

Abstract
Infection of a host cell by a retrovirus requires an initial interaction with a cellular receptor. For numerous gammaretroviruses, such as the gibbon ape leukemia virus, woolly monkey virus, feline leukemia virus subgroup B, feline leukemia virus subgroup T, and 10A1 murine leukemia virus, this receptor is the human type III sodium-dependent inorganic phosphate transporter, SLC20A1, formerly known as PiT1. Understanding the critical receptor functionalities and interactions with the virus that lead to successful infection requires that we first know the surface structure of the cellular receptor. Previous molecular modeling from the protein sequence, and limited empirical data, predicted a protein with 10 transmembrane helices. Here we undertake the biochemical approach of substituted cysteine accessibility mutagenesis to resolve the topology of this receptor in live cells. We discover that there are segments of the protein that are unexpectedly exposed to the outside milieu. By using information determined by substituted cysteine accessibility mutagenesis to set constraints in HMMTOP, a hidden Markov model-based transmembrane topology prediction method, we now propose a comprehensive topological model for SLC20A1, a transmembrane protein with 12 transmembrane helices and 7 extracellular regions, that varies from previous models and should permit approaches that define both virus interaction and transport function.
Nearly all of the gammaretroviral receptors identified to date are members of the major carrier facilitator class of proteins (1). The cellular role of these proteins is the transport of solutes essential for cell metabolism. SLC20A1 (PiT1), the receptor for gibbon ape leukemia virus (GALV)2 and feline leukemia virus subgroup B (FeLV-B), has been identified to normally function as the sodium-dependent inorganic phosphate (Na/Pi) transporter SLC20A1 (2–4). SLC20A1 is ubiquitously expressed and plays a major role in the housekeeping process of maintaining cell Pi homeostasis in transporting monovalent H2PO4 forms of Pi (5). This Pi transporter is important for chondroblastic and osteoblastic mineralization and vascular calcification (5).
Using Kyte Doolittle hydropathy plots, the structure of SLC20A1 (PiT1) was initially predicted to contain 10 transmembrane helices (TMHs), with both the N and C termini positioned intracellularly (6). The predicted structure of SLC20A1 (PiT1) has since been modified, based on experimental data that showed both ends of the protein are extracellular and that the protein contains an N-linked glycosylation site (7); however, the number and positions of the extracellular regions (ECRs) and transmembrane helices (TMHs) remain not experimentally validated.
A closely related phosphate transporter, SLC20A2 (PiT2), functions as a receptor for the murine amphotropic leukemia virus (A-MLV) (3, 8–10). Salaun et al. (11) experimentally assessed PiT2 topology using N- and C-terminal epitope tags, glycosylation studies, and in vitro translation of C-terminally epitope-tagged truncated mutants exposed to microsomal vesicles. However, as noted by these authors, C-terminally truncated PiT2 mutants in membrane vesicles can behave differently than PiT2 in the cell membrane, and thus assignment of hydrophilic loops to one side or the other of the membrane using this experimental approach provides ambiguous results (11).
High resolution structural analysis of proteins such as SLC20A1 (PiT1), which pass through the cell membrane multiple times, is impeded by difficulties in purification and crystallization. To circumvent these obstacles and to increase the understanding of structure-function relationships, multiple membrane-spanning proteins can be characterized through mutational analysis in the context of their normal lipid environment within living cells. In light of this consideration, we undertook cellular topological studies of the SLC20A1 (PiT1) protein using substituted cysteine accessibility mutagenesis (SCAM), a powerful method that has been successfully employed in establishing the topology of a large number of multiple membrane-spanning proteins because it minimally perturbs the structure and function of the target protein (12). Over 50 cysteine mutants were evaluated, allowing us to construct a model for the membrane topology of SLC20A1 (PiT1), in which the protein crosses the plasma membrane 12 times.
The A-MLV binding domain for its receptor, SLC20A2 (PiT2), has been experimentally determined (13); however, a region(s) within PiT1 that binds virus has not been resolved. It has been proposed that region A PiT1 residues 550–558 function as the virus-binding site, based on the loss of receptor function when the corresponding residues of PiT2 are substituted for those of PiT1 (14), and other experimental criteria that demonstrate region A is important in facilitating virus entry (15). However, it is important to note that these experiments did not include virus binding assays. Later we determined that substitution of PiT2 residues for PiT1 region A residues in fact did not abolish virus binding for FeLV-B, even though receptor function was inactivated (7). Thus, the virus-binding site for SLC20A1 (PiT1) remains unresolved. Now that we have established an experimentally validated topology for SLC20A1 (PiT1), the extracellular residues that are required for virus binding can be identified, and the effects of SLC20A1 (PiT1) receptor-inactivating mutations can be assessed with respect to whether or not they alter the ability of the virus to bind or enter cells at a post-binding stage of infection.
EXPERIMENTAL PROCEDURES
Cell Culture
Cell lines used in this study include Mus dunni tail fibroblasts (MDTF), obtained from Olivier Danos (Institute Pasteur) and originally derived by Lander and Chattopadhyay (16), and human embryonic kidney 293T cells (Cell Genesys, Inc.). MDTF and 293T cells were maintained in Dulbecco's modified Eagle's medium with Glutamax (Invitrogen) supplemented with 10% fetal bovine serum, 100 units of penicillin/ml, and 100 μg of streptomycin/ml. Retroviral vectors were produced as described previously (7). Cell lines expressing various cysteine mutant SLC20A1 (PiT1) receptor proteins were exposed to GALV-enveloped retroviral vectors with genomes encoding β-galactosidase to determine whether the modified proteins were functional for virus entry. Cells were histochemically analyzed for expression of β-galactosidase as described previously (7).
Mutagenesis
The cysteineless SLC20A1 (PiT1) cDNA was constructed in the pLNSPiT1-HA plasmid (7) using the QuikChange II XL site-directed mutagenesis kit (Stratagene), according to the manufacturer's instructions. Mutagenesis primers were synthesized and purified by high pressure liquid chromatography (Integrated DNA Technologies, Inc.). Plasmids containing mutant cDNAs were sequenced to confirm each mutation and to ensure that no unscheduled mutations were introduced.
Labeling Individual Cysteine Residues
Cells expressing HA epitope-tagged PiT1 (PiT1-HA) or mutant cysteineless PiT1 (PiT1-13-HA) were seeded in 6-cm tissue culture dishes at 2 × 106 cells/dish 1 day prior to labeling. Optimal conditions for 3-(N-maleimidylpropionyl)biocytin (biotin maleimide or B-mal) (Molecular Probes) reactivity was determined to occur at 250 μm with incubation for 30 min at room temperature. These conditions resulted in little permeabilization of the membrane as assessed for MDTF cells expressing a PiT1-13-HA cysteine mutant wherein the substituted cysteine was determined to be extracellular (supplemental figure, A). The next day, cells were washed with PBSCM (PBS containing 0.1 mm CaCl2 and 1 mm MgCl2) and then exposed to 250 mm B-mal in PBSCM for 30 min at room temperature. The maleimide portion of B-mal reacts with thiol groups such as cysteine, leaving the biotin portion of B-mal available for binding to avidin (Fig. 1A). After washing three times in PBSCM supplemented with 2% (v/v) 2-mercaptoethanol and once in PBSCM, cells were lysed on ice for 30 min in 25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40 lysis buffer, containing Complete protease inhibitor mixture tablets (Roche Applied Science). Cell lysates were centrifuged at 16,000 × g for 20 min at 4 °C to remove insoluble material, and supernatants were immunoprecipitated with UltraLink Immobilized NeutrAvidin Protein (Pierce) for at least 1 h at 4 °C; the immobilized avidin reacts with all biotinylated cell surface proteins. The precipitated protein was washed three times in lysis buffer and then resuspended in 30 μl of reducing protein loading buffer and heated to 95 °C for 5 min before SDS-PAGE followed by Western blotting.
FIGURE 1.

Methods used in SCAM are schematically presented. A, membrane-spanning protein containing a single introduced cysteine residue (yellow circle) is reacted with B-mal (step 1); after cell lysis (step 2), lysate is mixed with avidin-coated beads to isolate biotinylated proteins (step 3); only epitope-tagged, biotinylated proteins are detected by anti-HA antibody on a Western blot (step 4). Black line represents the protein; beige rectangle is the plasma membrane; yellow star represents the HA epitope tag; and white rectangle represents nitrocellulose membrane with tagged proteins. B, preincubation of cells with MTSET (red hexagon), a membrane-impermeant thiol-reactive compound, blocks B-mal reaction with the extracellular cysteine residue. C, permeabilization of cells with SLO creates pores in the plasma membrane, allowing B-mal access to intracellular cysteine residues.
Blocking of cysteine residues that reacted with B-mal was achieved by exposing cells to 1 mm 2-(trimethylammonium)ethyl methanethiosulfonate bromide (MTSET) (Toronto Research Chemicals) in PBS for 5 min at room temperature and then washing the cells in PBS. MTSET is membrane-impermeant (17) and reacts with thiol groups on the cell surface. The cells were then exposed to B-mal and lysed, as described above. B-mal was unable to react with cysteines bound to MTSET, and therefore pulldown of PiT1-HA with avidin was inhibited, thus reducing or eliminating the signal on a Western blot.
Cells that did not initially react with B-mal were permeabilized before exposing to B-mal in the following manner: after removal of medium, cells were washed twice with PBS and then incubated in 1 ml of 250 units of streptolysin O (SLO)/ml (Sigma) prepared in SLO buffer (2.5 mm MgCl2, 1 mm dithiothreitol, 115 mm potassium acetate, 2.5 mm HEPES) for 15 min on ice. Cells were washed twice with cold SLO buffer and then incubated at 37 °C for 30 min in 1 ml of SLO buffer pre-warmed to 37 °C; they were next washed in cold SLO buffer and then PBSCM before labeling with B-mal (18).
Western Blotting of Labeled Cell Lysates
Biotinylated protein samples were subjected to SDS-PAGE on 4–20% Tris-glycine gels (Invitrogen). Proteins were subsequently transferred to nitrocellulose membranes by electroblotting and detected with anti-HA monoclonal antibody HA.11 (Covance) and goat anti-mouse IgG conjugated to horseradish peroxidase (Pierce). Chemiluminescent horseradish peroxidase substrate (Immobilon, Millipore) was used to detect signals using a CoolSnap HQ2 camera and Ivision software (Biovision Technologies, Inc.).
Detection and Quantification of Epitope-tagged SLC20A (PiT1) Expression
MDTF cells expressing mutant SLC20A1(PiT1) proteins containing C-terminal HA epitope tags were analyzed using flow cytometry. First, cells were removed from flasks with Cell Stripper (Cellgro), after which they were washed in Hanks' buffered salt solution. One million cells for each cell line was placed in a tube containing 1 μg of anti-HA-Alexa Fluor 488 monoclonal antibody (Invitrogen) and incubated for 1 h at room temperature. Cells were washed in Hanks' buffered salt solution and then analyzed to determine the number of antibody-binding sites per cell. Controls for each sample cell were not reacted with antibody and counted to determine autofluorescence values. These values were subtracted from the values obtained for stained cells. Quantification of bound antibodies per cell was accomplished through use of Quantum Simply Cellular anti-mouse IgG quantitative beads (Bangs Laboratories), containing known numbers of antibody-binding sites, according to the manufacturer's instructions.
Topology Prediction
The protein sequence of the human SLC20A1 (PiT1) (SwissProt identifier, SLC20A1_HUMAN) was submitted to the HMMTOP server with the following constraints: 1-19-o, 88-97-o, 147-150-o, 222-228-o, 547-557-o, 613-621-o, and 677-E-o (19).
RESULTS
Cysteineless SLC20A1 (PiT1) Protein Functions as a GALV Receptor
SLC20A1 (PiT1) contains 13 cysteine residues, at positions 63, 132, 204, 238, 242, 251, 264, 403, 426, 468, 524, 573, and 625. To perform SCAM, it was first necessary to determine whether any one of these cysteines is critical for SLC20A1 (PiT1) virus receptor function. We therefore performed site-directed mutagenesis on a hemagglutinin epitope-tagged form of SLC20A1 (PiT1-HA) (7), in which we sequentially replaced each of the 13 cysteine residues with an alanine to produce a cysteineless protein, and we assessed receptor function on each of the 13 resultant PiT1-HA mutants lacking 1–13 of these cysteine residues. Infection results from MDTF cells expressing the cysteineless form of SLC20A1 (PiT1), PiT1-13-HA, show that replacement of all 13 cysteine residues in SLC20A1 (PiT1) with alanine does not compromise GALV receptor function (Table 1). The PiT1-13-HA plasmid was used as the template for mutagenesis, in which single cysteine residues were substituted for specific residues innate to SLC20A1 (PiT1) at selected positions in the protein. In this manner, multiple mutants containing individual cysteines at different positions were generated and tested. The epitope-tagged mutant proteins were stably expressed in GALV-resistant murine MDTF cells and analyzed with respect to their abilities to confer susceptibility to GALV vectors. All mutants functioned as GALV receptors when stably expressed in MDTF cells, producing titers within 1–2 orders of magnitude of the titers obtained with MDTF cells expressing wild-type SLC20A1 (PiT1) following exposure to GALV-enveloped retroviral vectors (Table 1).
TABLE 1.
Biochemical analysis of SLC20A1 (PiT1) cysteine mutants and HMMTOP-predicted location of residues using experimental constraints
| SLC20A1 receptor | B-mal | MTSET (block) | SLO (perm) | GALV functiona | Predicted location |
|---|---|---|---|---|---|
| PiT1-HA | − | + | + | NAb | |
| PiT1-13 | − | − | + | NA | |
| V19C | + | + | ECR 1 | ||
| A79C | + | + | + | ECR 2 | |
| G88C | + | + | + | ECR 2 | |
| L89C | + | + | + | ECR 2 | |
| V92C | + | + | + | ECR 2 | |
| N96C | + | + | +a | ECR 2 | |
| S97C | + | + | + | ECR 2 | |
| L101C | + | + | + | ECR 2 | |
| A104C | − | ± | + | TMH III | |
| A109C | − | + | + | TMH III | |
| A114C | − | + | + | TMH III | |
| G135C | − | − | + | TMH IV | |
| Q147C | + | + | + | ECR 3 | |
| V150C | + | + | + | ECR 3 | |
| L155C | − | + | + | ECR 3/TMH V | |
| S161C | − | + | + | TMH V | |
| L176C | − | + | + | ICR3c | |
| G222C | + | + | + | ECR 4 | |
| L228C | + | + | + | ECR 4 | |
| G230C | − | + | + | TMH VII | |
| L545C | + | + | + | ECR 5 | |
| Y546C | − | − | + | ECR 5 | |
| L547C | + | + | + | ECR 5 | |
| V548C | + | + | + | ECR 5 | |
| Y549C | + | + | + | ECR 5 | |
| D550C | + | + | + | ECR 5 | |
| V554C | + | + | + | ECR 5 | |
| K557C | + | + | + | ECR 5 | |
| L576C | − | + | + | TMH IX | |
| R581C | − | + | + | ICR 5 | |
| L591C | − | + | + | ICR 5 | |
| L592C | − | + | + | ICR 5 | |
| I594C | − | + | + | TMH X | |
| G599C | − | + | + | TMH X | |
| S606C | − | + | + | TMH X | |
| A613C | + | + | + | ECR 6 | |
| N615C | + | + | + | ECR 6 | |
| I616C | + | + | + | ECR 6 | |
| G617C | + | + | + | ECR 6 | |
| L618C | + | + | + | ECR 6 | |
| S621C | + | + | + | ECR 6 | |
| S661C | − | + | + | TMH XII | |
| I668C | − | + | + | TMH XII | |
| A670C | − | + | + | TMH XII | |
| L677C | + | + | + | ECR 7 |
a GALV titers of all mutant cell lines were within 10-fold of the titer obtained with MDTFPiT1-HA at 1 × 106 blue (β-galactosidase)-forming units/ml. Exceptions are N96C titers that were 2 orders of magnitude lower than PiT1-HA.
b NA indicates not applicable.
c ICR refers to intracellular region as determined by experimentally validated HMMTOP prediction.
SCAM Assessment of PiT1-13-HA
The first step in SCAM (reviewed in Refs. 20, 21) analysis is the construction of a cysteineless protein wherein all native cysteines are substituted by alanines. The next step is the introduction by site-directed mutagenesis of individual cysteine residues into the cysteineless protein as a means to assess the topology of a functional protein expressed in live cells. Cysteine residues are small, hydrophobic and well tolerated at most positions in polytopic proteins, thereby not disturbing the behavior of a protein (20). It is important that each mutated protein retain its given function. Once we had ascertained that each PiT1-13-HA mutant with a substituted cysteine retained virus receptor function, we exposed MDTF cells expressing the mutant to the thiol-reactive agent maleimide conjugated to biotin (B-mal). All B-mal-reactive cell surface proteins were precipitated using avidin-coated beads. Western blots of these B-mal-reactive proteins were then probed with anti-HA antibody to detect specific labeling of the HA epitope-tagged SLC20A1 (PiT1) cysteine mutant proteins (Fig. 1A). B-mal is a very large molecule and is membrane-impermeant except at high concentrations and for long incubation periods; thus it has limited access to intracellular cysteines or cysteines obscured because of protein folding or secondary structure. Nonetheless, to ensure that B-mal reactivity is confined to cysteines expressed in accessible extracellular portions of SLC20A1 (PiT1) and not attributable to B-mal accessing intracellular cysteines, we verified its reactivity by using the membrane-impermeant thiol-reactive sulfhydryl reagent MTSET (17). In cells pretreated with MTSET, extracellular cysteines are no longer available for labeling by B-mal, and subsequent isolation of immunoreactive proteins from cell lysates using avidin-coated beads is prevented (Fig. 1B). Therefore, preincubation of cells with MTSET prior to B-mal labeling results in a significantly weaker or absent signal on a Western blot (Fig. 1, A and B).
To confirm that cysteine residues that failed to label after exposure to B-mal were intracellular, cells were treated with SLO, a thiol-activated toxin that permeabilizes the mammalian cell membrane by binding to cholesterol and forming pores in the membrane, thus allowing B-mal improved access to residues inside the cell membrane (Fig. 1C). HA-tagged PiT1-13 mutants that can only be detected on Western blots after permeabilization with SLO or whose signal is markedly enhanced by SLO are not extracellularly localized. Cell lines that gave strong positive signals for B-mal and were blocked by treatment with MTSET were not subjected to permeabilization by SLO.
Not all extracellular or intracellular regions containing cysteines react with B-mal or B-mal with SLO equivalently. This is most likely because of conformational constraints that hinder access to some of these residues, thus generating variable immunoreactive HA antibody reactivity, as seen by band intensity on Western blots. Alternatively, the reduced band intensity may reflect reduced levels of mutant receptors on the cell. Cell lines that consistently produced weaker B-mal-reactive signals were analyzed by flow cytometry to enumerate the HA antibody-binding sites on the cell surface (Table 2) to determine the protein expression levels of the particular PiT1-13-HA cysteine mutant. If the expression level was comparable with that of PiT1-13-HA, the weaker B-mal signal was attributed to reduced accessibility of the residue because of protein folding or other forms of steric hindrance. If the number of HA antibody-binding sites on the cells was considerably less than that for control PiT1-HA cells, diminished signals were more likely to be a consequence of reduced protein expression.
TABLE 2.
Quantification of antibody binding sites on the cell surface of SLC20A1 (PiT1) using flow cytometry
| SLC20A1 receptor | Antibody-binding sites per cell (×1000) |
|---|---|
| PiT1-HA | 57.44 |
| PiT1-13-HA | 114.54 |
| V19C | 127.11 |
| N96C | 18.73 |
| S97C | 96.28 |
| L101C | 133.31 |
| Q147C | 96.24 |
| L155C | 42.72 |
| L545C | 58.31 |
| Y546C | 71.44 |
| V548C | 108.45 |
| L591C | 97.52 |
| N615C | 122.96 |
For cysteine mutants that contained a cysteine residue substituted for an intracellular residue of PiT1-13-HA, a light degree of B-mal permeabilization, as represented by faint bands, is obtained on Western blots, but this band is markedly enhanced in the presence of SLO (supplemental figure, B, compare lanes 1–4). B-mal reactivity with extracellular residues is blocked with MTSET (supplemental figure, B, lanes 5 and 6). The band intensity achieved with SLC20A1 (PiT1) cysteine residues reactive with B-mal are, as expected, diminished when cells are first incubated with the membrane-permeabilizing reagent SLO, as SLO permits B-mal to react with extraneous intracellular cysteines that would not be detected on a Western blot, thus ostensibly diluting the amount of B-mal available for reacting with SLC20A1 (PiT1) extracellular cysteines (supplemental figure, B, lanes 7 and 8).
Control MDTF cells and MDTF cells expressing HA epitope-tagged wild-type SLC20A1 (PiT1-HA) were exposed to B-mal, precipitated with avidin-coated beads, and analyzed by Western blotting. As expected, no proteins were detected using an HA antibody on control MDTF cells after exposure to B-mal (Fig. 2A, lane 1). Similarly, MDTF cells expressing PiT1-HA protein exposed to B-mal were also unreactive (Fig. 2B, lane 1), indicating that all 13 cysteine residues present in wild-type SLC20A1 (PiT1), at positions 63, 132, 204, 238, 242, 251, 264, 403, 426, 468, 524, 573, and 625, are inaccessible to B-mal and therefore not extracellularly localized. Exposing control MDTF cells or MDTF cells expressing PiT1-HA to B-mal after treatment with MTSET did not affect B-mal reactivity, as expected (Fig. 2, A and B, lanes 2). Permeabilization of cells with SLO rendered the internal cysteine residues present in PiT1-HA reactive (Fig. 2B, lane 3) but did not result in a signal on HA-negative MDTF control cells (Fig. 2A, lane 3). When MDTF or MDTFPiT1-HA cells were treated with MTSET before permeabilization with SLO and labeling with B-mal, there was no change, because MTSET is membrane-impermeant and therefore will not block residues that are not extracellular (Fig. 2, A and B, lanes 4).
FIGURE 2.

Western blot of cell lysates from MDTF cells (A), MDTFPiT1-HA cells (B), and MDTFPiT1-13S97C-HA cells (C) under different labeling conditions. Lane 1, cells were labeled with B-mal; lane 2, cells were pretreated with MTSET and then labeled with B-mal; lane 3, cells were permeabilized with SLO before B-mal labeling; lane 4, cells were first treated with MTSET, then permeabilized with SLO, and finally labeled with B-mal.
To determine which regions of SLC20A1 (PiT1) are accessible on the cell surface, we substituted selected residues with individual cysteines in PiT1-13-HA, which retains full GALV receptor function (Table 1). The first cysteine substitution in PiT1-13-HA was at position 97 (PiT1-13S97C-HA). Position 97 was chosen because it was previously demonstrated that SLC20A1 (PiT1) is an N-glycosylated protein and that glycosylation of SLC20A1 (PiT1) can be eliminated by either treating cells with the enzyme N-glycosidase-F or by substituting a cysteine for the asparagine at position 96 (7). Thus, we reasoned a residue adjacent to this asparagine residue is part of the glycosylation motif in ECR 2 and is predicted to be B-mal-reactive.
HA-immunoreactive bands were obtained in the Western blot for PiT1-13S97C-HA treated with B-mal, suggesting that this residue is indeed extracellular (Fig. 2C, lane 1). MTSET blocks the signal (Fig. 2C, lane 2), supporting its extracellular position. To systematically evaluate the membrane orientations of residues comprising the N-terminal region of SLC20A1 (PiT1), the following residue substitutions were made in PiT1-13-HA: V19C, A79C, G88C, L89C, V92C, N96C, S97C, L101C, A104C, A109C, and A114C; each mutant was evaluated for its ability to function as a GALV receptor. All mutants retained GALV receptor function similar to that of PiT1-HA with the exception of N96C (Table 1). MDTF cells expressing the PiT1-13N96C-HA were 2 orders of magnitude less susceptible to GALV vectors (data not shown), and there are a reduced number of HA antibody-binding sites on MDTF cells expressing PiT1-13N96C-HA compared with PiT1-HA and PiT1-13-HA (18,730 compared with 57,440 and 114,540, respectively; see Table 2). Cells expressing each of the above mutant receptors were exposed to B-mal alone or B-mal after exposure to MTSET or SLO, to determine the sidedness of the introduced cysteine residues. Western blots of lysates from B-mal-labeled cells expressing PiT1-13-HA mutants A79C, G88C, L89C, V92C, and S97C show strong signals (Fig. 3A, lanes 2–5 and 7); this reactivity was specifically blocked by pretreatment of the cells with MTSET (Fig. 3B, lanes 2–5 and 7), indicating that these cysteine residues are accessible on the outer surface of the plasma membrane. The intensities of the B-mal-reactive bands observed on cells expressing V19C, N96C, and L101C were reduced compared with the five other mutants shown to include the intracellular region 2 (Fig. 3A, lanes 1, 6, and 8), even though MTSET blocked signals obtained with B-mal (Fig. 3B, lanes 1, 6, and 8), and B-mal in the presence of SLO did not enhance reactivity (data not shown). The reduced signal intensity was reproducible from different lysate preparations and is therefore less likely to reflect the amount of protein loaded on the gel. The modest signal could result from reduced receptor expression or reduced accessibility to B-mal because of steric hindrance. To address these alternatives, protein expression levels on the cell surface were evaluated by measuring the number of HA antibody-binding sites per cell for each mutant cell line in question, as described under “Experimental Procedures.” As shown in Table 2, HA antibody-binding site numbers for MDTF cells expressing V19C and L101C (127,110 and 133,310, respectively) were greater than those obtained with receptors that reacted strongly with B-mal (e.g. S97C with 96,280 binding sites). However, N96C-HA showed only 18,730 binding sites compared with S97C (Table 2). By substituting a cysteine for the asparagine at position 96, glycosylation is blocked (7). Blocking glycosylation may result in a protein that is poorly processed, resulting in lower GALV titers (Table 1) and reduced B-mal reactivity (Fig. 3A). Thus, the reduced signals observed with V19C and L101C may be a consequence of steric hindrance, whereas the reduced B-mal reactivity observed with N96C most probably results from low protein expression levels (Table 2).
FIGURE 3.

A, representative Western blot of cell lysates from MDTF cells expressing PiT1-13-HA mutants containing individual cysteines V19C, A79C, G88C, L89C, V92C, N96C, S97C, and L101C reacted with B-mal, as described under “Experimental Procedures.” B, lysates of cells containing above mutant proteins blocked with 1 mm MTSET prior to B-mal labeling. C, representative Western blot of cell lysates from MDTF cells expressing PiT1-13-HA mutants containing individual cysteines A104C, A109C, and A114C reacted with B-mal. D, lysates of these cells permeabilized with SLO prior to B-mal labeling. PiT1-HA is the control for SLO.
The cysteine residues in PiT1-HA and the PiT1-13-HA mutants A104C, A109C, and A114C were not B-mal-reactive (Fig. 3C); however, after permeabilization of the membrane with SLO and subsequent exposure to B-mal, HA-tagged proteins from cells expressing these mutant receptors were detected on Western blots, suggesting that these residues lie within the membrane (Fig. 3D). Thus, ECR 2 of SLC20A1 (PiT1) minimally spans residues 79–101, and residues Ala-104, Ala-109, and Ala-114 probably make up part of TMH III.
Mutants containing individual cysteines Q147C or V150C were shown to react with B-mal, and the strength of their signals was reduced in the presence of MTSET, consistent with them being positioned extracellularly in ECR 3 (Fig. 4, A and B, lanes 1 and 2), whereas mutant L155C did not react strongly with B-mal in the absence of SLO (Fig. 4, A and B, lane 3). Residues G222C and L228C are in ECR 4, as predicted, and confirmed by B-mal reactivity (Fig. 4, A and B, lanes 4 and 5), and mutant G230C yields a faint signal with B-mal that is not blocked by MTSET (data not shown) but is enhanced in the presence of SLO (Fig, 4, lane 6) consistent with an intramembrane or cytoplasmic residue position. B-mal-reactive signals were blocked for Q147C, V150C, G222C, and L228C following pretreatment with MTSET, further validating their extracellular positions in ECR 3 and ECR 4 (Fig. 4, A and B, lanes 1 and 2 and 4 and 5). Quantification of protein levels by measuring HA antibody-binding sites showed L155C receptor levels only slightly lower than those obtained with Q147C (Table 2), suggesting that the reduced B-mal reactivity was most likely attributable to this residue being less accessible to B-mal than the flanking residues Val-150 and Gly-222. We constructed cysteine mutants to identify residues that comprise ECR 5. The cysteine residues at positions Leu-545, Tyr-546, Leu-547, Val-548, Tyr-549, Asp-550, Val-554, and Lys-557 react with B-mal and can be blocked by MTSET (Fig. 5, A and B), suggesting that these residues are part of ECR 5. Quantification of protein levels for Y546C (Fig. 5A, lane 2) was assessed, showing lower levels than those obtained with V548C (108,450) (Table 2) but higher than those obtained with L545C (58,310) (Table 2). Cells expressing SLC20A1 (PiT1) mutants R581C, L591C, and T592C do not react with B-mal (Fig. 5C); however, after these cells were permeabilized with SLO they showed positive signals for B-mal (Fig. 5D). These data indicate that these residues are not part of ECR 5 and not extracellularly localized. Finally, we have discovered a small, unanticipated sixth extracellular region (ECR 6) in SLC20A1 (PiT1), along with two additional TMHs (TMH X and XI). A Western blot of cell lysates from MDTF cells expressing PiT1-13A613C-HA exposed to B-mal showed that the residue at position 613 is B-mal-reactive (Fig. 6A, lane 1); reactivity is blocked in the presence of MTSET (Fig. 6B, lane 1). This finding indicates that residue 613 is accessible to B-mal and part of a new ECR 6, and not intracellular as predicted previously (7). Mutants N615C, I616C, G617C, L618C, and S621C also react with B-mal (Fig. 6A, lanes 2–6); reactivity is blocked in the presence of MTSET prior to B-mal labeling (Fig. 6B, lanes 2–6). Wild-type SLC20A1 (PiT1) contains a cysteine at position 625 that is unreactive with B-mal, suggesting that this residue is not extracellular. Cysteines positioned at residues 661, 668, and 670 are all nonreactive with B-mal, suggesting they are positioned inside the cell (Fig. 7, lanes 1–3). The last six residues of SLC20A1 (PiT1) minimally define the extracellular C terminus as demonstrated by the ability of B-mal to react with L677C, and this reactivity is blocked in the presence of MTSET (Fig. 7, A and B, lane 4), a finding consistent with this residue being extracellular.
FIGURE 4.

A, representative Western blot of MDTF cell lysates expressing PiT1-13-HA mutants containing individual cysteines Q147C, V150C, L155C, G222C, L228C, and G230C reacted with B-mal. B, lysates of cells expressing above mutant proteins blocked with MTSET or permeabilized with SLO prior to B-mal treatment.
FIGURE 5.

A, representative Western blot of lysates of MDTF cells expressing PiT1-13-HA mutants containing individual cysteines L545C, Y546C, L547C, V548C, Y549C, D550C, V554C, and K557C. B, lysates of cells expressing above mutant proteins treated with MTSET prior to B-mal labeling. C, lysates of MDTF cells expressing PiT1-13-HA mutants containing individual cysteines R581C, L591C, and T592C reacted with B-mal. D, lysates of cells expressing above mutants permeabilized with SLO before B-mal labeling. PiT1-HA is the positive control for SLO.
FIGURE 6.

Novel ECR 6 proximal to two incomplete TMHs is identified. A, representative Western blot of lysates of MDTF cells expressing PiT1-13-HA mutants containing individual cysteines A613C, N615C, I616C, G617C, L618C, and S621C reacted with B-mal. B, lysates of cells expressing above mutant proteins treated with MTSET prior to B-mal labeling.
FIGURE 7.
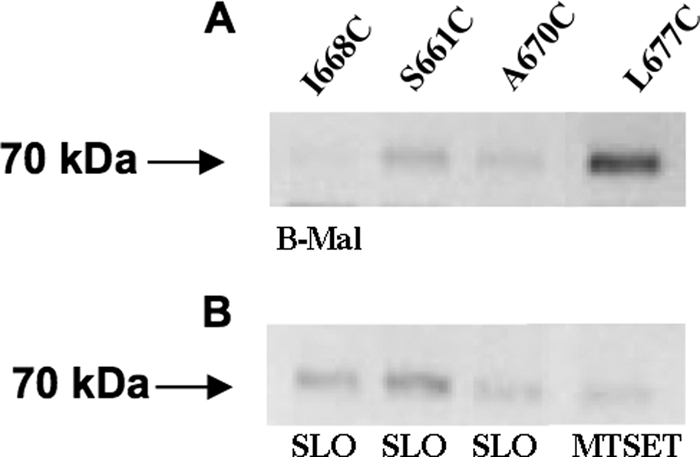
A, representative Western blot of lysates of MDTF cells expressing PiT1-13-HA mutants containing individual cysteine residues I668C, S661C, A670C, and L677C. B, lysates of cells expressing above mutant proteins treated with MTSET or permeabilized with SLO prior to B-mal labeling.
Modeling SLC20A1 (PiT1) Structure Using Experimental Data
A constrained prediction with the HMMTOP method (22, 23) has been made to create the final topology model of SLC20A1 (PiT1). We have applied only the positive results of our SCAM study, e.g. those positions where B-mal labeling was detected and could be blocked by MTSET pretreatment for the prediction, constraining these residues to be outside during the Baum-Welch optimization and the final topology prediction by the Viterbi algorithm (Fig. 8B). It should be noted that the predicted TMHs in our model may have some errors at their ends. This is a concession to predicting the structure of a multiple membrane-spanning protein that weighs the number of TMHs as more significant than the accuracy of TMH boundaries, as opposed to failing to predict the precise number of TMHs in the protein.
FIGURE 8.

A, previously proposed structure for SLC20A1 (PiT1). Residues are represented as circles. The lipid bilayer is represented as the region within the double line; residues above the line are extracellular, and residues below the line are intracellular. The extracellular regions are numbered 1–6. The first and last residues of each TMH are numbered; TMHs are numbered with roman numerals. B, comprehensive topological model for SLC20A1 (PiT1) using information experimentally derived by SCAM to set constraints for an HMMTOP-based prediction. Residues that are B-mal-accessible on the outside of the cell are shown as red circles. Green circles represent native cysteine residues inaccessible to B-mal. The extracellular regions are numbered 1–7 with the novel ECR 6 and TMHs X and XI shown in red.
DISCUSSION
For transporter proteins such as SLC20A1 (PiT1), the organization of the TMHs determines Pi transport function, while determining the positions of the ECRs with respect to the plane of the membrane bilayer is important for virus binding function. Definitive resolution of the ECRs is critical for resolving how these proteins regulate their very different functions as phosphate transporters or virus receptors. Topology models generated by the available prediction methods such as PredictProtein (24), TMHMM (25), and Phobius (26) do not address long range interactions between transmembrane helices, inter-protein and intra-protein interactions, or specific protein-lipid interactions (reviewed in Refs. 12, 21). It was shown earlier that incorporating prior knowledge about the topology of transmembrane proteins, such as domain locations or experimental results, into topology prediction methods highly increases their accuracies (19, 27). Submitting the sequence of SLC20A1 (PiT1) without applying experimental constraints to each of these three protein prediction websites results in a predicted transmembrane protein with 10 TMHs, not 12. After providing constraints based on our experimental cysteine analysis of the polytopic protein SLC20A1 (PiT1), the final topology model was produced, showing the newly identified ECR and two TMHs. This is the first evaluation of a retroviral receptor protein using an experimental method that assesses the topology of membrane proteins in the context of their normal lipid environment in concert with a topology prediction algorithm.
It is important to note that we did not authenticate the ability of the various cysteine mutants we constructed to function as phosphate transporters. The major thrust of this investigation was to use the topological information gleaned from these experiments to design experiments to resolve regions within SLC20A1 (PiT1) that are critical for GALV and FeLV-B entry.
We have previously determined that the first extracellular loop region of SLC20A2 (PiT2) is the A-MLV virus-binding site (13). The virus-binding site for SLC20A1 (PiT1) remains unresolved. Region A (residues 550–558) has been proposed as the SLC20A1 (PiT1) virus-binding site (14). However, we have more recently determined that region A alone is not sufficient to confer virus binding, because substitution of residues from SLC20A2 (PiT2) for the corresponding SLC20A1 (PiT1) region A residues blocks virus entry but does not prevent virus binding (7). Next, we have shown that a receptor lacking region A facilitates GALV and FeLV-B entry (28). Finally, Malhotra et al. (29) reported on a motif conserved among gammaretrovirus receptors and the receptor for avian leukosis virus (SLC20A1 (PiT1) Asp-550 and Val-554) (29). Here we show that when a cysteine residue is substituted for the asparagine residue at position 550 or the valine residue at position 554, GALV receptor function is unperturbed (Fig. 5 and Table 1). Thus this motif does not appear to be conserved in functional GALV receptors.
Prior topological annotation of human SLC20A1 (PiT1) provided by UniProtKB/TrEMBL (a computer-annotated protein sequence data base complementing the UniProtKB/Swiss-Prot Protein Knowledgebase) and the Kyte Doolittle-based structural proposal posited by O'Hara et al. (6) positioned the residues corresponding to the newly identified ECR 6 in the ninth transmembrane helix. The determination that the residues comprising ECR 6 are extracellular presents the possibility that this region may directly interact with virus. This possibility is particularly relevant in the context of our previous findings that mutations in region A (part of ECR 5) affect the topology of downstream SLC20A1 (PiT1) segments rendering the C terminus intracellular (7). Region A is a hypervariable segment of SLC20A1 (PiT1) (30), whereas ECR 6 is highly conserved among SLC20A1 (PiT1) orthologs that function as viral receptors. The conserved nature of ECR 6 among SLC20A1 (PiT1) viral receptors is more consistent with this region functioning as a virus-binding site. Resolution of the topology of SLC20A1 (PiT1) with a clear assignment of the transmembrane boundaries and the extracellular loops will accelerate the identification of the regions required for virus binding and entry.
Supplementary Material
Acknowledgments
We thank Claudia Han for expert technical assistance; Jim Nagle and Debbie Kauffmann at NINDS sequencing facility (National Institutes of Health), and Laura Li, Parisa Moghaddam-Taaheri, Kendra Leigh, Mandana Manzari, and Elizabeth Le for help constructing the cysteine mutants. We also thank Arlette Kouwenhoven, Wenqin Xu, Meihong Liu, and Dwayne Taliaferro for critical reading of the manuscript, and Jon Marsh and Nidia Oliveira for helpful discussions.
This work was authored, in whole or in part, by National Institutes of Health staff. This work was supported by a Bolyai János scholarship and Hungarian Research and Development Funds OTKA K75460 (to G. E. T.).

The on-line version of this article (available at http://www.jbc.org) contains a supplemental figure.
- GALV
- gibbon ape leukemia virus
- A-MLV
- amphotropic murine leukemia virus
- FeLV-B
- feline leukemia virus subgroup B
- TMH
- transmembrane helix
- ECR
- extracellular region
- SCAM
- substituted cysteine accessibility mutagenesis
- B-mal
- biotin maleimide
- SLO
- streptolysin O
- MTSET
- methanethiosulfonate
- PBS
- phosphate-buffered saline
- MDTF
- M. dunni tail fibroblast
- HA
- hemagglutinin.
REFERENCES
- 1.Hein S., Prassolov V., Zhang Y., Ivanov D., Löhler J., Ross S. R., Stocking C. (2003) J. Virol. 77, 5926–5932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kavanaugh M. P., Kabat D. (1996) Kidney Int. 49, 959–963 [DOI] [PubMed] [Google Scholar]
- 3.Kavanaugh M. P., Miller D. G., Zhang W., Law W., Kozak S. L., Kabat D., Miller A. D. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 7071–7075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olah Z., Lehel C., Anderson W. B., Eiden M. V., Wilson C. A. (1994) J. Biol. Chem. 269, 25426–25431 [PubMed] [Google Scholar]
- 5.Virkki L. V., Biber J., Murer H., Forster I. C. (2007) Am. J. Physiol. Renal Physiol. 293, F643–F654 [DOI] [PubMed] [Google Scholar]
- 6.O'Hara B., Johann S. V., Klinger H. P., Blair D. G., Rubinson H., Dunn K. J., Sass P., Vitek S. M., Robins T. (1990) Cell Growth Differ. 1, 119–127 [PubMed] [Google Scholar]
- 7.Farrell K. B., Russ J. L., Murthy R. K., Eiden M. V. (2002) J. Virol. 76, 7683–7693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Zeijl M., Johann S. V., Closs E., Cunningham J., Eddy R., Shows T. B., O'Hara B. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 1168–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller D. G., Miller A. D. (1994) J. Virol. 68, 8270–8276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson C. A., Eiden M. V., Anderson W. B., Lehel C., Olah Z. (1995) J. Virol. 69, 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salaün C., Rodrigues P., Heard J. M. (2001) J. Virol. 75, 5584–5592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Q., Casey J. R. (2007) Methods 41, 439–450 [DOI] [PubMed] [Google Scholar]
- 13.Feldman S. A., Farrell K. B., Murthy R. K., Russ J. L., Eiden M. V. (2004) J. Virol. 78, 595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johann S. V., van Zeijl M., Cekleniak J., O'Hara B. (1993) J. Virol. 67, 6733–6736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pedersen L., van Zeijl M., Johann S. V., O'Hara B. (1997) J. Virol. 71, 7619–7622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lander M. R., Chattopadhyay S. K. (1984) J. Virol. 52, 695–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmgren M., Liu Y., Xu Y., Yellen G. (1996) Neuropharmacology 35, 797–804 [DOI] [PubMed] [Google Scholar]
- 18.Grunewald M., Bendahan A., Kanner B. I. (1998) Neuron 21, 623–632 [DOI] [PubMed] [Google Scholar]
- 19.Tusnády G. E., Kalmár L., Simon I. (2008) Nucleic Acids Res. 36, D234–D239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loo T. W., Clarke D. M. (1995) J. Biol. Chem. 270, 843–848 [DOI] [PubMed] [Google Scholar]
- 21.Bogdanov M., Zhang W., Xie J., Dowhan W. (2005) Methods 36, 148–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tusnády G. E., Simon I. (1998) J. Mol. Biol. 283, 489–506 [DOI] [PubMed] [Google Scholar]
- 23.Tusnády G. E., Simon I. (2001) Bioinformatics 17, 849–850 [DOI] [PubMed] [Google Scholar]
- 24.Rost B. (2001) J. Struct. Biol. 134, 204–218 [DOI] [PubMed] [Google Scholar]
- 25.Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. (2001) J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 26.Käll L., Krogh A., Sonnhammer E. L. (2004) J. Mol. Biol. 338, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 27.Bernsel A., Von Heijne G. (2005) Protein Sci. 14, 1723–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eiden M. V., Farrell K. B., Wilson C. A. (1996) J. Virol. 70, 1080–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malhotra S., Scott A. G., Zavorotinskaya T., Albritton L. M. (1996) J. Virol. 70, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaudry G. J., Eiden M. V. (1997) J. Virol. 71, 8078–8081 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.