

Abstract
The Zap1 transcription factor is a central player in the response of yeast to changes in zinc status. Previous studies identified over 80 genes activated by Zap1 in zinc-limited cells. In this report, we identified 36 genes repressed in a zinc- and Zap1-responsive manner. As a result, we have identified a new mechanism of Zap1-mediated gene repression whereby transcription of the MET3, MET14, and MET16 genes is repressed in zinc-limited cells. These genes encode the first three enzymes of the sulfate assimilation pathway. We found that MET30, encoding a component of the SCFMet30 ubiquitin ligase, is a direct Zap1 target gene. MET30 expression is increased in zinc-limited cells, and this leads to degradation of Met4, a transcription factor responsible for MET3, MET14, and MET16 expression. Thus, Zap1 is responsible for a decrease in sulfate assimilation in zinc-limited cells. We further show that cells that are unable to down-regulate sulfate assimilation under zinc deficiency experience increased oxidative stress. This increased oxidative stress is associated with an increase in the NADP+/NADPH ratio and may result from a decrease in NADPH-dependent antioxidant activities. These studies have led to new insights into how cells adapt to nutrient-limiting growth conditions.
Zinc is an essential nutrient for all organisms because it is required as a structural or catalytic cofactor by many proteins. It was recently estimated that about 10% of the ∼30,000 proteins encoded by the human genome need zinc for their function (1). Zinc deficiency perturbs a wide variety of processes and is associated with many disease symptoms in mammals (2). Excess zinc can also be toxic to cells (3). Thus, organisms have evolved with mechanisms to tightly control intracellular zinc levels. We have examined cellular responses to zinc deficiency in the yeast Saccharomyces cerevisiae (4, 5). In this yeast, the Zap1 transcription factor is a central player in their response to zinc deficiency (6). For many of its target genes, Zap1 acts as an activator of transcription and increases gene expression when zinc levels are low. To perform this function, Zap1 binds to one or more zinc-responsive elements (ZREs)2 in the promoters of its target genes. The consensus sequence for a ZRE is ACCTTNAAGGT (4, 7). Previous studies have identified a large number of Zap1 target genes in the yeast genome (4, 5, 8, 9). Many activated Zap1 targets contribute to zinc homeostasis. For example, Zap1 induces its own expression by positive autoregulation (10). In addition, ZRT1, ZRT2, and FET4 encode transporter proteins responsible for zinc uptake and are targets of Zap1 activation (11–13). Zap1 also induces expression of ZRT3 and ZRC1, which encode vacuolar zinc transporters involved in controlling zinc storage in the vacuole (14). In addition to maintaining zinc homeostasis, Zap1 regulates the expression of genes that play a more adaptive role in zinc-limited cells. For example, Zap1 controls the level of several lipid biosynthetic enzymes (DPP1, PIS1, EKI1, and CKI1) to maintain the levels of some membrane phospholipids and to alter the levels of others (15). Thus, Zap1 mediates both homeostatic and adaptive responses to zinc limitation.
Zap1 also activates expression of an antioxidant gene, TSA1, which encodes the major cytosolic thioredoxin-dependent peroxidase, to combat the oxidative stress of zinc deficiency (16). Studies of mammalian cells have shown that zinc deficiency causes the increased accumulation of reactive oxygen species (17). We have observed that yeast also experience increased oxidative stress when grown under low zinc conditions (16). The source of this oxidative stress is unknown. Reactive oxygen species (ROS), including the superoxide anion, hydrogen peroxide (H2O2), and hydroxyl radical, can cause various types of biological damage. Zinc deficiency is associated with increased levels of lipid and protein oxidation (18, 19). In addition, the oxidative stress associated with zinc deficiency leads to increased levels of DNA damage (19–21). For these reasons, zinc deficiency has been proposed to be an important risk factor for cancer and other human diseases (22, 23).
Although the more recognized role of Zap1 is to activate gene expression, this protein can also repress transcription of some target genes. Previous studies have identified two different mechanisms of Zap1-mediated repression. The first example, ZRT2, encodes a low affinity zinc uptake transporter, and this gene is induced in moderate zinc limitation but repressed in severe zinc deficiency (24). This paradoxical pattern of regulation is because of the presence of three ZREs in the ZRT2 promoter. Two ZREs, ZRE1 and ZRE2, are located upstream of the TATA box, and these elements mediate Zap1-dependent activation of gene expression. The third ZRE, ZRE3, is located downstream of the TATA box and is essential for repression of ZRT2 expression. Under moderate conditions of zinc deficiency, Zap1 binds to ZRE1 and ZRE2 and activates gene expression. Under more severe zinc deficiency, Zap1 also binds to ZRE3 and interferes with ZRT2 expression perhaps by blocking transcription initiation.
The ADH1 and ADH3 genes provide examples of a second mechanism of Zap1-mediated repression. ADH1 and ADH3 encode two major isoforms of the zinc-dependent alcohol dehydrogenases. These genes are highly expressed in zinc-replete cells but are repressed in zinc-deficient cells (25). Zap1 mediates ADH1 and ADH3 repression in low zinc by inducing intergenic transcription through the ADH1 and ADH3 promoters. These intergenic transcripts do not encode protein products but rather their synthesis results in the transient displacement of the Rap1 and Gcr1 transcription factors normally required for ADH1 and ADH3 expression. This loss of Rap1 and Gcr1 binding results in the reduced expression of two highly abundant zinc-binding proteins in the cell. Thus, by decreasing expression of zinc-dependent ADH isozymes, the cell may conserve zinc for other uses.
Transcriptome analysis with DNA microarrays identified over 80 potential targets of Zap1 activation in the yeast genome (4, 5). In this study, we further analyzed these microarray data and identified genes repressed in a zinc- and Zap1-responsive manner. As a result, we have uncovered a new mechanism for how Zap1 can repress transcription of the MET3, MET14, and MET16 genes. These genes encode the first three enzymes on the sulfate assimilation pathway (26). This pathway mediates the conversion of SO42− to methionine, S-adenosylmethionine, cysteine, and glutathione. Expression of MET3, MET14, and MET16 is normally activated by the Met4 transcription factor when the levels of these sulfur-containing compounds are low (26, 27). Met4 is inactivated by the SCFMet30 ubiquitin ligase via the ubiquitin-proteasome system when these compounds accumulate to high levels. Met30, an F-box protein of the SCFMet30 complex, is required for down-regulation of Met4 activity. Here we find that Zap1 promotes degradation of the Met4 protein in zinc-limited cells by activating expression of the MET30 gene. In this way, Met4 protein accumulation, expression of MET3, MET14, and MET16, and sulfate assimilation all decrease in zinc-limited cells. We further address the physiological significance of this repression. Our results suggest that down-regulated sulfate assimilation conserves NADPH for use in combating the oxidative stress of zinc deficiency.
EXPERIMENTAL PROCEDURES
Growth Conditions and Strains
Yeast cells were grown in YPD (YP medium + 2% glucose) and in synthetic defined SD medium with 2% glucose or 2% galactose and any necessary auxotrophic requirements. YPD and SD are zinc-replete media because they contain micromolar levels of zinc and lack strong zinc chelators. Yeast were made zinc-limited by culturing in low zinc medium (LZM) prepared as described previously (28). LZM is zinc-limiting because it contains 1 mm EDTA and 20 mm citrate to buffer metal availability. Zinc was added to LZM as ZnCl2. Strains used in this study were DY1457 (MATα ade6 can1 his3 leu2 trp1 ura3), ZHY6 (DY1457 zap1Δ::TRP1), CWY21 (DY1457 pdr5Δ::KanMX), WCG4a (MATa his3 leu2 ura3), WCG4-11/21a (WCG4a pre1-1pre2-1), RHY2862 (MATα ade2 his3 leu2 lys2 met2 trp1 ura3), RHY2938 (RHY2862 hrd2-1), W303 (MATα ade2 can1 his3 leu2 trp1 ura3), and TAL31 (W303 met4Δ::TRP1).
Microarray Data
The microarray data used are from Wu et al. (5) (Gene Expression Omnibus series GSE11983). In the first set of experiments (experiment 3, E3) (5), wild type (DY1457) cells were transformed with the vector (pYef2) or a plasmid (pYef2-Zap1TC) encoding a constitutive allele of Zap1 under the regulation of the galactose-inducible GAL1 promoter. The plasmid pYef2 and pYef2-Zap1TC constructs were described previously (29). These transformants were inoculated into zinc-replete SD medium + 2% galactose + 1 μm ZnCl2 and grown for 20–24 h before harvesting at an absorbance measured at 600 nm (A600) of ∼0.8. In the second set of experiments (experiment 4, E4) (5), wild type (DY1457) cells were grown in a zinc-limiting medium (LZM + 3 μm ZnCl2) and in a zinc-replete medium (LZM + 3000 μm ZnCl2) for 14–16 h and harvested at an A600 of ∼0.7. Genes exhibiting fold changes above the cutoff value were selected for subsequent analysis. We chose an arbitrary cutoff value with a fold change ≥1.5 based on the average of two independent microarrays with the provision that both arrays showed a fold change of at least 1.4.
Promoter Motif Analysis
ZREs were identified using a position-specific probability matrix generated from the ZREs of 46 potential Zap1 targets identified previously (4) and regulatory sequence analysis tools (30).
RNA and Protein Analysis
S1 nuclease protection assays were performed with total RNA as described (31). The oligonucleotide probes used for these experiments are described in supplemental Table 1. For each reaction, 15 μg of total RNA was hybridized to 32P-end-labeled DNA oligonucleotide probes before digestion with S1 nuclease and separation on a 10% polyacrylamide gel containing 5 m urea. Band intensities were quantified by PhosphorImager analysis (PerkinElmer Life Sciences). Crude protein extracts were generated by lysis in trichloroacetic acid, and immunoblot analysis was performed as described previously (29). The primary antibodies used were anti-Met4 polyclonal antibody (1:5000; a gift from Dr. Traci Lee, University of Wisconsin, Park side) and anti-Pgk1 monoclonal antibody (1:5000; Invitrogen).
Electrophoretic Mobility Shift Assays
The Zap1 DNA binding domain (Zap1DBD, residues 687–880) was expressed in Escherichia coli as a fusion to glutathione S-transferase and purified, and then the glutathione S-transferase tag was removed as described previously (7). Electrophoretic mobility shift assays were performed using purified Zap1DBD protein and radiolabeled ZRE oligonucleotides (supplemental Table 2). Binding reactions were prepared containing 0.5 pmol of radiolabeled DNA oligonucleotide (20,000 cpm/pmol), 10 mm Tris-HCl (pH 8.0), 10 mm MgCl2, 50 mm KCl, 1 mm dithiothreitol, 0.02 mg/ml poly(dI-dC), 0.2 mg/ml bovine serum albumin, 0.04% Nonidet P-40, 10% glycerol, and the indicated concentrations of purified Zap1DBD. After incubation for 1 h at room temperature, the samples were resolved on 6% polyacrylamide gels. Gels were dried onto blotting paper, and the signals were visualized by autoradiography.
Plasmid Constructs
The reporter plasmid pMET30-lacZ was constructed in YEp353 (32) by homologous recombination (33). PCR products were generated from genomic DNA that contained 1000 bp of MET30 promoter sequence (bases −1000 to +1) flanked by sequence with homology to the vector. This fragment was gel-purified and transformed with EcoRI- and BamHI-digested YEp353; transformants were selected for URA3 prototrophy. The mutant allele of MET30 ZRE (pMET30mZRE-lacZ) was constructed in a similar fashion after generation of the mutant promoter fragment by overlap PCR (34). All plasmid constructs were confirmed by DNA sequencing. Plasmids RGS-His-tagged wild type Met4 (Pp323), mutant Met4K163R (Pp326), and vector control (Pp264) driven by the inducible GAL1 promoter are gifts from Dr. Peter Kaiser, University of California, Irvine. Met4K163R is a mutant allele in which lysine 163 of Met4 is replaced with arginine (35). We used GEV system to drive expression of Met4 and Met4K163R. The GEV system uses a hybrid transcription factor that contains the Gal4 DNA binding domain, a β-estradiol-responsive domain, and the VP16 activation domain (36). GEV allows dose-dependent activation of the expression of Gal4 target genes in glucose-grown cells in the presence of β-estradiol. In this study, we used 10−6 m β-estradiol to express moderate levels of Met4 and Met4K163R in a met4Δ strain. Higher expression levels of Met4K163R, e.g. in 2% galactose mediated by endogenous Gal4, is toxic to cells.
β-Galactosidase Assays
Cells were grown for 15–20 h to exponential phase in the indicated media. β-Galactosidase activity was measured in permeabilized cells as described previously (37).
Assay of Free Amino Acid Pools
Sixty-ml cultures of yeast cells in zinc-replete (LZM + 1000 μm zinc) or zinc-limited media (LZM + 0.3 μm zinc) were harvested at an absorbance of 0.6–1 and washed five times with chilled medium without zinc and auxotrophic supplements. After removing all standing liquid, samples were weighed to determine their fresh weights. The cell pellets were then resuspended in 600 μl of the extraction solvent (methanol/chloroform/water (3:5:12, v/v)) to lyse the cells and extract the amino acids. After pelleting cellular debris by centrifuging for 20 min at 18,000 × g, 150 μl of chloroform and 225 μl of water were added to the supernatant to split the phases. The upper methanol/water phase was dried down in a vacuum concentrator, resuspended in 200 μl of water, and used to quantify the soluble amino acids. The amino acids were derivatized into fluorescent adducts with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AccQ·FluorTM reagent kit, Waters) following the manufacturer's protocol with minor modifications as indicated below. To the deproteinated assay mixture (2.5 μl) was added 17.5 μl of AccQ·Fluor borate buffer and 5 μl of AccQ·Fluor reagent. The resulting mixture was incubated at 55 °C for 10 min in a heating block. Prior to high pressure liquid chromatography separation, the derivatized samples were diluted to 50 μl with water to increase the volume and were filtered through a 0.22-μm polyvinylidene difluoride membrane. The resulting fluorescent amino acid derivatives were separated on a SunFire C18 column (4.6 × 150 mm, 3.5 μm) using a Waters Alliance 2695 separations module and were measured by a Waters 2475 fluorescence detector using an excitation wavelength of 250 nm and an emission wavelength of 395 nm. Solvent composition consisted of the following: A, water; B, acetonitrile; C1, 140 mm sodium acetate and 17 mm triethylamine (pH 5.05); C2, 100 mm sodium acetate and 5.6 mm triethylamine (pH 5.7); D, 100 mm sodium acetate and 5.6 mm triethylamine (pH 6.8). Chromatographic conditions are described in supplemental Table 3. The volume of sample injected was 10 μl. The 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate-amino acid adducts were identified and quantified by comparison with standards. To determine free cysteine levels, cells were extracted in similar manner except that samples were deproteinated with 2.5% 5-sulfosalicylic acid and analyzed by using L-8800 amino acid analyzer.
DCF Assays
Measurement of the accumulation of ROS used 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) (Calbiochem) (38). DCFH-DA is membrane-permeable and is trapped intracellularly following deacetylation. The resulting compound, DCFH, reacts with ROS (primarily H2O2 and hydroxyl radicals) to produce the oxidized fluorescent form 2,7-dichlorofluorescein (DCF). ROS analysis using DCFH-DA was performed as follows. Yeast cells were treated with 10 μm DCFH-DA in culture media for 1 h prior to harvesting. Cells were then washed twice in ice-cold phosphate-buffered saline, resuspended in phosphate-buffered saline, and disrupted by vortexing with glass beads. Following centrifugation at 14,000 × g for 10 min at 4 °C, the supernatant was collected, and protein concentration was measured by the Bradford method. DCF fluorescence intensity was measured with a fluorescence spectrophotometer at an excitation wavelength of 504 nm and an emission wavelength of 524 nm and then normalized to protein level.
Glutathione Assay
Glutathione levels were determined using the method described by Vandeputte et al. (39). Cells were grown to exponential phase (∼1 × 107 cells/ml), washed twice with distilled deionized H2O, and resuspended in 250 μl of cold 1% 5-sulfosalicylic acid. Cells were disrupted by vortexing with glass beads and incubated at 4 °C for 15 min to precipitate protein prior to centrifugation for 10 min at 14,000 × g. The supernatants were used to determine glutathione levels. Total glutathione was determined by adding 10 μl of lysate to 150 μl of assay mixture (0.1 m potassium phosphate (pH 7.0), 1 mm EDTA, 0.03 mg/ml 5,5′-dithiobis(2-nitrobenzoic acid), 0.12 unit of glutathione reductase). The samples were mixed and incubated for 5 min at room temperature, and 50 μl of NADPH (0.16 mg/ml) was then added. The formation of thiobis(2-nitrobenzoic acid) was monitored spectrophotometrically at 420 nm over a 5-min period. The reaction rate was proportional to the concentration of glutathione. Standard curves were generated for each experiment using 0–0.5 nmol of glutathione in 1% 5-sulfosalicylic acid. To measure GSSG alone, 100-μl lysate samples were derivatized by adding 2 μl of 97% 2-vinylpyridine, and the pH was adjusted by adding 2 μl of 25% triethanolamine. These samples were then incubated at room temperature for 60 min. The samples were then assayed as described above for total glutathione. GSSG standards (0–0.1 nmol) were also treated with 2-vinylpyridine in an identical manner to the samples. Subtraction of the amount of GSSG in the lysate from the total glutathione allowed a determination of GSH levels present in each sample.
NADPH and NADP+ Measurements
Intracellular NADPH and NADP+ levels were measured using a 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (Sigma) and phenazine ethosulfate (Sigma) cycling assay described by Gibon and Larher (40). Fifteen-ml cultures of yeast cells were harvested at A600 of ∼0.8, washed twice with distilled deionized H2O, and resuspended in 250 μl of either alkaline (0.1 m KOH) or acidic solutions (0.1 m HCl) to measure NADPH and NADP+ levels, respectively. Cells were then broken by vortexing with glass beads. Extracts were heated at 90 °C for 3 min, chilled on ice for 5 min, and centrifuged at 14,000 × g for 10 min at 4 °C. Ten μl of the supernatant added to 150 μl of assay mixture contained 260 mm triethanolamine-HCl (pH 7.4), 4 mm EDTA, 0.1 mm phenazine ethosulfate, 0.5 mm 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide, 2 mm glucose 6-phosphate, and 1 unit of yeast glucose-6-phosphate dehydrogenase (Sigma). The color development was monitored spectrophotometrically at 595 nm. The concentrations of cellular NADPH and NADP+ were determined using the linear range of the standard curve of NADPH (0–6 μm) and normalized to the cell density of the harvested culture.
Statistical Analysis
Statistical analyses were performed using the paired, two-tailed Student's t test.
RESULTS
Identifying Genes Repressed by Zap1 Activity
Our studies have shown that Zap1 can act as a repressor as well as an activator of gene expression (24, 25). Therefore, we examined our previously obtained microarray data (5) for genes that are repressed in a zinc- and Zap1-responsive manner. In the first set of these microarray experiments, we had examined expression of genes in cells expressing the Zap1TC allele versus wild type cells grown under zinc-replete conditions. The Zap1TC allele contains mutations that render Zap1 constitutively active (29). In the second set of experiments, we had compared gene expression in wild type cells grown in low and high zinc. We have retained our previous designations for these experimental conditions, i.e.“experiment 3” (E3) and “experiment 4” (E4), respectively, to be consistent with our previous report (5). We predicted that genes repressed by Zap1 would be expressed at lower levels in ZapTC-expressing cells as well as in zinc-limited wild type cells. From our results, we noted that several genes met these criteria. Specifically, 413 genes were repressed an average of ≥1.5-fold in Zap1TC-expressing cells, and 123 genes were repressed an average of ≥1.5-fold in wild type cells grown in low zinc; the overlap between these two sets was 36 genes (Fig. 1). These genes were potentially repressed by Zap1 activity either directly or indirectly, and Table 1 summarizes the microarray results obtained. Included among these genes are ADH1 and ADH3, which were previously shown to be negatively regulated by Zap1 (25). In addition, we observed that ADH2 was similarly affected by these perturbations. Other genes showing potential repression by Zap1 include those involved in sulfur metabolism, mitochondrial function, nucleotide and amino acid metabolism, and protein synthesis.
FIGURE 1.

Complementary microarray experiments to identify genes repressed by Zap1. A Venn diagram is shown describing our methods of identifying genes repressed by Zap1. Genes showing decreased expression in zinc-replete cells expressing a constitutive Zap1 allele (Zap1TC) and in zinc-limited wild type (WT) cells were identified. E3, experiment 3; E4, experiment 4.
TABLE 1.
Genes potentially repressed by Zap1
| ORF | Genea | Function | Fold repressionb |
|||
|---|---|---|---|---|---|---|
| E3+ Zn; WT/Zap1TC |
E4 WT; +Zn /−Zn |
|||||
| Exp. A | Exp. B | Exp. A | Exp. B | |||
| Zinc homeostasis | ||||||
| YOL086C | ADH1 | Alcohol dehydrogenase I | 2.5 | 2.1 | 4.1 | 3.3 |
| YMR083W | ADH3 | Alcohol dehydrogenase III | 2.9 | 2.5 | 1.9 | 1.9 |
| YMR303C | ADH2 | Alcohol dehydrogenase II | 2.9 | 2.0 | 4.3 | 3.4 |
| Sulfur metabolism | ||||||
| YGL254W | FZF1 | Transcription factor involved in sulfite metabolism | 1.6 | 1.6 | 1.6 | 1.6 |
| YJR010W | MET3 | Sulfate adenylyltransferase, sulfate assimilation | 2.3 | 2.1 | 1.6 | 1.5 |
| YKL001C | MET14 | Adenylylsulfate kinase, sulfate assimilation | 2.3 | 2.3 | 1.9 | 2.1 |
| YPR167C | MET16 | Phosphoadenylyl-sulfate reductase, sulfate assimilation | 2.1 | 2.0 | 1.6 | 2.0 |
| Mitochondrial function | ||||||
| YDR194C | MSS116 | RNA helicase, mitochondrial RNA splicing | 1.8 | 1.7 | 1.5 | 1.6 |
| YJL166W | QCR8 | Ubiquinol-cytochrome c reductase | 1.7 | 1.6 | 1.5 | 1.6 |
| YJR122W | CAF17 | Mitochondrial protein, function unknown | 1.6 | 1.7 | 1.5 | 1.5 |
| Nucleotide metabolism | ||||||
| YJR025C | BNA1 | 3-Hydroxyanthranilic acid dioxygenase, nicotinic acid synthesis | 2.2 | 1.9 | 2.6 | 2.4 |
| YPR062W | FCY1 | Cytosine deaminase, uracil synthesis | 1.8 | 1.7 | 1.6 | 1.6 |
| Amino acid metabolism | ||||||
| YDR380W | ARO10 | Phenylpyruvate decarboxylase | 2.7 | 2.7 | 1.6 | 1.5 |
| YGL009C | LEU1 | 3-Isopropylmalate dehydratase, leucine synthesis | 1.8 | 1.7 | 3.2 | 3.1 |
| YHR208W | BAT1 | Transaminase, branched-chain amino acid synthesis and degradation | 2.3 | 2.2 | 2.2 | 2.5 |
| YPR060C | ARO7 | Chorismate mutase, aromatic amino acid synthesis | 1.7 | 1.5 | 1.5 | 1.6 |
| Protein synthesis | ||||||
| YDR471W | RPL27B | Large (60 S) ribosomal subunit protein | 2.2 | 2.6 | 1.4 | 1.6 |
| YER056C-A | RPL34A | Large (60 S) ribosomal subunit protein | 1.8 | 2.0 | 1.4 | 1.7 |
| YGR084C | MRP13 | Mitochondrial small ribosomal subunit protein | 1.7 | 1.9 | 1.5 | 1.5 |
| YLR448W | RPL6B | Large (60 S) ribosomal subunit protein | 2.0 | 1.9 | 1.4 | 1.6 |
| YML073C | RPL6A | Large (60 S) ribosomal subunit protein | 1.9 | 1.9 | 1.4 | 1.6 |
| YOL040C | RPS15 | Small (40 S) ribosomal subunit protein | 2.3 | 1.9 | 1.7 | 2.1 |
| YOR091W | TMA46 | Ribosome-associated protein | 1.7 | 1.6 | 1.5 | 1.6 |
| Others | ||||||
| YBR158W | CST13 | Protein required for daughter cell separation | 1.6 | 1.5 | 1.5 | 1.8 |
| YDR177W | UBC1 | E2 ubiquitin-conjugating enzyme | 1.6 | 1.6 | 1.5 | 1.5 |
| YEL040W | UTR2 | Cell wall protein, putative chitin transglycosidase | 2.5 | 2.1 | 2.3 | 2.5 |
| YGL089C | MFα2 | α-Factor mating pheromone | 3.1 | 2.8 | 9.3 | 8.7 |
| YIL118W | RHO3 | Small GTPase protein, control of cell polarity | 1.8 | 1.7 | 1.6 | 1.5 |
| YIL158W | Function unknown | 1.6 | 1.6 | 1.4 | 1.6 | |
| YKL029C | MAE1 | Malate dehydrogenase | 2.0 | 1.6 | 1.6 | 1.7 |
| YKL178C | STE3 | α-Factor mating pheromone receptor | 2.1 | 2.2 | 2.0 | 1.8 |
| YML040W | Ty1 Gag nucleocapsid protein | 1.9 | 2.0 | 1.4 | 1.8 | |
| YMR051C | Ty1 Gag nucleocapsid protein | 2.3 | 2.3 | 1.4 | 1.6 | |
| YNL051W | COG5 | Involved in intra-Golgi vesicle trafficking | 1.9 | 1.6 | 2.4 | 2.1 |
| YNL255C | GIS2 | Involved in the RAS/cAMP signaling pathway | 2.6 | 2.3 | 2.7 | 2.5 |
| YOR315W | SFG1 | Putative transcription factor for pseudohyphal growth | 1.7 | 1.8 | 2.4 | 2.4 |
a Results obtained for the genes in boldface type were confirmed independently by S1 nuclease protection assay in Fig. 2.
b Fold repression is reported for each gene in duplicate experiments A and B.
To confirm the microarray results, we selected 11 genes from the 36 total for analysis by S1 nuclease protection assay of additional RNA samples not used for the microarray analyses (Fig. 2A). CMD1, encoding calmodulin, is not zinc-regulated and was included as a loading control. In each case, these assays confirmed the microarray results. This included ADH1 and ADH3, which were used as positive controls for Zap1-dependent transcriptional repression. Intriguingly, we noted that the genes encoding the first three enzymes in the sulfate assimilation pathway, MET3, MET14, and MET16 (26), were down-regulated in zinc-deficient cells and in zinc-replete cells expressing the Zap1TC allele. We further characterized the zinc dose dependence of this regulation (Fig. 2B, lanes 1–4) and showed its Zap1 dependence; zap1Δ cells did not show repression under zinc-limiting conditions (Fig. 2B, lanes 5 and 6).
FIGURE 2.
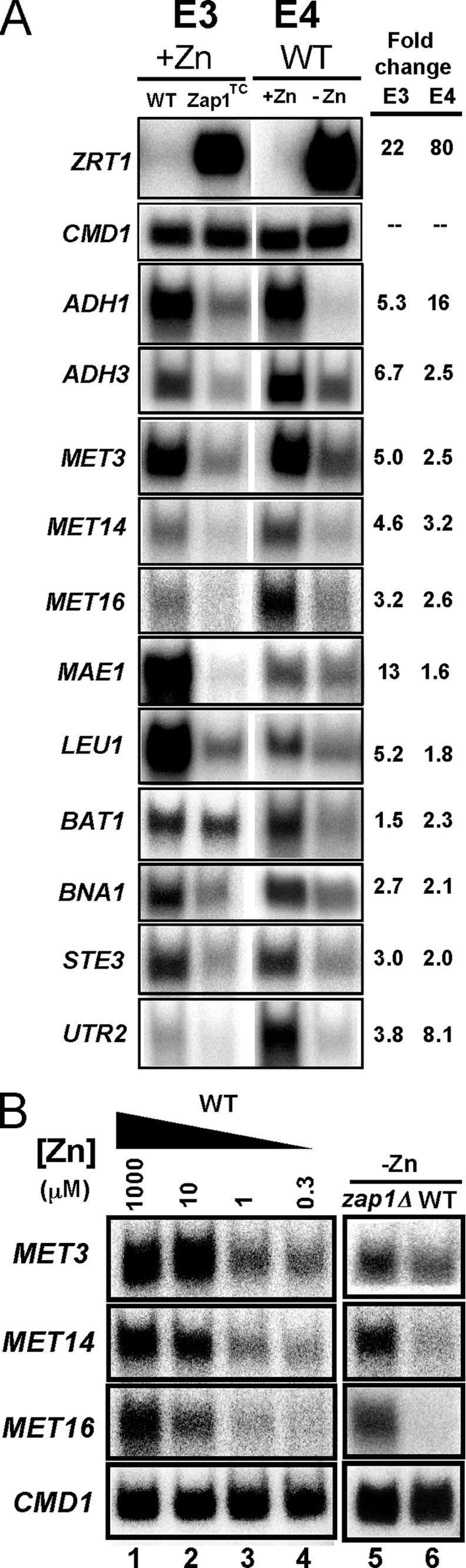
Confirmation of the microarray results for potential Zap1-repressed genes. A, S1 nuclease protection assays were performed using RNA isolated from cells grown under the same conditions as the two sets of microarray experiments. Controls for induced genes (ZRT1), repressed genes (ADH1 and ADH3), and for equal loading (calmodulin, CMD1) were included. In addition, candidate genes selected from Table 1 were also tested. The band intensities were quantified, and the fold changes are reported. These data confirmed the microarray results for these genes. E3, experiment 3; E4, experiment 4. B, zinc dose-dependent and Zap1-dependent repression of MET3, MET14 and MET16. Wild type (DY1457) cells were grown in LZM supplemented with a range of added zinc, and mRNA levels of MET3, MET14, MET16, and CMD1 were analyzed by S1 nuclease protection assay (lanes 1–4). In addition, wild type (WT) and zap1Δ mutant cells (ZHY6) were grown in low zinc (−Zn, LZM + 1 μm ZnCl2), and RNA was isolated and analyzed by S1 nuclease protection assay (lanes 5 and 6).
These observations suggested that Zap1 mediates the repression of sulfate assimilation. Consistent with this hypothesis, the free pool of methionine measured in zinc-limited cells was below the detection limit of the assay (i.e. <10% of the levels in zinc-replete cells) (Table 2). Free cysteine levels also showed a significant decrease in zinc-limited cells. Although the free pools of some amino acids (e.g. tyrosine and lysine) increased and other amino acids (e.g. leucine and valine) decreased, the most dramatic effect was that observed for methionine. These data are consistent with repression of sulfate assimilation in zinc-limited cells.
TABLE 2.
Effects of zinc status on the free amino acid pools in wild type cells
| +Zna | − Zna | p valueb | % of +Zn levels | |
|---|---|---|---|---|
| nmol/g fresh wt | nmol/g fresh wt | |||
| Methionine | 0.05 ± 0.01 | NDc | 4.5 ×10−4 | <10 |
| Cysteined | 0.13 ± 0.01 | 0.05 ± 0.01 | 3.1 ×10−5 | 38 |
| Leucine | 2.39 ± 0.24 | 0.45 ± 0.04 | 5.2 ×10−5 | 19 |
| Valine | 0.08 ± 0.01 | 0.05 ± 0.01 | 5.0 ×10−5 | 62 |
| Arginine | 4.23 ± 0.14 | 2.87 ± 0.15 | 1.6 ×10−4 | 68 |
| Proline | 0.38 ± 0.05 | 0.31 ± 0.01 | 0.20 | 82 |
| Phenylalanine | 0.16 ± 0.01 | 0.14 ± 0.01 | 0.08 | 88 |
| Threonine | 0.73 ± 0.08 | 0.71 ± 0.03 | 0.87 | 97 |
| Tyrosine | 0.44 ± 0.04 | 1.43 ± 0.10 | 1.7 ×10−5 | 325 |
| Lysine | 0.89 ± 0.07 | 2.93 ± 0.04 | 6.6 ×10−9 | 329 |
| Alanine | 1.42 ± 0.04 | 2.89 ± 0.06 | 3.6 ×10−8 | 204 |
| Isoleucine | 0.09 ± 0.01 | 0.14 ± 0.01 | 1.0 ×10−2 | 156 |
a Cells were grown in high zinc (+Zn, LZM + 1000 μm ZnCl2) or low zinc (− Zn, LZM + 0.3 μm ZnCl2) for 10 generations prior to analysis. Values are means ± S.E., n = 4 or 5.
b p value results were determined using Student’s t test.
c ND means not detectable.
d Cysteine levels were measured in separate assays; see “Experimental Procedures.”
Role for Zap1 in Regulating Sulfur Metabolism in Yeast
The sulfate assimilation pathway is regulated at the transcriptional level by the Met4 transcription factor (26). When levels of intracellular organic sulfur compounds (e.g. cysteine) are low, Met4 is active in promoting transcription of sulfur assimilation genes, including MET3, MET14, and MET16 (Fig. 3A). When the levels of sulfur-containing compounds are high, Met4 is ubiquitinated by the multisubunit SCFMet30 ubiquitin ligase (41–43). Ubiquitination inactivates Met4 and, under some growth conditions, leads to Met4 degradation by the cytosolic proteasome (43–45). We reasoned that the zinc- and Zap1-responsive effects on MET3, MET14, and MET16 expression could result from Zap1 increasing expression of MET30 in zinc-limited cells thereby increasing the abundance of the SCFMet30 complex (Fig. 3A). It has been shown previously that Met30 is the rate-limiting component of SCFMet30 (46), so its regulation by Zap1 could greatly alter the activity of the complex.
FIGURE 3.

MET30 has zinc- and Zap1-responsive gene expression. A, proposed regulatory circuitry controlling MET3, MET14, and MET16 expression in response to zinc. B, transcriptional regulation of MET30 in response to zinc and Zap1 activity. Wild type (DY1457) cells were grown in LZM supplemented with a range of added zinc, and mRNA levels of MET30 and CMD1 were analyzed by S1 nuclease protection assay (lanes 1–7). In addition, wild type and zap1Δ mutant cells (ZHY6) were grown in low zinc (−Zn, LZM + 1 μm ZnCl2) (lanes 8 and 9), and wild type cells with and without expressing the constitutive Zap1TC allele were grown in zinc-replete SD medium (+Zn, lanes 10 and 11). RNA was isolated and analyzed by S1 nuclease protection assay. C, met4Δ mutant strain (TAL31) was grown in LZM supplemented with methionine and with a range of added zinc. RNA was isolated and analyzed by S1 nuclease protection assay.
Initial support for this hypothesis came from the previous observation that MET30 transcription is up-regulated in zinc-limited cells, although the role of Zap1 was not determined (47). To confirm this result and specifically test the role of Zap1 in MET30 regulation, we first examined the effects of zinc and Zap1 alleles on MET30 expression. By S1 nuclease protection assay, we confirmed that MET30 mRNA levels are elevated ∼3-fold in zinc-limited cells (Fig. 3B, lanes 1–7). Moreover, we found that the increase in MET30 expression in zinc-limited cells was at least partially Zap1-dependent; MET30 mRNA levels were higher (1.5 ± 0.1-fold, n = 3) in zinc-limited wild type cells than in zap1Δ mutant cells (Fig. 3B, lanes 8 and 9). MET30 expression was also elevated (2.2 ± 0.4-fold, n = 3) in zinc-replete cells expressing the constitutive Zap1TC allele (Fig. 3B, lanes 10 and 11). Thus, MET30 expression responds to these conditions as expected for a direct target of Zap1 activation.
MET30 expression is regulated by Met4 via a negative feedback loop (42) (Fig. 3A). To assess whether the changes in MET30 expression in response to zinc were due to changes in Met4 activity, we assessed MET30 zinc responsiveness in a met4Δ mutant strain. Increased MET30 expression in zinc-limited cells was observed in the met4Δ mutant indicating that the effects of zinc on MET30 expression did not require transcriptional activation by Met4 (Fig. 3C).
MET30 Is a Zap1 Direct Target
These results suggested that MET30 is a direct target of Zap1 regulation. To identify potential Zap1-binding sites (ZREs) in the MET30 promoter, a motif identification algorithm (regulatory sequence analysis tools) was used to scan the sequence of the intergenic region upstream of the MET30 open reading frame. This analysis identified a potential ZRE (AACTGCAGGGT) ∼400 bp upstream of the MET30 ORF (Fig. 4A). This site is also upstream of the Cbf1 and Met31/Met32-binding sites to which Met4 is recruited (48). Electrophoretic mobility shift assays indicated that the purified Zap1 DNA binding domain (Zap1DBD) binds specifically to this MET30 candidate ZRE in vitro. Oligonucleotides containing the wild type TSA1 ZRE or a nonfunctional mutant TSA1 ZRE (TSA1m) (16) were used as positive and negative controls in this experiment (Fig. 4B, lanes 2 and 3). Zap1DBD-DNA complexes were detected when the MET30 ZRE fragment was used as probe (Fig. 4B, lanes 4 and 5). In addition, a binding competition experiment also indicated that Zap1DBD binds specifically to the MET30 ZRE; a 200-fold excess of unlabeled TSA1 ZRE or of unlabeled MET30 ZRE oligonucleotides (Fig. 4C, lanes 2 and 4, respectively) effectively competed for Zap1DBD binding to the labeled TSA1 ZRE probe, whereas the mutant TSA1 ZRE did not (Fig. 4C, lane 3).
FIGURE 4.

MET30 is a Zap1 direct target. A, diagram of the MET30 promoter. Met4 is recruited to this promoter by binding to the Cbf1, Met31, and Met32 proteins bound at the indicated sites. A potential ZRE is located upstream of these sites of Met4 recruitment. B, Zap1 binds specifically to the MET30 ZRE in vitro as assessed by electrophoretic mobility shift assay. Radiolabeled double-stranded oligonucleotides (0.5 pmol, 10,000 cpm) containing ZRE-like sequences from the indicated promoters were used as probes. The probes were mixed with 0 (−), 0.2 (±) or 0.4 μg (+) per reaction of purified Zap1 DNA binding domain (Zap1DBD). FP denotes the free probe, and the arrow indicates the Zap1DBD-DNA complex. The wild type TSA1 ZRE and mutant TSA1 ZRE (TSA1m) were included as positive and negative controls, respectively. The MET30 ZRE was used as probe in lanes 4 and 5. C, electrophoretic mobility shift assays were performed using the wild type TSA1 ZRE as the probe. Nonradiolabeled oligonucleotides were used as competitors at a 200-fold excess of the labeled probe concentration. Wild type TSA1 ZRE and MET30 ZRE fragments competed effectively for Zap1 binding, whereas the mutant TSA1m ZRE did not. D, MET30 ZRE is required for induction of gene expression in low zinc in vivo. Wild type (WT) or zap1Δ mutant cells bearing the pMET30-lacZ promoter fusion or the MET30 promoter fusion with a mutated ZRE (pMET30mZRE-lacZ) were grown in high zinc (+Zn, LZM + 1000 μm ZnCl2) and low zinc (−Zn, LZM + 1 μm ZnCl2) for 14–20 h. Cells were then harvested and assayed for β-galactosidase activity. The vector YEp353 was used as negative control, and the Zap1-responsive pZRC1-lacZ served as a positive control. Shown are the means from three independent experiments, and the error bars indicate ± S.D.
To determine whether the MET30 ZRE is functional in vivo, each of the 11 bases of the ZRE were altered by transversion mutations in the MET30 promoter and fused to the E. coli lacZ gene. Expression of wild type pMET30-lacZ and ZRE mutant pMET30mZRE-lacZ reporters was then assayed in zinc-replete and zinc-deficient cells. The vector YEp353 alone showed little expression in both zinc-replete and zinc-limited conditions (Fig. 4D, upper left). A control Zap1-regulated reporter containing the ZRC1 promoter, pZRC1-lacZ, showed low expression in zinc-replete wild type cells and high expression in zinc-limited cells (Fig. 4D, upper right). The increased expression of pZRC1-lacZ in low zinc was eliminated in a zap1Δ mutant strain. The wild type pMET30-lacZ reporter in wild type cells also showed a marked increase in expression in low zinc (Fig. 4D, lower left). These data also indicated that the expression of the pMET30-lacZ reporter in low zinc conditions is largely dependent on Zap1 because little expression was observed in the zap1Δ mutant strain. Expression from the MET30 promoter in zinc-limited wild type cells was greatly diminished by mutating the ZRE (pMET30mZRE-lacZ) (Fig. 4D, lower right) indicating the importance of the Zap1-binding site for MET30 up-regulation in zinc-deficient cells. Taken together, these results strongly suggest that MET30 is a direct target of Zap1 activation.
Zinc and Zap1 Affect Met4 Protein Accumulation Post- transcriptionally
Another prediction of our hypothesis was that up-regulation of MET30 expression in zinc-limited cells would result in decreased Met4 protein level and/or activity (Fig. 3A). As noted above, ubiquitination of Met4 inactivates the protein directly and can also signal its degradation by the cytosolic proteasome under some growth conditions. To test the effects of zinc and Zap1 on Met4, we assessed Met4 protein levels by immunoblotting. In zinc-replete cells, multiple Met4 bands were detected, and previous studies have shown that these represent the various degrees of Met4 ubiquitination (Fig. 5A, lane 2) (42, 43). The form with the lowest molecular mass represents unmodified, active Met4. Consistent with the hypothesis that zinc deficiency inhibits Met4 function, Met4 protein level decreased to very low levels in zinc-limited cells (Fig. 5A, lanes 2–6). The decrease in Met4 levels was dependent on Zap1 and did not occur in a zinc-limited zap1Δ mutant strain (Fig. 5A, lanes 7 and 8). It was notable that only the active form of Met4 accumulated in the zap1Δ mutant indicating that Zap1 function is required for efficient ubiquitination in zinc-limited cells. Furthermore, Met4 protein levels were reduced in zinc-replete cells expressing the Zap1TC allele (Fig. 5A, lanes 9 and 10). S1 nuclease protection assay indicated that these changes in Met4 protein level were not due to alterations in MET4 mRNA abundance (Fig. 5B). Thus, the effects of zinc and Zap1 on Met4 levels are mediated post-transcriptionally. Finally, we note that the zinc dose dependence of changes in MET30 mRNA (Fig. 3B) and Met4 protein levels (Fig. 5A) correlated well with changes in MET3, MET14, and MET16 mRNA levels (Fig. 2B). Specifically, these effects were all apparent in cells grown in LZM medium containing less than 10 μm zinc. These data suggested that Zap1 promotes degradation of Met4 by directly activating expression of MET30 gene expression.
FIGURE 5.

Met4 protein level is regulated post-transcriptionally by zinc and by Zap1. A, immunoblot analysis of Met4 protein levels in wild type cells grown in LZM supplemented with a range of added zinc (lanes 2–6). A met4Δ strain (TAL31) was used as a control (lane 1). In addition, wild type and zap1Δ mutant cells (ZHY6) were grown in low zinc (−Zn, LZM + 1 μm ZnCl2) (lanes 7 and 8) and wild type cells with and without expressing the constitutive Zap1TC allele were grown in zinc-replete SD medium (+Zn, lanes 9 and 10). The multiple Met4 bands represent differences in ubiquitination and/or phosphorylation states of the protein. 3-Phosphoglycerate kinase (Pgk1) protein was used as a loading control. B, cells were grown under the same conditions as in A. RNA was isolated and was then analyzed by S1 nuclease protection assay. No change in MET4 mRNA was detected indicating the changes in protein level are because of post-transcriptional effects.
Low Met4 Protein Accumulation Requires the Ubiquitin-Proteasome System
Does the lower accumulation of Met4 protein in zinc-limited cells require proteasome function? To address this question, we first examined Met4 protein levels in proteasome-defective pre1-1pre2-1 and hrd2-1 mutants grown in zinc-replete and zinc-limited media. Met4 protein levels accumulated to higher levels in the proteasome-defective mutants compared with their congenic wild type strains during zinc deficiency (Fig. 6A). Specifically, although Met4 protein levels in zinc-limited wild type cells were reduced by 55–66% of zinc-replete levels, Met4 levels decreased by only 21–29% in the proteasome mutant strains. The degree of regulation observed in these other wild type strain backgrounds was less than that observed in the W303-related wild type strain (DY1457) used in our other experiments (e.g. see Fig. 5A). Nonetheless, the effect of disrupted proteasome function on Met4 accumulation was clear.
FIGURE 6.

Decreased Met4 protein accumulation in zinc-limited cells requires the ubiquitination-proteasome degradation pathway. A, immunoblot analysis of Met4 protein from wild type (WT), pre1-1pre2-1, or hrd2-1 mutant cells. Cells were grown in high zinc (+, LZM + 1000 μm ZnCl2) and low zinc (−, LZM + 1 μm ZnCl2) prior to immunoblot analysis. Pgk1 is shown as a loading control. B, immunoblot analysis of Met4 protein levels from wild type (WT, DY1457) and pdr5Δ (CWY21) mutant cells grown in high zinc (+, LZM + 1000 μm ZnCl2) and low zinc (−, LZM + 1 μm ZnCl2) supplemented with MG132 (0.2 mg/ml in DMSO) or DMSO alone as a control. C, wild type Met4 and the Met4K163R allele were expressed in a met4Δ strain (TAL31) from the GAL1 promoter using the GEV system (see “Experimental Procedures”). Cells were grown in high zinc (+, LZM + 1000 μm ZnCl2) or low zinc (−, LZM + 1 μm ZnCl2) in the presence of the inducer, β-estradiol, prior to immunoblotting. D, immunoblot analysis of Met4 protein levels in wild type cells grown in high zinc (+, LZM + 1000 μm ZnCl2) or low zinc (−, LZM + 1 μm ZnCl2) and in cells with or without the Zap1TC allele in high zinc (LZM + 1000 μm ZnCl2). The cells were treated with 1 mm cadmium for the indicated times prior to harvest. The cells in lanes 3, 9, and 10 were treated with cadmium for 8 h prior to harvest.
To examine the role of the proteasome in our strain background, we assessed the accumulation of Met4 protein in the presence of MG132, a proteasome inhibitor. A strain carrying a deletion of the PDR5 multidrug efflux transporter gene was used in these experiments to increase MG132 sensitivity. The effect of zinc status on Met4 protein accumulation in pdr5Δ cells without MG132 treatment was similar to that observed in wild type cells (Fig. 6B, lanes 1–4). Consistent with the hypothesis that Met4 protein degradation is mediated by the proteasome, MG132 prevented the decreased accumulation of Met4 protein in zinc-limited cells (Fig. 6B, lane 4 versus 6). It was previously shown that ubiquitination of a lysine residue at position 163 of the Met4 protein is required for proteasome degradation (35). Replacing that lysine with arginine in the Met4K163R allele resulted in defective ubiquitination and degradation, which in turn leads to constitutive Met4 activity. Consistent with our hypothesis, the Met4K163R protein failed to be ubiquitinated and accumulated to high levels in zinc-limited cells (Fig. 6C).
Cells mutant for met30 are inviable (49). Therefore, to test whether the zinc-dependent effects on Met4 protein accumulation are also controlled by the SCFMet30 complex, we used cadmium to perturb SCFMet30 activity. Previous studies have shown that cadmium treatment prevents Met4 protein ubiquitination and subsequent degradation by dissociating the Met30 subunit from the SCFMet30 complex (50). We predicted that cadmium treatment of zinc-limited cells would block Met4 ubiquitination and degradation. When zinc-replete cells were treated with cadmium for 1 h prior to harvest, Met4 protein accumulated primarily in its under-ubiquitinated forms (Fig. 6D, lanes 1 and 3). The broad Met4 band observed following cadmium treatment is likely due to some of the protein being phosphorylated as has been observed previously (43, 50). Treatment of zinc-limited cells with cadmium resulted in increased accumulation of Met4 protein in under-ubiquitinated forms (Fig. 6D, lanes 2 and 4–6). Moreover, similar results were seen in zinc-replete cells expressing the Zap1TC-allele (Fig. 6D, lanes 7–10); cadmium treatment of Zap1TC-expressing cells increased Met4 accumulation. These data suggested that Met4 degradation caused by zinc deficiency and by the Zap1TC allele is mediated by the SCFMet30 activity.
As shown in Fig. 6, Met4 protein levels can be maintained in zinc-limited cells by inhibiting ubiquitination or proteasomal degradation of Met4. To determine whether these perturbations also prevent repression of MET3, MET14, and MET16, we examined the effects of the proteasome inhibitor MG132 and the Met4K163R allele (Fig. 7) on their repression in response to zinc. When proteasome activity was inhibited in zinc-limited cells with MG132, we found no repression of MET3 mRNA (Fig. 7A, lanes 5 and 6). MET30 transcriptional regulation by Zap1 was unaffected by MG132 treatment, and constitutive MET4 expression was also unchanged. These results are consistent with our model that regulation of MET3 requires proteasomal degradation of the Met4 protein. In addition, inhibition of Met4 degradation in zinc-limited cells with the Met4K163R allele also blocked repression of MET3, MET14, and MET16 (Fig. 7B, lanes 5 and 6). The results indicated that disruption of Met4 ubiquitination or proteasome degradation did indeed prevent repression of MET3, MET14, and MET16 genes.
FIGURE 7.

Derepression of MET3, MET14, and MET16 expression in cells with disrupted ubiquitination and proteasome degradation of Met4. A, wild type (WT, DY1457) and pdr5Δ (CWY21) mutant cells were grown in the same conditions as Fig. 6B. RNA was isolated and analyzed by S1 nuclease protection assay. CMD1 serves as a loading control. B, RNA samples for S1 nuclease protection assay were prepared from wild type (WT, DY1457) cells bearing the vector control and met4Δ cells (TAL31) expressing wild type Met4 or Met4K163R as described in Fig. 6C.
Physiological Significance of Repressing Sulfate Assimilation in Zinc-limited Cells
The ability to specifically perturb repression of the sulfate assimilation pathway in zinc-limited cells with the Met4K163R allele allowed us to address directly the importance of this regulation to cell physiology under low zinc conditions. One hypothesis we addressed was whether repression of sulfate assimilation in zinc-limited cells occurred to spare NADPH levels for combating the oxidative stress of zinc deficiency. Sulfate assimilation consumes considerable amounts of NADPH (26), and NADPH is also an important cofactor for oxidative stress defense (51). Given that zinc-limited cells experience increased oxidative stress, we reasoned that down-regulation of sulfate assimilation might occur in zinc-limited cells to ensure ample supplies of NADPH are available for ROS elimination.
This hypothesis predicted that cells unable to repress MET3, MET14, and MET16 expression in low zinc would have increased oxidative stress because of limiting NADPH levels. Consistent with this prediction, we found that zinc-limited met4Δ cells expressing the Met4K163R allele were hypersensitive to exogenous H2O2 relative to met4Δ cells expressing wild type Met4 (Fig. 8A). No difference in H2O2 sensitivity was observed between these strains when they were grown under zinc-replete conditions. In addition, we used the fluorescent probe DCFH-DA to measure the levels of ROS in these cells. This probe can detect superoxide, hydrogen peroxide, and hydroxyl radical to varying degrees (38). As was seen previously (16), ROS levels in zinc-replete cells were low, and there was no difference between cells expressing wild type Met4 or Met4K163R (Fig. 8B). ROS levels rose in zinc-limited cells expressing wild type Met4 but were even higher (∼2-fold) in the Met4K163R-expressing cells. Third, we measured the levels of total glutathione, reduced glutathione (GSH), and oxidized glutathione (GSSG) in these cells. Consistent with the increase in sulfate assimilation that we predicted would occur in Met4K163R-expressing cells, we observed that the levels of all three were elevated when compared with cells expressing wild type Met4 (Fig. 8C). Evidence of increased oxidative stress in the Met4K163R-expressing cells was apparent in the ∼10-fold increase in the ratio of oxidized to reduced (GSSG/GSH) glutathione.
FIGURE 8.

Derepression of sulfate assimilation increases oxidative stress and the NADP+/NADPH ratio in zinc-limited cells. A, met4Δ mutant cells expressing wild type Met4 (WT) or Met4K163R were inoculated at low density (A600 = 0.1) in high or low zinc media as described in Fig. 6C in the presence of the indicated concentration of H2O2. Cell densities were then measured after 38 h of culturing at 30 °C. Cell densities are reported as percent of the corresponding untreated controls. B, met4Δ mutant cells expressing wild type Met4 (WT) or Met4K163R were grown in high or low zinc media as described in Fig. 6C and then assayed for reactive oxygen species using the fluorescent probe DCFH-DA. The values are means of three independent experiments, and the error bars represent ± S.D. C, met4Δ mutant cells expressing wild type Met4 (WT) or Met4K163R were grown in low zinc medium (LZM + 1 μm ZnCl2) prior to assay of total glutathione (Total GSH), reduced glutathione (GSH), and oxidized glutathione (GSSG). The ratios of GSSG/GSH ratios are shown in the right panel. The plotted values are the means of 12 independent cultures, and the error bars represent ±1 S.E. D, met4Δ mutant cells expressing wild type Met4 (WT) or Met4K163R were grown in high or low zinc media as described in Fig. 6C prior to analysis of total NADP(H), reduced NADPH, and oxidized NADP+. The NADP+/NADPH ratio is reported in the right panel. The data plotted are the means of six independent cultures, and the error bars indicate 1 S.D. p values were determined using the paired, two-tailed Student's t test.
Taken together, the results shown in Fig. 8, A–C, indicate that oxidative stress is increased in cells that are unable to repress sulfate assimilation because of the Met4K163R mutation. Our hypothesis would predict that these effects are due, at least in part, to depletion of NADPH and an increase in the NADP+/NADPH ratio. To test this prediction, we assayed total NADPH, oxidized NADP+, and reduced NADPH in these cells. No differences in these levels were observed in zinc-replete cells (Fig. 8D). Consistent with our hypothesis, the level of NADPH decreased, and the level of NADP+ increased in cells expressing Met4K163R. These results are consistent with the hypothesis that repression of sulfate assimilation during zinc deficiency occurs to conserve NADPH levels to deal with oxidative stress. Unexpectedly, total NADP(H) levels increased in zinc-limited cells expressing Met4K163R. This effect may be due to feedback regulation of NADP(H) synthesis mediated by NAD kinase (see “Discussion”).
DISCUSSION
In this report, we have systematically identified targets of Zap1-mediated repression. We previously identified ZRT2, ADH1, and ADH3 as direct targets of Zap1 repression, and their examples motivated us to search for additional Zap1-repressed genes. As a result, we identified 34 other genes that have lower expression in zinc-limited cells and in zinc-replete cells expressing a constitutive allele of Zap1 (Table 1). We are in the process of elucidating the various mechanisms regulating these genes. Our previous studies established two possible mechanisms for Zap1-mediated gene repression. First, Zap1-repressed genes may be down-regulated in a manner similar to ZRT2 where binding of Zap1 to a ZRE located downstream of the TATA box of the gene interferes with transcription initiation (24). Motif analysis suggests that some repressed genes may be regulated in this manner. For example, one gene showing zinc- and Zap1-responsive repression, CAF17, has a potential ZRE (TCCTTAAAGGT) at position −35 upstream of the translation initiation codon. The CAF17 transcriptional start site was mapped to position −51 relative to the start codon (52) suggesting that the mechanism of its repression in zinc-limited cells may be similar to ZRT2.
A second mechanism of Zap1-mediated repression was established by our prior characterization of ADH1 and ADH3. These genes are repressed by Zap1 via intergenic transcription disrupting activator binding in their promoters (25). Some of the newly identified repressed genes may be regulated similarly. For example, ADH2 has a potential ZRE (TCCCTGAGGGA) upstream of its promoter at position −880. In contrast to ADH1 and ADH3, however, the candidate ADH2 ZRE is located within the adjacent ORF rather than in an intergenic region. The RPL6B gene has a potential intergenic ZRE at position −888 (CCCTTTAAGGT) that could be involved in its Zap1-mediated repression. A third candidate for this type of regulation is FZF1. FZF1 is located adjacent to ZRT1, a highly induced Zap1 target gene. The FZF1 promoter lies downstream of the ZRT1 ORF, and the intergenic region between the two ORFs is only 196 bp long. Therefore, we hypothesize that high level transcription of the ZRT1 mRNA into the FZF1 promoter in zinc-limited cells disrupts FZF1 expression. Given that Fzf1 regulates expression of a sulfite transporter, this regulation may be relevant to changes in sulfur metabolism (see below).
We have now uncovered a third mechanism of transcriptional repression mediated by Zap1. Our data indicate that Zap1 directly induces expression of the MET30 gene in zinc-deficient cells. We proposed that increased Met30 expression leads to increased SCFMet30 ubiquitin ligase activity, increased ubiquitination of the Met4 transcription factor, and degradation of Met4 by the cytosolic proteasome. Several predictions of this model were confirmed here experimentally. A particularly key finding was that mutation of the lysine residue in Met4 that is the site of ubiquitination completely abolished the repression observed in zinc-limited cells. Furthermore, this hypothesis is consistent with the previous observation that the level of the Met30 subunit can control the overall activity of the SCFMet30 complex (46).
It has previously been shown that cadmium treatment also regulates Met4 activity through the SCFMet30 complex (50, 53). Specifically, cadmium inhibits the SCFMet30 activity by dissociating the Met30 protein from the complex. The consequent decrease in ubiquitin ligase function results in increased Met4 activity and up-regulation of the sulfate assimilation pathway. This mechanism is distinct from the effects of zinc on the SCFMet30 activity indicating that our findings reveal a second mechanism for the regulation of sulfur metabolism by metal ions.
Down-regulation of sulfate assimilation in zinc-limited cells would likely reduce the free pools of sulfur-containing amino acids. As predicted, we found that the free pools of both methionine and cysteine were reduced significantly in low zinc (Table 2). In fact, free methionine levels were reduced to less than 10% of the levels measured in zinc-replete cells. Zinc-limited cells grow more slowly than do zinc-replete cells. Could the reduced pools of methionine and/or cysteine be responsible for restricting the growth of zinc-limited cells? The answer to this question appears to be no; methionine and/or cysteine added to the medium failed to increase the growth rate of cells in low zinc media.3 Thus, although the pools of methionine and cysteine are reduced under low zinc conditions, sufficient levels of these amino acids must still be produced to supply the demands of the cell. Decreased activity of other zinc-dependent processes is likely responsible for the slower growth of zinc-limited cells. It is unclear at this time why the effects of zinc status on methionine levels were more pronounced than those observed for cysteine pools. Differences in the flux of organic sulfur through the two branches of this pathway is a likely explanation.
Consistent with a decrease in sulfate assimilation in zinc-limited cells, a previous study suggested that SAM3 is a potential target of Zap1 activation (5) (Fig. 9). SAM3 encodes an S-adenosylmethionine permease that can transport extracellular S-adenosylmethionine into the cell (54). Imported S-adenosylmethionine could then be used as a source of methionine or as a source of homocysteine for cysteine and glutathione synthesis (26). Thus, increased uptake of extracellular S-adenosylmethionine by Sam3 can bypass the decreased assimilation of sulfate that occurs in zinc-limited cells. It is also interesting to note that the MUP1 gene was identified previously as a potential Zap1 activation target (9) (Fig. 9). MUP1 encodes an amino acid permease that mediates the uptake of methionine and cysteine (55, 56). Induction of MUP1 expression by Zap1 under low zinc conditions is also consistent with our model for the overall remodeling of sulfur metabolism in zinc-deficient cells. The resulting increased capacity for uptake of exogenous methionine and cysteine would bypass any shortage in these amino acids imposed by decreased sulfate assimilation.
FIGURE 9.

Role of Zap1 regulation in sulfate metabolism. S. cerevisiae genes whose expression is activated in low zinc are indicated in yellow, and genes negatively regulated in low zinc are indicated in blue. S. pombe genes up-regulated under zinc deficiency are marked in red, and those down-regulated in low zinc are denoted in green.
We found two additional links between zinc and sulfur metabolism among the results of this and previous studies. First, the Fzf1 transcription factor activates expression of the Ssu1 sulfite efflux transporter (57) (Fig. 9). As noted above, Zap1 may repress FZF1 expression perhaps by induction of the adjacent ZRT1 gene. Because Fzf1 normally activates SSU1 expression, decreased Fzf1 activity would be expected to decrease the capacity of the cell for sulfite efflux. Thus, repression of FZF1 may help conserve intracellular SO32−. Given that SO32− production is likely to be reduced in zinc-limited cells because of repressed sulfate assimilation, this conservation would help ensure that whatever sulfate is assimilated is used for methionine and cysteine synthesis rather than exported. Finally, a potential direct target of Zap1 activation, YPR003C (5), encodes a potential sulfate transporter that localizes to the ER (58, 59). Its role in sulfate metabolism is unknown.
An intriguing question raised by these observations is what is the physiological relevance of repressing sulfate assimilation in zinc-limited cells? One potential reason for this control is that the yeast Met6 enzyme, methionine synthase, is zinc-dependent (60). Met6 methylates homocysteine to generate methionine (Fig. 9). If Met6 activity were compromised under low zinc conditions without a decrease in sulfate assimilation, homocysteine levels would be predicted to rise. Homocysteine is damaging to cells because it can modify cysteine residues in proteins, i.e. protein S-homocysteinylation, and disrupt their function (61). In addition, homocysteine can be converted into homocysteine thiolactone, an even more potent toxin (61). Consistent with this notion that zinc-limited cells are at risk of elevated homocysteine thiolactone levels, we found that the gene encoding the enzyme responsible for hydrolyzing this toxic by-product, the Lap3 homocysteine thiolactonase (62), is induced by zinc deficiency (5) (Fig. 9). Our results indicated that LAP3 is a direct target of Zap1 activation. Although these observations make this model appealing, it was not supported by direct measurements of Met6 enzyme activity. In cell lysates assayed under reducing or nonreducing conditions, Met6 activity was unaffected by zinc status.4 Thus, a decrease in Met6 activity in vivo may not be the explanation for repression of sulfate assimilation.
A second possible explanation comes from our previous finding that zinc-deficient yeast experience increased oxidative stress and that Zap1 activates expression of the TSA1 gene as a defense response. TSA1 encodes the major cytosolic peroxiredoxin, which metabolizes H2O2 (63). Cysteine residues in Tsa1 become oxidized during this reaction and require thioredoxin to be reduced back to their active state. Thioredoxin, in turn, requires thioredoxin reductase to be restored to its active, reduced state. Thioredoxin reductase requires NADPH to perform this function. Thus, the NADPH pool is clearly important for this antioxidant mechanism. NADPH is also required for the function of other antioxidant mechanisms as well (51). Sulfate assimilation requires considerable amounts of NADPH; 6 mol of NADPH are oxidized to NADP+ to generate 1 mol of homocysteine from 1 mol of SO42−. Based on these issues, we reasoned that repression of sulfate assimilation might occur to ensure that an abundant supply of NADPH is available for ROS defense. Our results are consistent with this hypothesis. In cells that are unable to down-regulate sulfate assimilation under zinc deficiency because of mutation of lysine 163, oxidative stress increased as did the NADP+/NADPH ratio. This hypothesis is also supported by previous observations of a significant degree of competition for NADPH between sulfate assimilation pathway and oxidative stress tolerance mechanisms (64).
We were initially surprised to observe that total NADP(H) levels increased in cells that are unable to repress sulfate assimilation in low zinc. One possible explanation for this result comes from studies of feedback inhibition of NAD kinase (NadK) of Salmonella enterica (65). The activity S. enterica NadK is inhibited by NADPH. Therefore, when NADPH levels drop in these cells, NadK responds by increasing synthesis of NADP+. If one or more of the yeast NAD kinases (Pos5, Utr1, and Yef1) are regulated in the same fashion, we would predict precisely the results we obtained.
Finally, it is worth noting that sulfate assimilation in Schizosaccharomyces pombe may be regulated by zinc status via a mechanism similar to what we have uncovered in S. cerevisiae. In S. pombe, the SPAC869.05c gene encodes a sulfate uptake transporter similar to SUL1 and SUL2 of S. cerevisiae, sua1+ encodes the ortholog of Met3, and SPBC428.11 encodes the ortholog of MET17 (Fig. 9). Expression of these genes is reduced in zinc-limited cells (66). In contrast, expression of the MET30 ortholog, pof1+, is up-regulated in low zinc. Although the zinc-responsive transcription factor in S. pombe that is analogous to Zap1 is unknown, the similar effects of zinc deficiency seen with these two distantly related fungal species suggest that repression of sulfate assimilation is an evolutionarily conserved response to zinc deficiency.
Supplementary Material
Acknowledgments
We thank Dr. Traci Lee (University of Wisconsin, Parkside) for providing the anti-Met4 antibody, Dr. Peter Kaiser (University of California, Irvine) for providing plasmids, and Dr. Randy Hampton (University of California, San Diego) for strains. We also thank Dr. Hieronim Jakubowski for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant GM56285 (to D. J. E.). This work was also supported by National Science Foundation Grant MCB-0429968 (to S. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1–3.
C. Wu, unpublished results.
C. Wu, F. J. Sandoval, and S. Roje, unpublished results.
- ZRE
- zinc-responsive element
- LZM
- low zinc medium
- DCF
- 2,7-dichlorofluorescein
- DCFH-DA
- 2,7-dichlorodihydrofluoroscein diacetate
- ROS
- reactive oxygen species
- ORF
- open reading frame.
REFERENCES
- 1.Andreini C., Banci L., Bertini I., Rosato A. (2006) J. Proteome Res. 5, 196–201 [DOI] [PubMed] [Google Scholar]
- 2.Hambidge M. (2000) J. Nutr. 130, 1344S–1349S [DOI] [PubMed] [Google Scholar]
- 3.Cai L., Li X. K., Song Y., Cherian M. G. (2005) Curr. Med. Chem. 12, 2753–2763 [DOI] [PubMed] [Google Scholar]
- 4.Lyons T. J., Gasch A. P., Gaither L. A., Botstein D., Brown P. O., Eide D. J. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 7957–7962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu C. Y., Bird A. J., Chung L. M., Newton M. A., Winge D. R., Eide D. J. (2008) BMC Genomics 9, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao H., Eide D. J. (1997) Mol. Cell. Biol. 17, 5044–5052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bird A., Evans-Galea M. V., Blankman E., Zhao H., Luo H., Winge D. R., Eide D. J. (2000) J. Biol. Chem. 275, 16160–16166 [DOI] [PubMed] [Google Scholar]
- 8.Higgins V. J., Rogers P. J., Dawes I. W. (2003) Appl. Environ. Microbiol. 69, 7535–7540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Nicola R., Hazelwood L. A., De Hulster E. A., Walsh M. C., Knijnenburg T. A., Reinders M. J., Walker G. M., Pronk J. T., Daran J. M., Daran-Lapujade P. (2007) Appl. Environ. Microbiol. 73, 7680–7692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao H., Butler E., Rodgers J., Spizzo T., Duesterhoeft S., Eide D. (1998) J. Biol. Chem. 273, 28713–28720 [DOI] [PubMed] [Google Scholar]
- 11.Waters B. M., Eide D. J. (2002) J. Biol. Chem. 277, 33749–33757 [DOI] [PubMed] [Google Scholar]
- 12.Zhao H., Eide D. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 2454–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao H., Eide D. (1996) J. Biol. Chem. 271, 23203–23210 [DOI] [PubMed] [Google Scholar]
- 14.MacDiarmid C. W., Gaither L. A., Eide D. (2000) EMBO J. 19, 2845–2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carman G. M., Han G. S. (2007) Biochim. Biophys. Acta 1771, 322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu C. Y., Bird A. J., Winge D. R., Eide D. J. (2007) J. Biol. Chem. 282, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 17.Powell S. R. (2000) J. Nutr. 130, 1447S–1454S [DOI] [PubMed] [Google Scholar]
- 18.Burke J. P., Fenton M. R. (1985) Proc. Soc. Exp. Biol. Med. 179, 187–191 [DOI] [PubMed] [Google Scholar]
- 19.Oteiza P. I., Olin K. L., Fraga C. G., Keen C. L. (1995) J. Nutr. 125, 823–829 [DOI] [PubMed] [Google Scholar]
- 20.Ho E., Ames B. N. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16770–16775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho E., Courtemanche C., Ames B. N. (2003) J. Nutr. 133, 2543–2548 [DOI] [PubMed] [Google Scholar]
- 22.Ho E. (2004) J. Nutr. Biochem. 15, 572–578 [DOI] [PubMed] [Google Scholar]
- 23.Ames B. N., Wakimoto P. (2002) Nat. Rev. Cancer 2, 694–704 [DOI] [PubMed] [Google Scholar]
- 24.Bird A. J., Blankman E., Stillman D. J., Eide D. J., Winge D. R. (2004) EMBO J. 23, 1123–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bird A. J., Gordon M., Eide D. J., Winge D. R. (2006) EMBO J. 25, 5726–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas D., Surdin-Kerjan Y. (1997) Microbiol. Mol. Biol. Rev. 61, 503–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaiser P., Su N. Y., Yen J. L., Ouni I., Flick K. (2006) Cell Div. 1, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gitan R. S., Luo H., Rodgers J., Broderius M., Eide D. (1998) J. Biol. Chem. 273, 28617–28624 [DOI] [PubMed] [Google Scholar]
- 29.Herbig A., Bird A. J., Swierczek S., McCall K., Mooney M., Wu C. Y., Winge D. R., Eide D. J. (2005) Mol. Microbiol. 57, 834–846 [DOI] [PubMed] [Google Scholar]
- 30.van Helden J. (2003) Nucleic Acids Res. 31, 3593–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dohrmann P. R., Butler G., Tamai K., Dorland S., Greene J. R., Thiele D. J., Stillman D. J. (1992) Genes Dev. 6, 93–104 [DOI] [PubMed] [Google Scholar]
- 32.Myers A. M., Tzagoloff A., Kinney D. M., Lusty C. J. (1986) Gene 45, 299–310 [DOI] [PubMed] [Google Scholar]
- 33.Ma H., Kunes S., Schatz P. J., Botstein D. (1987) Gene 58, 201–216 [DOI] [PubMed] [Google Scholar]
- 34.Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989) Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 35.Flick K., Ouni I., Wohlschlegel J. A., Capati C., McDonald W. H., Yates J. R., Kaiser P. (2004) Nat. Cell Biol. 6, 634–641 [DOI] [PubMed] [Google Scholar]
- 36.Gao C. Y., Pinkham J. L. (2000) BioTechniques 29, 1226–1231 [DOI] [PubMed] [Google Scholar]
- 37.Guarente L. (1983) Methods Enzymol. 101, 181–191 [DOI] [PubMed] [Google Scholar]
- 38.Halliwell B., Whiteman M. (2004) Br. J. Pharmacol. 142, 231–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vandeputte C., Guizon I., Genestie-Denis I., Vannier B., Lorenzon G. (1994) Cell Biol. Toxicol. 10, 415–421 [DOI] [PubMed] [Google Scholar]
- 40.Gibon Y., Larher F. (1997) Anal. Biochem. 251, 153–157 [DOI] [PubMed] [Google Scholar]
- 41.Thomas D., Kuras L., Barbey R., Cherest H., Blaiseau P. L., Surdin-Kerjan Y. (1995) Mol. Cell. Biol. 15, 6526–6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rouillon A., Barbey R., Patton E. E., Tyers M., Thomas D. (2000) EMBO J. 19, 282–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaiser P., Flick K., Wittenberg C., Reed S. I. (2000) Cell 102, 303–314 [DOI] [PubMed] [Google Scholar]
- 44.Kuras L., Rouillon A., Lee T., Barbey R., Tyers M., Thomas D. (2002) Mol. Cell 10, 69–80 [DOI] [PubMed] [Google Scholar]
- 45.Chandrasekaran S., Deffenbaugh A. E., Ford D. A., Bailly E., Mathias N., Skowyra D. (2006) Mol. Cell 24, 689–699 [DOI] [PubMed] [Google Scholar]
- 46.Smothers D. B., Kozubowski L., Dixon C., Goebl M. G., Mathias N. (2000) Mol. Cell. Biol. 20, 7845–7852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan D. S. (2000) Genetics 156, 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiang D. Y., Nix D. A., Shultzaberger R. K., Gasch A. P., Eisen M. B. (2006) BMC Mol. Biol. 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patton E. E., Peyraud C., Rouillon A., Surdin-Kerjan Y., Tyers M., Thomas D. (2000) EMBO J. 19, 1613–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barbey R., Baudouin-Cornu P., Lee T. A., Rouillon A., Zarzov P., Tyers M., Thomas D. (2005) EMBO J. 24, 521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grant C. M. (2001) Mol. Microbiol. 39, 533–541 [DOI] [PubMed] [Google Scholar]
- 52.Zhang Z., Dietrich F. S. (2005) Nucleic Acids Res. 33, 2838–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yen J. L., Su N. Y., Kaiser P. (2005) Mol. Biol. Cell 16, 1872–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rouillon A., Surdin-Kerjan Y., Thomas D. (1999) J. Biol. Chem. 274, 28096–28105 [DOI] [PubMed] [Google Scholar]
- 55.Isnard A. D., Thomas D., Surdin-Kerjan Y. (1996) J. Mol. Biol. 262, 473–484 [DOI] [PubMed] [Google Scholar]
- 56.Kosugi A., Koizumi Y., Yanagida F., Udaka S. (2001) Biosci. Biotechnol. Biochem. 65, 728–731 [DOI] [PubMed] [Google Scholar]
- 57.Avram D., Leid M., Bakalinsky A. T. (1999) Yeast 15, 473–480 [DOI] [PubMed] [Google Scholar]
- 58.Paulsen I. T., Sliwinski M. K., Nelissen B., Goffeau A., Saier M. H., Jr. (1998) FEBS Lett. 430, 116–125 [DOI] [PubMed] [Google Scholar]
- 59.Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., O'Shea E. K. (2003) Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 60.Zhou Z. S., Peariso K., Penner-Hahn J. E., Matthews R. G. (1999) Biochemistry 38, 15915–15926 [DOI] [PubMed] [Google Scholar]
- 61.Perła-Kaján J., Twardowski T., Jakubowski H. (2007) Amino Acids 32, 561–572 [DOI] [PubMed] [Google Scholar]
- 62.Zimny J., Sikora M., Guranowski A., Jakubowski H. (2006) J. Biol. Chem. 281, 22485–22492 [DOI] [PubMed] [Google Scholar]
- 63.Rhee S. G., Chae H. Z., Kim K. (2005) Free Radic. Biol. Med. 38, 1543–1552 [DOI] [PubMed] [Google Scholar]
- 64.Slekar K. H., Kosman D. J., Culotta V. C. (1996) J. Biol. Chem. 271, 28831–28836 [DOI] [PubMed] [Google Scholar]
- 65.Grose J. H., Joss L., Velick S. F., Roth J. R. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 7601–7606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dainty S. J., Kennedy C. A., Watt S., Bähler J., Whitehall S. K. (2008) Eukaryot. Cell 7, 454–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.