced by AKRs can cause ROS-mediated genotoxicity via an AhR-dependent mechanism, and this might contribute to PAH-mediated carcinogenesis.
EXPERIMENTAL PROCEDURES
Materials
(±)-B[a]P-7,8-dione was synthesized according to published methods (33). The purity and identity of all PAH metabolites were checked by LC/MS before use. Cell culture media, reagents, and DNAzol BD were obtained from Invitrogen. HiPerFect transfection reagent was acquired from Qiagen. hOGG1 was obtained from New England BioLabs Inc. (Beverly, MA). Bovine pancreas DNase I was purchased from Calbiochem Co. (San Diego, CA), and phosphodiesterase I was acquired from Worthington (Lakewood, NJ). All other chemicals were of the highest grade available and all solvents were HPLC grade. B[a]P-7,8-dione is potentially hazardous and should be handled in accordance with the National Institutes of Health Guidelines for the Laboratory Use of Chemical Carcinogens.
Cell Culture
Human bronchoalveolar H358 cells (ATCC number CRL-5807) were maintained in RPMI 1640 with 10% heat-inactivated fetal bovine serum, 1% l-glutamine, and 100 unit/ml penicillin/streptomycin. Murine Hepa1c1c4 (ATCC no. CRL-2717), Hepa1c1c7 (ATCC number CRL-2026), and Hepa1c1c12 (ATCC number CRL-2710) cells were maintained in α-minimal essential medium (without nucleosides) supplemented with 10% fetal bovine serum, 1% l-glutamine, and 100 unit/ml penicillin/streptomycin. The cells were incubated at 37 °C in a humidified atmosphere of 5% CO2 and passaged every 4 days at a dilution of 1:5.
siRNA of AhR
H358 cells were seeded in a 12-well plate (2.5 × 105 cells) or 60-mm culture dish (1.4 × 106 cells) 24 h before introducing small interfering AhR-specific RNA duplexes. The sense and antisense sequences of AhR siRNA were 5′-GGAUUAAAUUAGUUUGUGAdTdT-3′ and 5′- UCACAAACUAAUUUAAUCCdAdA-3′, respectively. The siRNA was purchased from Qiagen. AhR-specific siRNA oligonucleotide (50 nm) was then transfected into cells with HiPerFect Transfection reagent according to the manufacturer's instructions. The knockdown of AhR with the siRNA duplexes was repeated 24 h after the initial transfection. The siRNA-transfected cells were incubated for a further 24 h before use.
Treatment of Cells with Hydrogen Peroxide, Potassium Bromate, and B[a]P-7,8-dione
Confluent (90–100%) H358 and Hepa cells were plated in either a 12-well plate (∼1.3 × 106) or a 60-mm culture dish (∼7 × 106) depending on the assay. The cells were washed with HBSS containing Mg2+ and Ca2+. H358 cells were then treated with HBSS buffer containing 0–500 μm H2O2 for 40 min, 2.5 mm KBrO3 for 3 h, or 20 μm B[a]P-7,8-dione for 3, 6, 9, and 12 h, respectively. Hepa cells were treated with HBSS containing 5% α-minimal essential medium with and without fetal bovine serum and treated similarly with H2O2, KBrO3, or B[a]P-7,8-dione. The cells were treated with 0 or 3% DMSO as negative controls. To investigate the effects of ROS attenuators, desferal (an iron chelator and antioxidant) and α-tocopherol (an antioxidant) were added to the media to attain final concentrations of 1 and 0.25 mm, respectively, 2 h prior to treatment of the cells with B[a]P-7,8-dione. α-Naphthoflavone (α-NF) was also used at a final concentration of 1 μm and was added to the cells 1 h before B[a]P-7,8-dione treatment. At the end of PAH treatment, the cells were washed with cold phosphate-buffered saline and subsequently treated with 50 μl (12-well plate) or 200 μl (60-mm culture dish) of proteinase K (200 μg/ml in phosphate-buffered saline) for each of 5 min. The cells were suspended in 150 μl (12-well plate) or 600 μl (60-mm culture dish) of cold culture medium and kept on ice until used. The cells (1 × 105 cells) were used for the comet assay to measure DNA strand breaks. In addition one set of cells (1.2 × 106) from a 12-well plate was used to prepare a total cell lysate for Western blot analysis and another set of cells (6.9 × 106) from a 60-mm culture dish were subjected to LC/ESI/MRM/MS analysis for quantification of 8-oxo-dGuo in the DNA pellet. Cell viability was checked by the trypan blue exclusion method. Unless otherwise stated, cell viability was >95%. All of the experiments were repeated at least three times.
Detection of DNA Strand Breaks by Comet Assay
DNA strand breaks in cells were measured using a hOGG1-coupled comet assay as previously published (25, 34). The cells (1 × 105 cells) were mixed with 85 μl of 0.7% low melting point agarose. The agarose cell suspensions were immediately embedded onto an agarose-precoated fully frosted microscope glass slide (70 × 25 mm; Fischer) using a coverslip (No 1, 24 × 50 mm; Corning Inc., Corning, NY) to spread the mixture. The mixture was allowed to form a gel on an ice bath for 5 min. A second agarose gel layer was used to cover the surface of the layer of cells embedded in agarose by using 85 μl of the same low melting point agarose. The slides were then soaked with cell lysis buffer (10 mm Tris, 2.5 m NaCl, 100 mm Na2-EDTA, and 1% sodium sarcosinate plus 10% DMSO and 1% Triton X-100, pH 10.0) for at least 1 h to remove all cellular components and to leave behind the nucleus and genomic DNA. At the end of the cell lysis, the slides were placed in a horizontal electrophoretic apparatus with fresh alkaline electrophoretic buffer (300 mm NaCl and 1 mm Na2-EDTA, pH > 12.0) for 20 min to allow the DNA to unwind and to reveal the alkali labile damage. Electrophoresis was conducted for 20 min at 25 V and 300 mA at a temperature of 4 °C. When the comet assay was coupled to hOGG1, the assay was performed as follows. After cell lysis, the slides were washed three times with hOGG1 enzyme buffer (40 mm HEPES, 0.1 m KCl, 0.5 mm Na-EDTA, and 0.2 mg/ml bovine serum albumin, pH 8.0) for 5 min and treated with 60 μl of hOGG1 (0.08 unit in phosphate-buffered saline) for 20 min at 37 °C in a humidified atmosphere. hOGG1-treated slides were processed as described above to permit DNA unwinding and exposure of the alkali labile damage. After electrophoresis, the slides were neutralized with Tris-HCl buffer (0.4 m Tris, pH 7.4) for 5 min and stained with 80 μl of ethidium bromide (2 μg/ml in H2O).
Quantification of DNA Strand Breaks
Ethidium bromide-stained nucleoids were examined using an epifluorescence microscope (Nikon Eclipse E600; Nikon Instrument Inc.) equipped with a excitation filter (535–550 nm) and a barrier filter (590–650 nm). To quantify the amount of DNA damage, 100 comets/slide were scored visually and classified according to tail intensity and assigned a value of 0, 1, 2, 3, or 4 (0 indicates undamaged,1 indicates slightly damaged, 2 indicates moderately damaged, 3 indicates severely damaged, and 4 indicates very severely damaged). The total score for 100 comets was again divided by factor of 5 to yield an arbitrary value ranging from 0 (if the counted hundred comets are all undamaged) to 80 (if the counted 100 comets are all very severely damaged).
DNA Extraction and Hydrolysis
Extraction and enzymatic hydrolysis of genomic DNA was performed as published (25, 34). The DNA was extracted from 6.9 × 106 cells using DNAzol BD (Invitrogen) and then quantitatively digested by the addition of 556 units of DNase I, one unit of phosphodiesterase I (from Crotalus adamanteus venom), 6 units of shrimp alkaline phosphatase and spiked with an internal standard [15N5]8-oxo-dGuo. The resultant DNA digests were filtered through 0.22-μm nylon filters before analysis. The DNA samples were divided into two aliquots. One aliquot was used for the isolation and detection of 8-oxo-dGuo via immunoaffinity capture stable isotope dilution LC/ESI/MRM/MS analyses. The remaining aliquot was used for DNA base analysis.
Immunoaffinity Purification of 8-Oxo-dGuo from Cellular DNA Digests
DNA hydrolysates were diluted with 50 mm phosphate buffer (pH 7.4) containing 0.02% sodium azide and loaded onto an 8-oxo-dGuo mAb 4E9 coupled CNBr-activated Sepharose 4B column. The column was washed with phosphate buffer followed by a further wash with water. 8-Oxo-dGuo was eluted with 50% methanol. The samples were dried under vacuum, and the residue was dissolved in Chelex-treated HPLC water.
Detection of 8-Oxo-dGuo by LC-ESI/MS
The analysis of 8-oxo-dGuo was conducted on API 4000 (Applied Biosystems/MDS Sciex) equipped with positive ESI source, CTC autosampler (CTC Analytics), and Analyst 1.4.1 software as described previously (34). Chromatography was performed with a Phenomenex Luna C18 column (3 μm; 2.0 × 150 mm) with a C18 guard column cartridge (4 × 2.0 mm) using a gradient (2–15% acetonitrile in 0.02% formic acid (v/v) for the first 15 min, 15% acetonitrile for 1 min, and then a gradient of 15–2% acetonitrile for 1 min and 2% acetonitrile for the last 10 min, at a flow rate of 0.2 ml/min). During sample analysis, the column effluent was diverted to waste for the first 5 min and the last 12 min to prevent extraneous material from entering the mass spectrometer. For detecting 8-oxo-dGuo, MRM was conducted in the positive ESI mode on m/z 284 (MH+, 8-oxo-dGuo) → m/z 168 (MH+-deoxyribose+H) and m/z 289 (MH+, [15N5]8-oxo-dGuo) → m/z 173 (MH+-deoxyribose+H). Quantification of 8-oxo-dGuo was achieved by using Analyst 1.4.1 from raw spectral data. A calibration curve (range, 50 pg/ml to 5 ng/ml) was obtained by plotting the ratio of the peak area of 8-oxo-dGuo to the internal standard [15N5]8-oxo-dGuo obtained by LC-MRM/MS against known amounts of 8-oxo-dGuo (limit of detection = 0.353 fmol).
DNA Base Analysis by HPLC
The base analysis of DNA samples was conducted with a Hitachi HPLC system equipped with an L-2130 pump, L-2200 autosampler, and L-2450 diode array detector. DNA base separation was conducted with a Phenomenex Luna C18 column (5 μm; 4.6 × 250 mm) with a C18 guard column cartridge (4 × 3.0 mm) using a gradient (5–15% acetonitrile in water (v/v) for the first 12 min, 15–80% acetonitrile for 2 min, and then 80% acetonitrile for 6 min and 5% acetonitrile for the last 10 min; the flow rate was 0.8 ml/min). A calibration curve (range, 0–50 μg/ml) was obtained with a linear regression analysis of the peak area of known amounts of DNA base. Unknown amounts of DNA base levels in the DNA samples were calculated by interpolation from the calibration curve.
Cellular Cytosolic and Nuclear Protein Preparation
The cells were washed twice with cold phosphate-buffered saline (pH 7.4) and lysed with cold radioimmune precipitation assay buffer (1% Triton X-100 (v/v), 1% sodium deoxycholate (w/v), 0.1% SDS (w/v), 150 mm NaCl, 1 mm EDTA, 1 mm Na3VO4, 10 mm NaF, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor cocktails). The lysates were kept on ice for 30 min and centrifuged at 15,000 × g for 30 min at 4 °C, and the resultant supernatants were saved as cytosolic protein extracts at −80 °C until used. Nuclear proteins from the precipitated pellets were extracted with a nuclear extraction kit (Marligen Bioscience, Urbana Pike, MD). Briefly, the pellets were washed twice with complete wash solution and suspended in the mixture of same volume of complete extraction buffer A and B. Then the samples were incubated on ice for 30 min to extract nuclear proteins. After centrifugation at 15,000 × g for 30 min at 4 °C, the supernatants were stored at −80 °C until used.
Western Blot Analysis
The protein concentrations of cytosolic and nuclear extracts were measured using the Bradford assay (35), and Western blot analysis was performed as previously described (36). Proteins (50 μg) were separated by SDS-PAGE and electrotransferred to nitrocellulose filters (Bio-Rad). The filters were incubated with rabbit polyclonal anti-mouse AhR antibody at a 1:500 dilution (SA-210; Biomol), rabbit polyclonal anti-human AhR antibody (Sc-5579; Santa Cruz), or goat polyclonal anti-murine ARNT antibody (sc-Sc-8078; Santa Cruz). The immunoblots were developed by incubation with either goat anti-rabbit IgG-horseradish peroxidase or donkey anti-goat IgG-horseradish peroxidase as the secondary antibody followed by ECL detection (GE Healthcare).
RESULTS
DNA Strand Breaks Produced by Hydrogen Peroxide and Potassium Bromate in Hepa and H358 Cells
To examine the role of AhR in enhancing PAH o-quinone-mediated DNA damage, we took advantage of three Hepa cell lines: ARNT-deficient Hepa1c1c4 cells, AhR-deficient Hepa1c1c12 cells, and their wild type counterpart Hepa1c1c7 cells. The phenotype of these cells was checked by immunoblot analysis (Fig. 2A). The Hepa1c1c4 cells yield an immunopositive band for ARNT because this subline expresses a point mutation G326D that prevents binding of the AhR·ARNT heterodimer to the XRE (37).
FIGURE 2.
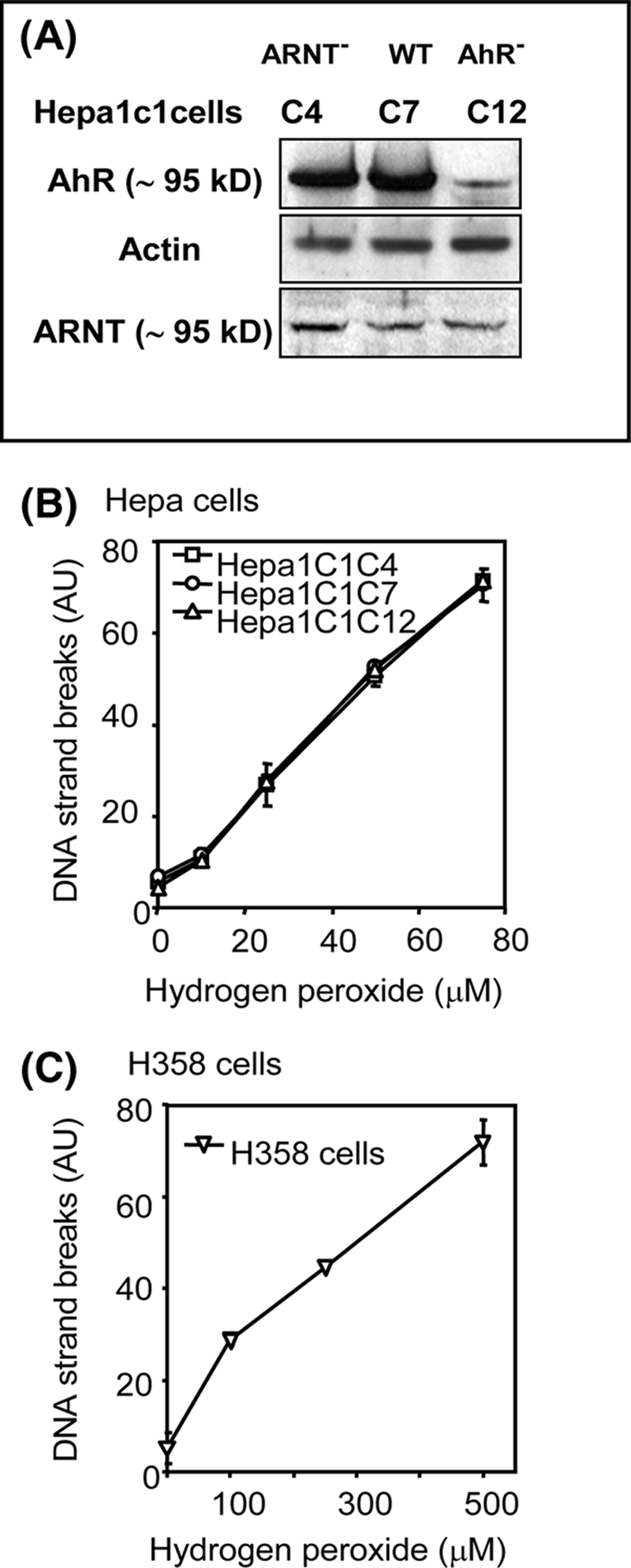
H2O2-mediated DNA strand breaks in murine Hepa1c1c7 cells and its ARNT-deficient Hepa1c1c4 and AhR-deficient Hepa1c1c12 variant cells and human H358 cells. A, immunoblot analysis of AhR and ARNT expression in Hepa1c1c cell lines. B and C, Hepa cells (B) and H358 cells (C) were treated with different concentrations of hydrogen peroxide for 40 min, harvested, and used for the detection of DNA strand breaks by the comet assay. WT, wild type; AU, arbitrary units.
We next calibrated our comet assay protocols in the three Hepa cell clones as well as human bronchoalveolar H358 cells using hydrogen peroxide and potassium bromate. The results showed that the comet assay detected hydrogen peroxide-mediated DNA strand breaks in all cell lines in a concentration-dependent manner (Fig. 2, B and C). Levels of DNA strand breaks formed in Hepa cells treated with 80 μm of hydrogen peroxide were almost the same as those produced in H358 cells-treated with 500 μm of hydrogen peroxide, suggesting that Hepa cells were more sensitive to oxidative DNA damage than H358 cells. The levels of DNA strand breaks formed between the three Hepa cell lines were similar, showing identical susceptibility to hydrogen peroxide.
The cell lines were then treated with potassium bromate, an oxidant that is known to produce 8-oxo-dGuo (38). The 8-oxo-dGuo levels were estimated using the comet assay coupled with hOGG1, which catalyzes the excision of 8-oxo-Gua to yield DNA strand breaks that are dependent upon this lesion (39). Potassium bromate did not induce significant levels of DNA strand breaks in either Hepa or H358 cells unless hOGG1 was included in the assay (Fig. 3). Once hOGG1 was used in the assay, significant levels of DNA breakage were observed in all cell types. The levels of DNA strand breaks induced by potassium bromate in the coupled comet assay in the three Hepa cell sublines were the same (Fig. 3A). These data suggest that these sublines could be used to measure DNA strand breaks and 8-oxo-dGuo formation that might be mediated by PAH o-quinones without being concerned about differences in susceptibility to oxidative DNA damage.
FIGURE 3.
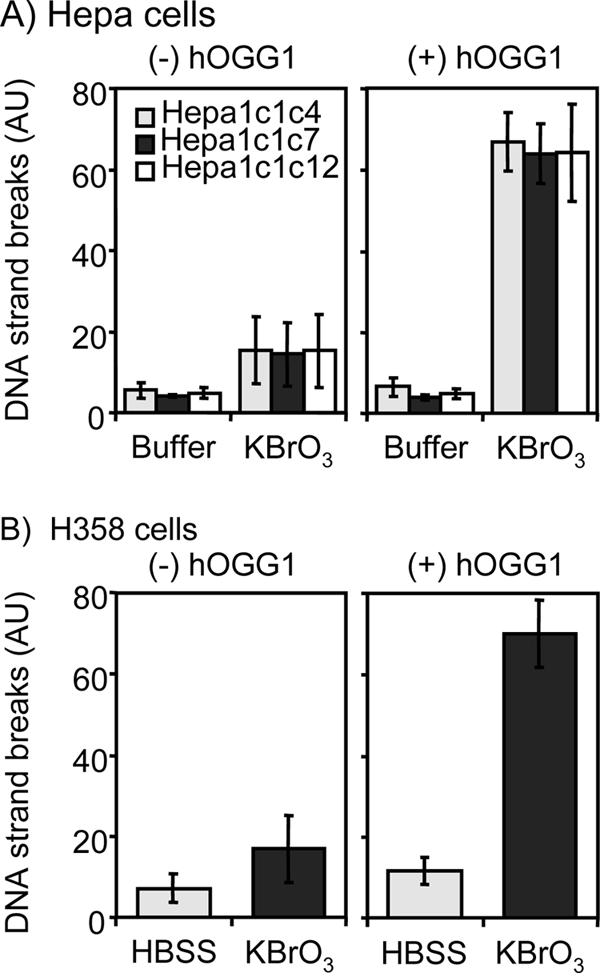
Analysis of hOGG1-dependent DNA strand breaks in both KBrO3-treated Hepa and H358 cells. Three Hepa cell sublines and H358 cells were treated with 2.5 mm of potassium bromate for 3 h. The cells were harvested and used for the detection of DNA strand breaks by hOGG1-coupled comet assay. A, Hepa cells. B, H358 cells. AU, arbitrary units.
B[a]P-7,8-dione-mediated DNA Strand Breaks and 8-Oxo-dGuo Formation in Hepa1c1c4, Hepa1c1c7, and Hepa 1c1c12 Cells
We next treated Hepa cells with B[a]P-7,8-dione for 6 h. B[a]P-7,8-dione produced DNA strand breaks in all three Hepa cells, and the use of hOGG1 further increased B[a]P-7,8-dione-mediated DNA strand breaks (Fig. 4A). Importantly, AhR-deficient Hepa1c1c12 cells formed fewer DNA strand breaks with B[a]P-7,8-dione treatment, irrespective of whether hOGG1 was included in the assay. This was not the case for the ARNT-deficient Hepa1c1c4 cells, which gave the same response as wild type cells.
FIGURE 4.

AhR-dependent DNA strand breaks and 8-oxo-dGuo formation in Hepa cells-treated with B[a]P-7,8-dione. A, B[a]P-7,8-dione-mediated DNA strand breaks in Hepa cells. AhR-deficient Hepa1c1c12 cells, ARNT-deficient Hepa1c1c4, and their wild type Hepa1c1c7 cells were treated with 20 μm B[a]P-7,8-dione for 6 h. The cells were harvested and used for the detection of DNA strand breaks by the hOGG1-coupled comet assay. Significant effects were observed at different incubation times and after hOGG1 treatment following B[a]P-7,8-dione treatment. Significant effects were also observed between AhR-deficient Hepa1c1c12 cells and ARNT-deficient Hepa 1c1c4 cells and their wild type Hepa 1c1c7 cells. *, p < 0.05. B, LC/MS detection of 8-oxo-dGuo in Hepa cells-treated with B[a]P-7,8-dione. Significant effects were observed again when AhR-deficient Hepa1c1c12 cells were compared with ARNT-deficient Hepa 1c1c4 cells and wild type Hepa 1c1c7 cells. *, p < 0.05. AU, arbitrary units.
These experiments were then repeated to measure levels of 8-oxo-dGuo by immunoaffinity capture stable isotope dilution LC-ESI/MRM/MS in Hepa cells treated with B[a]P-7,8-dione. Treatment of B[a]P-7,8-dione increased the levels of 8-oxo-dGuo in the Hepa cells when compared with cells treated with DMSO alone. Significantly lower 8-oxo-dGuo levels were observed in the AhR-deficient Hepa1c1c12 cells (p < 0.05) but not in the ARNT-deficient Hepa1c1c4 cells (Fig. 4B).
AhR-dependent DNA Strand Breaks in B[a]P-7,8-dione-treated H358 Cells
To confirm that AhR is involved in the formation of PAH o-quinone-mediated DNA strand breaks, H358 cells were transfected with the AhR siRNA and incubated with B[a]P-7,8-dione for up to 12 h. Western blot analysis revealed that AhR protein levels were significantly reduced after AhR siRNA transfection, indicating that the knockdown was successful (Fig. 5A, lane 3), whereas the levels were not changed in nontransfected cells (Fig. 5A, lane 1). Treatment with B[a]P-7,8-dione also gradually decreased AhR protein levels in nontransfected cells in a time-dependent manner. Significant reduction of AhR protein levels were observed as early as 3 h (Fig. 5A, lane 2). These data support our previous findings that PAH o-quinones promote the translocation of AhR to the nucleus in Hepa1c1c7 cells (30).
FIGURE 5.

AhR expression and DNA strand break formation in AhR siRNA-transfected and nontransfected H358 cells treated with B[a]P-7,8-dione. H358 cells were transfected with AhR-siRNA and then treated with B[a]P-7,8-dione for 3, 6, or 12 h. The cells were then harvested and were divided into two aliquots. One was used for Western blot analysis, and other was used for the hOGG1-coupled comet assay. A, AhR expression in both AhR siRNA-transfected and nontransfected H358 cells treated with B[a]P-7,8-dione. Lane 1, non-AhR siRNA-transfected cells; lane 2, nontransfected cells treated with B[a]P-7,8-dione; lane 3, AhR-siRNA transfected cells; lane 4, AhR siRNA-transfected cells-treated with B[a]P-7,8-dione. B, AhR-dependent DNA strand break formation in AhR siRNA-transfected H358 cells treated with DMSO. C, AhR-dependent DNA strand break formation in AhR siRNA-transfected H358 cells treated with B[a]P-7,8-dione. Significant effects were observed after hOGG1 treatment. *, p < 0.05. Significant effects was observed between AhR siRNA-transfected cells and nontransfected cells-treated with B[a]P-7,8-dione for 6 h. *, p < 0.05. A detailed description can be found under “Experimental Procedures.” WT, wild type; AhR-k/d, aryl hydrocarbon receptor knock down cells; AU, arbitrary units.
Using these cells, PAH o-quinone-mediated DNA strand breaks were measured both in AhR siRNA-transfected and nontransfected H358 cells. B[a]P-7,8-dione increased DNA strand breaks in both AhR siRNA-transfected cells and nontransfected cells in a time-dependent manner when compared with DMSO-negative controls, and these effects were increased further by the treatment with hOGG1 (Fig. 5, B and C). Importantly, treatment with B[a]P-7,8-dione for 6 h caused significantly fewer DNA strand breaks in AhR siRNA-transfected cells compared with nontransfected cells, indicating that AhR enhanced the formation of B[a]P-7,8-dione-mediated DNA breakage at these time points (Fig. 5C). This effect was observed in the presence and absence of hOGG1. Longer treatments with B[a]P-7,8-dione for 12 h, however, produced the same level of DNA strand breaks in the AhR siRNA-transfected cells and wild type cells, suggesting that this PAH metabolite can eventually cause DNA strand breaks independently of the AhR.
DNA Strand Breaks Induced by B[a]P-7,8-dione in H358 Cells and the Involvement of ROS
To determine whether ROS is responsible for the AhR-dependent DNA strand breaks in H358 cells, cells were treated with B[a]P-7,8-dione for up to 12 h following pretreatment with the ROS attenuators, e.g. the antioxidant α-tocopherol (250 μm) and the iron chelator/antioxidant desferal (1 mm). These agents effectively suppressed the B[a]P-7,8-dione-mediated DNA strand breaks, suggesting that B[a]P-7,8-dione-mediated DNA strand breaks were ROS-dependent (Fig. 6).
FIGURE 6.
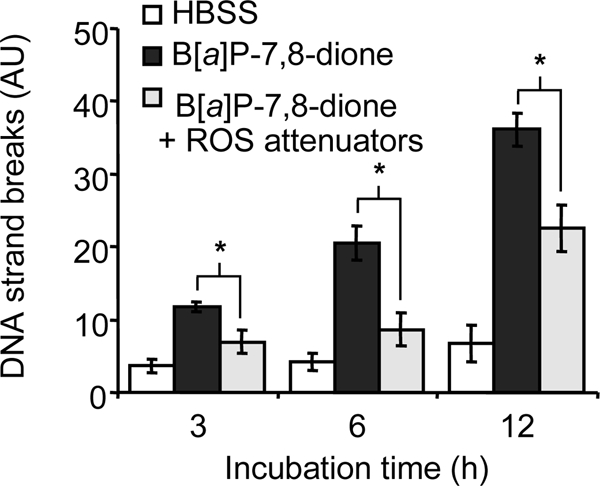
B[a]P-7,8-dione-mediated DNA strand breaks in H358 cells and the effect of ROS attenuators. The cells were treated with 20 μm B[a]P-7,8-dione in HBSS buffer for 3, 6, and 12 h. To determine the effects of ROS attenuators, desferal (1 mm) and α-tocopherol (250 μm) was added to the cells 2 h prior to B[a]P-7,8-dione treatment. At each time point, the cells were harvested and were used for the comet assay. Significant effects were observed after the treatment of ROS attenuators. *, p < 0.05. AU, arbitrary units.
AhR-dependent 8-Oxo-dGuo Formation in B[a]P-7,8-dione-treated H358 Cells
The experiments were replicated to measure 8-oxo-dGuo by LC-ESI/MRM/MS in H358 cells transfected with AhR siRNA. A typical MRM chromatogram from the analysis of 8-oxo-dGuo in H358 cells-treated with B[a]P-7,8-dione for 6 h was obtained and normalized to the heavy isotope internal standard [15N5]8-oxo-dGuo for quantification (Fig. 7A). The data showed that there was a difference in the formation of 8-oxo-dGuo between AhR siRNA-transfected cells and nontransfected cells. The basal level of 8-oxo-dGuo was 7.0 8-oxo-dGuo adducts/107 dGuo in untreated H358 cells, and this increased to 17.0 adducts/107 dGuo in cells treated with B[a]P-7,8-dione. This value was reduced to 13.0 adducts/107 dGuo in AhR knockdown cells, indicating that 8-oxo-dGuo formed by B[a]P-7,8-dione was again AhR-dependent (Fig. 7B).
FIGURE 7.

LC/MS detection of 8-oxo-dGuo in AhR siRNA-transfected and nontransfected H358 cells-treated with B[a]P-7,8-dione. AhR siRNA-transfected H358 cells and nontransfected H358 cells were treated with B[a]P-7,8-dione for 6 h. The cellular DNA was then extracted and digested. The levels of 8-oxo-dGuo in samples were determined by LC-ESI/MS/MRM. A, LC/MS chromatograms of 8-oxo-dGuo in AhR siRNA-transfected H358 cellular DNA. The upper two panels show ion-chromatograms for the detection of endogenous 8-oxo-dGuo and the deuterated [15N5]8-oxo-dGuo internal standard obtained from parental H358 cells-treated with B[a]P-7,8-dione. The lower two panels show ion chromatograms for the detection of 8-oxo-dGuo and the heavy isotope [15N5]8-oxo-dGuo internal standard obtained from AhR siRNA-transfected H358 cells treated with B[a]P-7,8-dione. B, levels of 8-oxo-dGuo in both AhR siRNA-transfected and nontransfected H358 cells-treated with B[a]P-7,8-dione. Significant effects were found between AhR siRNA-transfected H358 cells and nontransfected H358 cells. *, p < 0.05. A detailed description can be found under “Experimental Procedures.”
AhR-dependent DNA Strand Break Formation Modulated by α-NF in B[a]P-7,8-dione-treated H358 Cells
H358 cells were also incubated with B[a]P-7,8-dione for 6 h in the presence and absence of α-NF, which is known to be an AhR antagonist (40). α-NF failed to cause any significant DNA strand breaks by itself in the presence or absence of hOGG1. When cells were treated with either 5 or 10 μm B[a]P-7,8-dione, the DNA strand breaks were decreased in the presence of α-NF irrespective of whether hOGG1 was present or absent, showing that DNA strand breaks caused by B[a]P-7,8-dione were AhR-dependent (Fig. 8).
FIGURE 8.
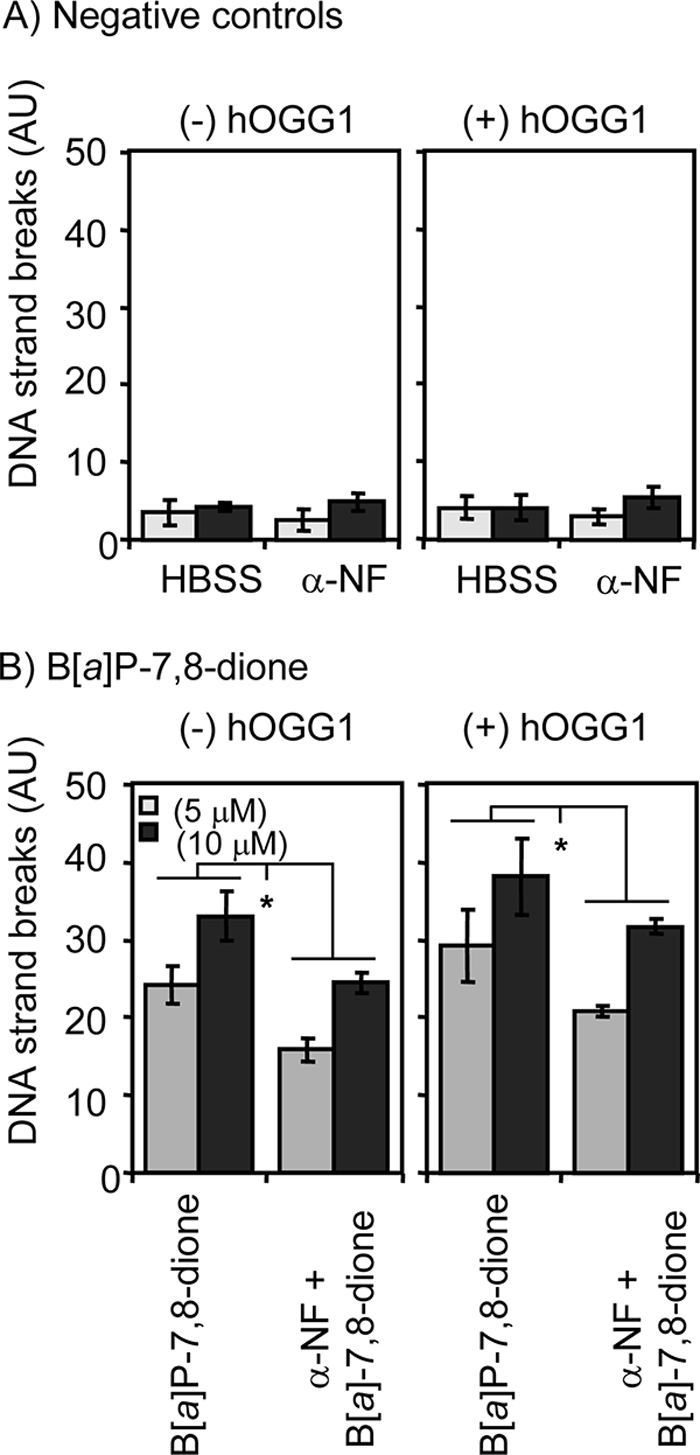
α-NF-dependent DNA strand break formation in B[a]P-7,8-dione-treated H358 cells. H358 cells were pretreated with 1 μm α-NF 1 h before B[a]P-7,8-dione treatment. The cells were then treated with B[a]P-7,8-dione for 6 h. At the end of the incubation, the cells were harvested and were used for the hOGG1-coupled comet assay. A, effects of 1 μm α-NF by itself. B, effects of 5.0 or 10 μm B[a]P-7,8-dione in the presence of 1 μm α-NF. Significant effects were found after α-NF treatment. *, p < 0.05. AU, arbitrary units.
DISCUSSION
In this study we provide evidence that the AhR acts as a carrier to transport and concentrate PAH o-quinones to the nucleus where they can cause oxidative DNA damage in the form of DNA strand breaks and the generation of the mutagenic lesion 8-oxo-dGuo. The evidence for this conclusion comes from multiple sources. First, using murine hepatoma cells in which important target genes are mutated, e.g. ARNT-deficient Hepa1c1c4 cells, AhR-deficient Hapa1c1c12 cells, and their wild type Hepa1c1c7 cells (41, 42), only those cells that are AhR-deficient produce fewer DNA strand breaks and lower amounts of 8-oxo-dGuo when treated with B[a]P-7,8-dione. Second, siRNA knockdown of the AhR leads to less oxidative DNA damage and 8-oxo-dGuo in human bronchoalveolar H358 cells treated with B[a]P-7,8-dione. Third, the use of an AhR antagonist such as α-NF effectively blocks the ability of B[a]P-7,8-dione to cause oxidative DNA damage.
For this study we used B[a]P-7,8-dione, a representative PAH o-quinone produced by the AKR-mediated NADP+-dependent oxidation of PAH-trans-dihydrodiols. We ensured that the there were no differences in the susceptibility of Hepa cell sublines to oxidants, e.g. H2O2 or KBrO3, and we measured 8-oxo-dGuo by two methods: a hOGG1 coupled comet assay and a highly sensitive immunocapture stable isotope dilution LC/ESI/MRM/MS assay. The latter method can detect a basal level of 1.0–2.0 8-oxo-dGuo/107-dGuo and is an order of magnitude more sensitive than other LC/MS assays reported thus far (34). We also demonstrated that DNA strand breaks and 8-oxo-dGuo levels could be attenuated by the use of anti-oxidants, e.g. desfereal and α-tocopherol, showing that the effects were mediated by ROS produced through redox cycling of the PAH o-quinone.
These data are in accord with a previous study from our group, which showed that B[a]P-7,8-dione is a ligand for the AhR receptor (30). In this earlier study we showed that B[a]P-7,8-dione induced P4501A1 expression in the human hepatocellular liver carcinoma HepG2 cell line, it had an estimated affinity for the AhR of 1.0 μm, it translocated the AhR to the nucleus as measured by immunocytochemistry and electrophoretic mobility shift assays, and it induced gene transcription in a HepG2 cell line stably transfected with a XRE-luciferase reporter gene construct (30). More recently, using a dichlorofluorescein diacetate assay, we showed that the metabolic activation of B[a]P-7,8-trans-dihydrodiol to B[a]P-7,8-dione caused ROS generation and oxidative DNA damage in human lung adenocarcinoma A549 cells and that the B[a]P-7,8-dione-mediated ROS production occurred in the nuclear compartment of the cells (25). These studies suggested the existence of cellular transport mechanism for B[a]P-7,8-dione to the nucleus. We now propose that the AhR is responsible for this transport.
Each of the individual approaches used in our experiments deserve some additional comment. The attenuation of DNA strand breaks and 8-oxo-dGuo formation in AhR-deficient cells (but not in cells in which ARNT is mutated) supports a role for the AhR in transporting PAH o-quinones to the nucleus because ARNT is localized in the nucleus (32, 43). AhR and wild type ARNT form a heterodimeric complex that binds to XREs. The mutated ARNT present in Hepa1c1c4 cells still forms a heterodimeric complex with the AhR, but the complex is incapable of binding to an XRE and driving gene expression (37). Because the level of oxidative DNA damage is the same irrespective of whether ARNT is mutated or not suggests that AhR can facilitate DNA damage without binding of the AhR·ARNT heterodimeric complex to an XRE. These data suggest that the AhR might serve as a nuclear transporter of the genotoxin but is not required to target the genotoxin to specific DNA sequences, e.g. those containing an XRE to mediate the DNA damage observed. Interestingly, B[a]P-7,8-dione will not cause CYP1A1 induction in ARNT negative cells. These data suggest that the genotoxic effects of B[a]P-7,8-dione mediated by the AhR can be separated from the inductive effects mediated by the same receptor. Whether normal AhR·ARNT heterodimers deliver B[a]P-7,8-dione to target specific DNA sequences for oxidative DNA damage could be further investigated using a ligation-mediated PCR strategy.
The siRNA knockdown of the AhR leads to only a transient reduction in DNA strand breaks and 8-oxo-dGuo formation, i.e. 6 h post-treatment. This is likely due to the fact that there are two competing processes by which B[a]P-7,8-dione can gain access to the nucleus, transport by the AhR and passive entry into the nucleus over time. Over prolonged time courses both processes lead to similar amounts of oxidative DNA damage, and this is what is observed in H358 cells. However, we cannot rule out the possibility that a long-lived quinone metabolite of B[a]P-7,8-dione also causes oxidative DNA damage at later time points. AhR delivery of PAH o-quinones to the nucleus likely affects the rate of oxidative DNA damage rather than the total amount observed. This kinetic phenomenon could be important because rapid formation of oxidative DNA damage could overwhelm protective mechanisms that might proceed at a slower rate.
The concentration of B[a]P-7,8-dione used in these studies at 20 μm is high. However, the requirement for this high concentration probably reflects the sensitivity of the various assays being used to measure an effect. In contrast, 1 μm B[a]P-7,8-dione can cause a robust induction of P4501A1 in HepG2 cells (30), and submicromolar concentrations can cause induction of P4501B1 in H358 cells (36). In addition, nanomolar concentrations of PAH o-quinones can cause oxidative DNA damage in bulk DNA and mutation of p53, provided redox cycling conditions are optimal (NADH and CuCl2) (24, 26–28).
Distinct parallels exist between the translocation of PAH o-quinones by the AhR to cause oxidative damage of DNA in human lung cells and the transport of genotoxic equine catechol estrogens, e.g. 4-hydroxyequilenin by estrogen receptor (ER) α to cause oxidative DNA damage in human mammary MCF-7 cells (44, 45). In the latter study, modest increases in DNA strand breaks and 8-oxo-dGuo formation were observed in the ER-positive cells versus the ER-negative cells. These data again support the concept that both nuclear receptor transport and passive diffusion of o-quinone genotoxins play a role in the oxidative DNA damage observed with catechol estrogens. This previous study suggested that a kinetic issue is at play because methods for oxidative DNA damage did not have the sensitivity to measure early and dramatic increases in intracellular ROS production detected by DCFA-DA fluorescence changes (45). Our observations are in agreement with these conclusions. It was also proposed that the delivery of 4-hydroxyequilenin by ERα to the nucleus is an example of the ER acting as a “Trojan Horse” to direct site-specific DNA damage (45). Our work with the AhR·ARNT mutant complex suggests that with PAH o-quinones, oxidative DNA damage can occur without specifically targeting XRE DNA sequences.
It is worthy of mention that 8-oxo-dGuo may be an intermediate oxidation product of guanine because further oxidation to spiroiminohydantoin and gaunidohydantoin adducts may occur, leading to an underestimate of the 8-oxo-dGuo formed (46, 47). Of these, spiroiminohydantoin adducts have been detected in vivo by Sugden and co-workers (48) and have been found to be more mutagenic than 8-oxo-dGuo (49).
Our proposed mechanism to account for B[a]P-7,8-dione enhancement of DNA strand breaks and 8-oxo-dGuo formation in an AhR-dependent manner is summarized in Fig. 9. This mechanism accounts for all of the observations made in the present study: 1) B[a]P-7,8-dione binds to the AhR in the cytosol, this induces a conformational change in the receptor leading to the dissociation of Hsp90, and AhR is translocated to the nucleus with B[a]P-7,8-dione bound; 2) the formation of B[a]P-7,8-dione·AhR·ARNT wild type complex then forms to mediate XRE-dependent gene transcription and may even target XRE dependent oxidative DNA damage; 3) formation of a B[a]P-7,8-dione·AhR·ARNT mutant complex results in the same amount of DNA damage without binding to the XRE; 4) passive diffusion of B[a]P-7,8-dione into the nucleus also results in oxidative DNA damage, but this takes longer to occur; and 5) B[a]P-7,8-dione bound to AhR·ARNT complexes and free B[a]P-7,-dione redox cycle to produce ROS and cause DNA strand breaks and 8-oxo-dGuo, and this is predicted to preferentially damage unraveled DNA, i.e. DNA undergoing active gene transcription.
FIGURE 9.

Mechanism of 8-oxo-dGuo formation by B[a]P-7,8-dione through ligand-mediated activation of AhR. The upper panel shows the mechanism with wild type AhR·ARNT complex, and the lower panel shows the mechanism with a wild type AhR·mutant·ARNT complex.
In conclusion, our studies have demonstrated that B[a]P-7,8-dione (produced by AKRs) causes enhanced DNA strand breaks and 8-oxo-dGuo formation in murine Hepa cells and human bronchoalveolar H358 cells via an AhR-dependent mechanism. AhR can act as the transport mechanism that is needed to deliver B[a]P-7,8-dione to the nucleus to cause oxidative DNA damage. This represents an example whereby a nuclear receptor transports a genotoxin to the nuclear DNA.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1-CA39504 and P30-ES013508 (to T. M. P.) and RO1-CA130038 (to I. A. B.).
- PAH
- polycyclic aromatic hydrocarbon
- AhR
- aryl hydrocarbon receptor
- ARNT
- aryl hydrocarbon receptor nuclear translocator
- AKR
- aldo-keto-reductase
- B[a]P-7,8-trans-dihydrodiol
- trans-7,8- dihydroxy-7,8-dihydrobenzo[a]pyrene-7,8-diol
- B[a]P-7,8-dione
- benzo[a]pyrene-7,8-dione
- comet assay
- single cell gel electrophoresis assay
- HBSS
- Hanks' balanced salt solution
- hOGG1
- human 8-oxoguanine glycosylase
- 8-oxo-dGuo
- 8-oxo-7,8-dihydro-2′-deoxyguanosine
- α-NF
- α-naphthoflavone
- P450
- cytochrome P-450
- ROS
- reactive oxygen species
- XRE
- xenobiotic response element
- LC/ESI/MRM/MS
- liquid chromatography/electrospray ionization/multiple reaction monitoring/mass spectrometry
- siRNA
- small interfering RNA
- LC/MS
- liquid chromatography/mass spectrometry
- HPLC
- high pressure liquid chromatography
- DMSO
- dimethyl sulfoxide
- ER
- estrogen receptor.
REFERENCES
- 1.Grimmer G., Böhnke H. (1975) J. Assoc. Off. Anal. Chem. 58, 725–733 [PubMed] [Google Scholar]
- 2.Dipple A. (1983) Cancer Res. 43, 2422s–2425s [PubMed] [Google Scholar]
- 3.Rothman N., Poirier M. C., Baser M. E., Hansen J. A., Gentile C., Bowman E. D., Strickland P. T. (1990) Carcinogenesis 11, 1241–1243 [DOI] [PubMed] [Google Scholar]
- 4.Hecht S. S. (1999) J. Natl. Cancer Inst. 91, 1194–1210 [DOI] [PubMed] [Google Scholar]
- 5.Pfeifer G. P., Denissenko M. F., Olivier M., Tretyakova N., Hecht S. S., Hainaut P. (2002) Oncogene 21, 7435–7451 [DOI] [PubMed] [Google Scholar]
- 6.Conney A. H. (1982) Cancer Res. 42, 4875–4917 [PubMed] [Google Scholar]
- 7.Shimada T., Gillam E. M., Oda Y., Tsumura F., Sutter T. R., Guengerich F. P., Inoue K. (1999) Chem Res. Toxicol. 12, 623–629 [DOI] [PubMed] [Google Scholar]
- 8.Shimada T., Oda Y., Gillam E. M., Guengerich F. P., Inoue K. (2001) Drug. Metab. Dispos. 29, 1176–1182 [PubMed] [Google Scholar]
- 9.Koreeda M., Moore P. D., Wislocki P. G., Levin W., Yagi H., Jerina D. M. (1978) Science 199, 778–781 [DOI] [PubMed] [Google Scholar]
- 10.Jennette K. W., Jeffrey A. M., Blobstein S. H., Beland F. A., Harvey R. G., Weinstein I. B. (1977) Biochemistry 16, 932–938 [DOI] [PubMed] [Google Scholar]
- 11.Marshall C. J., Vousden K. H., Phillips D. H. (1984) Nature 310, 586–589 [DOI] [PubMed] [Google Scholar]
- 12.Denissenko M. F., Pao A., Tang M., Pfeifer G. P. (1996) Science 274, 430–432 [DOI] [PubMed] [Google Scholar]
- 13.Shibutani S., Margulis L. A., Geacintov N. E., Grollman A. P. (1993) Biochemistry, 32, 7531–7541 [DOI] [PubMed] [Google Scholar]
- 14.Xie Z., Braithwaite E., Guo D., Zhao B., Geacintov N. E., Wang Z. (2003) Biochemistry 42, 11253–11262 [DOI] [PubMed] [Google Scholar]
- 15.Palackal N. T., Burczynski M. E., Harvey R. G., Penning T. M. (2001) Biochemistry 40, 10901–10910 [DOI] [PubMed] [Google Scholar]
- 16.Palackal N. T., Lee S. H., Harvey R. G., Blair I. A., Penning T. M. (2002) J. Biol. Chem. 277, 24799–24808 [DOI] [PubMed] [Google Scholar]
- 17.Penning T. M., Burczynski M. E., Hung C. F., McCoull K. D., Palackal N. T., Tsuruda L. S. (1999) Chem. Res. Toxicol. 12, 1–18 [DOI] [PubMed] [Google Scholar]
- 18.Shou M., Harvey R. G., Penning T. M. (1993) Carcinogenesis 14, 475–482 [DOI] [PubMed] [Google Scholar]
- 19.Balu N., Padgett W. T., Lambert G. R., Swank A. E., Richard A. M., Nesnow S. (2004) Chem. Res. Toxicol. 17, 827–838 [DOI] [PubMed] [Google Scholar]
- 20.Balu N., Padgett W. T., Nelson G. B., Lambert G. R., Ross J. A., Nesnow S. (2006) Anal. Biochem. 355, 213–223 [DOI] [PubMed] [Google Scholar]
- 21.McCoull K. D., Rindgen D., Blair I. A., Penning T. M. (1999) Chem. Res. Toxicol. 12, 237–246 [DOI] [PubMed] [Google Scholar]
- 22.Ohnishi S., Kawanishi S. (2002) Biochem. Biophys. Res. Commun. 290, 778–782 [DOI] [PubMed] [Google Scholar]
- 23.Seike K., Murata M., Oikawa S., Hiraku Y., Hirakawa K., Kawanishi S. (2003) Chem. Res. Toxicol. 16, 1470–1476 [DOI] [PubMed] [Google Scholar]
- 24.Park J. H., Gopishetty S., Szewczuk L. M., Troxel A. B., Harvey R. G., Penning T. M. (2005) Chem. Res. Toxicol. 18, 1026–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park J. H., Mangal D., Tacka K. A., Quinn A. M., Harvey R. G., Blair I. A., Penning T. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 6846–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park J. H., Troxel A. B., Harvey R. G., Penning T. M. (2006) Chem. Res. Toxicol. 19, 719–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu D., Berlin J. A., Penning T. M., Field J. (2002) Chem. Res. Toxicol. 15, 832–842 [DOI] [PubMed] [Google Scholar]
- 28.Park J. H., Gelhaus S., Vedantam S., Oliva A. L., Batra A., Blair I. A., Troxel A. B., Field J., Penning T. M. (2008) Chem. Res. Toxicol. 21, 1039–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flowers L., Bleczinski W. F., Burczynski M. E., Harvey R. G., Penning T. M. (1996) Biochemistry 35, 13664–13672 [DOI] [PubMed] [Google Scholar]
- 30.Burczynski M. E., Penning T. M. (2000) Cancer Res. 60, 908–915 [PubMed] [Google Scholar]
- 31.Hoffman E. C., Reyes H., Chu F. F., Sander F., Conley L. H., Brooks B. A., Hankinson O. (1991) Science 252, 954–958 [DOI] [PubMed] [Google Scholar]
- 32.Bohonowych J. E., Denison M. S. (2007) Toxicol. Sci. 98, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harvey R. G., Dai Q., Ran C., Penning T. M. (2004) J. Org. Chem. 69, 2024–2032 [DOI] [PubMed] [Google Scholar]
- 34.Mangal D., Vudathala D., Park J. H., Lee S. H., Penning T. M., Blair I. A. (2009) Chem. Res. Toxicol. 22, 788–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 36.Jiang H., Vudathala D. K., Blair I. A., Penning T. M. (2006) Chem. Res. Toxicol. 19, 68–78 [DOI] [PubMed] [Google Scholar]
- 37.Numayama-Tsuruta K., Kobayashi A., Sogawa K., Fujii-Kuriyama Y. (1997) Eur. J. Biochem. 246, 486–495 [DOI] [PubMed] [Google Scholar]
- 38.Smith C. C., O'Donovan M. R., Martin E. A. (2006) Mutagenesis 21, 185–190 [DOI] [PubMed] [Google Scholar]
- 39.Bjorâs M., Luna L., Johnsen B., Hoff E., Haug T., Rognes T., Seeberg E. (1997) EMBO J. 16, 6314–6322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merchant M., Arellano L., Safe S. (1990) Arch. Biochem. Biophys. 281, 84–89 [DOI] [PubMed] [Google Scholar]
- 41.Wood S. M., Gleadle J. M., Pugh C. W., Hankinson O., Ratcliffe P. J. (1996) J. Biol. Chem. 271, 15117–15123 [DOI] [PubMed] [Google Scholar]
- 42.Zhang J., Watson A. J., Probst M. R., Minehart E., Hankinson O. (1996) Mol. Pharm. 50, 1454–1462 [PubMed] [Google Scholar]
- 43.Soshilov A., Denison M. S. (2008) J. Biol. Chem. 283, 32995–33005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X., Yao J., Pisha E., Yang Y., Hua Y., van Breemen R. B., Bolton J. L. (2002) Chem. Res. Toxicol. 15, 512–519 [DOI] [PubMed] [Google Scholar]
- 45.Wang Z., Wijewickrama G. T., Peng K. W., Dietz B. M., Yuan L., van Breeman R. B., Bolton J. L., Thatcher G. R. (2009) J. Biol. Chem. 284, 8633–8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo W., Muller J. G., Rachlin E. M., Burrows C. J. (2001) Chem. Res. Toxicol. 14, 927–938 [DOI] [PubMed] [Google Scholar]
- 47.Burrows C. J., Muller J. G., Kornyushyna O., Luo W., Duarte V., Leipold M. D., David S. S. (2002) Environ. Health. Persp. 110, (Suppl. 5) 713–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hailer M. K., Slade P. G., Martin B. D., Sugden K. D. (2005) Chem. Res. Toxicol. 18, 1378–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neeley W. L., Essigmann J. M. (2006) Chem. Res. Toxicol. 19, 491–505 [DOI] [PubMed] [Google Scholar]