

Abstract
Mutations in the TNF family ligand EDA1 cause X-linked hypohidrotic ectodermal dysplasia (XLHED), a condition characterized by defective development of skin appendages. The EDA1 protein displays a proteolytic processing site responsible for its conversion to a soluble form, a collagen domain, and a trimeric TNF homology domain (THD) that binds the receptor EDAR. In-frame deletions in the collagen domain reduced the thermal stability of EDA1. Removal of the collagen domain decreased its activity about 100-fold, as measured with natural and engineered EDA1-responsive cell lines. The collagen domain could be functionally replaced by multimerization domains or by cross-linking antibodies, suggesting that it functions as an oligomerization unit. Surprisingly, mature soluble EDA1 containing the collagen domain was poorly active when administered in newborn, EDA-deficient (Tabby) mice. This was due to a short stretch of basic amino acids located at the N terminus of the collagen domain that confers EDA1 with proteoglycan binding ability. In contrast to wild-type EDA1, EDA1 with mutations in this basic sequence was a potent inducer of tail hair development in vivo. Thus, the collagen domain activates EDA1 by multimerization, whereas the proteoglycan-binding domain may restrict the distribution of endogeneous EDA1 in vivo.
Tumor necrosis factor (TNF)7 family ligands spontaneously form homotrimers that can bind three individual receptors (1). For some family members, these trimeric complexes are biologically active. For example, trimers of TNF and TWEAK (TNF homologue with weak apoptosis-inducing activity) signal cell death in their respective target cells, and cross-linking of several trimers, via the action of an anti-Flag antibody, does not result in increased activity (2). For other ligands, the trimeric, soluble complexes are not or only poorly biologically active, but their membrane-bound forms are. This is the case, among others, for FasL and CD40L. These ligands can however become active in a soluble form if trimers are cross-linked, either with antibodies, or by fusion with oligomerizing proteins, such as the Fc portion of an immunoglobulin, or the collagen domain of ACRP/adiponectin (2, 3).
Ectodysplasin A (EDA) is a TNF family ligand involved in the development of various structures derived from the ectoderm, such as hair, teeth, and sweat glands, and EDA loss of function in mouse and human is associated with X-linked hypohidrotic ectodermal dysplasia (OMIM 305100). Of the several EDA isoforms that have been described, two of which comprise the TNF homology domain, only EDA1 has been implicated in the development of ectodermal appendages (4, 5). Activation of the NF-κB transcription factor through the EDA1 receptor (EDAR) is a central signaling event as suggested by the ectodermal dysplasia phenotype of NF-κB-compromised mice (6), and by the fact that early events of placode formation can be initiated by EDAR-independent NF-κB signals (7). One relevant NF-κB target downstream of EDAR is CTGF/CCN2 (connective tissue growth factor/CCN family protein2) that locally counteracts inhibitors of placode fate, thus allowing placode formation at the site of EDAR signaling in embryonic skin (8, 9).
Several lines of evidence indicate that EDA1 trimers must probably be aggregated in order to signal. First, EDA1 possesses a collagen domain that may potentially aggregate the TNF homology domain (10). Second, inactive trimeric FasL is partially activated by fusion with the collagen domain of EDA1 (11). Third, a number of X-linked hypohidrotic ectodermal dysplasia (XL-HED) patients have point mutations or in-frame deletions in the collagen domain that suggest this domain fulfills a functional role (11). Fourth, fusion of the EDA1 receptor-binding domain to the Fc portion of an immunoglobulin yields a biologically active ligand (12). However, it has never been formally demonstrated that EDA1 needs to be cross-linked to be active, and that the collagen domain of EDA1 fulfills this cross-linking function.
Using an EDA1-dependent NF-κB activation assay in HaCat keratinocytes and a specific biological assay to measure oligomerization of EDA1 trimers, we demonstrate here that the collagen domain is required for EDA1 to signal efficiently through EDAR. This function of the collagen domain can be mimicked by antibody-mediated cross-linking, or by fusion of the TNF homology domain of EDA1 to the Fc portion of human immunoglobulin G1. In vivo, the biological activity of exogenously administered EDA1 is also dependent on multimerization, but to a lesser extent than predicted by in vitro results, and is additionally negatively regulated by a newly identified, evolutionarily conserved heparan sulfate proteoglycan (HSPG)-binding region that may restrict biodistribution of the protein.
MATERIALS AND METHODS
Cells
293T cells were grown in DMEM supplemented with 10% of heat-inactivated fetal calf serum (FCS). HaCat cells were grown in DMEM:NutMix-F12 (1:1, v/v), 5% FCS, 50 units/ml penicillin and streptomycin, 9 ng/ml of cholera toxin (Sigma), 5 μg/ml of insulin (Sigma), 24.3 μg/ml of adenosine (Sigma), 10 ng/ml of epidermal growth factor (Sigma), and 0.5 μg/ml of hydroxycortisone (Sigma). Jurkat and Jurkat-EDAR:Fas cells were grown in RPMI supplemented with 10% FCS.
Cells were passaged twice weekly. HaCat cells were trypsinized. When cells were analyzed by FACS, however, they were detached with PBS, 1 mm EDTA.
Generation of EDAR:Fas Jurkat Cells
Retroviruses were produced essentially as described previously (13). Briefly, 293T cells were transiently transfected with pMSCVpuro-EDAR:Fas and co-transfected with the pHIT60 and VSV-G plasmids, containing the sequences for gag-pol and VSV-G, respectively. pMSCVpuro-EDAR:Fas encodes the extracellular domain of human EDAR (amino acids 1–183), amino acids VD and the transmembrane and intracellular domains of human Fas (amino acids 169–335). After transfection, 293T cells were incubated for 24 h in RPMI supplemented with 10% FCS. Fas-deficient Jurkat-JOM2 cells were a kind gift of Olivier Micheau (University of Dijon, France). Jurkat-JOM2 cells (106 cells in 1 ml) were mixed with virus-containing supernatants (3 ml) supplemented with 8 μg/ml of polybrene, left for 15 min at 37 °C, and centrifuged for 1 h at 37 °C and at 450 × g (1500 rpm). Cells were selected with 5 μg/ml of puromycin and cloned. About 40 clones were tested for their sensitivity to Fc-EDA1 (12, 14), and one of the sensitive clones (Jurkat-2199 clone 23) was selected for further experimentation. HaCat IκBα-DN cells were generated in a similar way using pMSCVpuro-IκBα-S32G vector, except that cells were not cloned but used as a population.
Expression Constructs
Expression constructs were cloned into the PCR3 mammalian expression vector (Invitrogen) according to standard molecular biology techniques. Vector for expression of Flag-tagged ligands, ACRP-ligands, Fc-ligands, receptors-Fc, receptors-GPI, and receptors-COMP-Flag have been described previously (3, 14–16). Fc-ligands with a PreScission protease cleavage site were constructed by insertion of the Prescission site in the Fc-ligand vector. Details regarding plasmids used in this study and the proteins they encode are provided in supplemental Table S1 and Fig. 1, respectively.
FIGURE 1.

Schematic structure of the EDA1 constructs and related proteins used in this study.
Calcium Phosphate Transfection of 293T Cells
106 293T cells were seeded in 8 ml of medium in a 10-cm diameter dish and transfected ∼8 h later. The transfection was performed by mixing 7 μg of plasmid of interest and 1 μg of EGFP tracer plasmid with 50 μl of 2.5 m CaCl2. Sterile water was then added to 500 μl. While vortexing the DNA mix, 500 μl of 2× HeBS buffer was added dropwise (16.4 g of NaCl, 11.9 g of Hepes, acid form, 0.21 g of Na2HPO4, 800 ml of H2O, adjusted to pH 7.05 with NaOH, and finally brought to 1 liter and filtrated). The transfection mix was added to cells within 1 min after mixing. The next day, cells were washed with PBS and 8 ml of fresh medium was added (DMEM, 10% fetal calf serum or serum-free Opti-MEM1 medium).
HaCat Cell Stimulation Assay
Two days before stimulation, cells were seeded in 24-well plates (105 cells/well). The day before stimulation, cells were starved overnight in serum-free DMEM medium, 50 units/ml penicillin, and streptomycin. On the day of stimulation, medium was removed, and 0.5–1 ml of fresh DMEM (no serum) was added that contained the ligand of interest either in a purified form (between 0.05 and 1 μg/ml) or as Opti-MEM supernatants.
Immunoprecipitations of Recombinant EDA1 from Cell Culture Supernatant
Conditioned Opti-MEM supernatant was concentrated 20 times in Amicon Ultra filter devices (molecular mass cutoff of 10,000 Da). Immunoprecipitations were performed on 200–400 μl of Opti-MEM supernatant (or 10–20 μl of concentrated Opti-MEM) with 10 μl of protein A-Sepharose beads. 1 μg of hEDAR-Fc was added if the target EDA1 protein did not contain an Fc moiety. PBS was added to a final volume of 400 μl, and the mixture was incubated for 1 h at 4 °C on a rotating wheel. Beads were washed twice with 1 ml of PBS, and transferred into a mini column made of a 200-μl tip plugged with a 1-mm diameter “stopper,” eluted with 15–20 μl of 100 mm sodium citrate pH 4, and neutralized with the appropriate volume of 1 m Tris-HCl, pH 9.
For heparin-Sepharose pull-downs (GE Healthcare), 10 μl of beads freshly rehydrated in PBS were mixed with 750 μl of Opti-MEM supernatant in PBS (1:1 v/v), incubated for 1 h at 4 °C on a rotating wheel, transferred into a mini column, washed with PBS, and eluted with 15 μl of PBS supplemented with 0.8 m NaCl.
Cell Lysis
HaCat cells were harvested at defined time points. Plates were put on ice, cells were washed once with ice-cold PBS and lysed in 50 μl of lysis buffer (1% Triton X-100, 5 mm 2-glycerophosphate, 10 mm NaF, 1 mm Na3VO4, 1 mm dithiothreitol, and one tablet/50 ml of Complete protease inhibitor (Roche, Basel, Switzerland)). Plates were briefly vortexed, cells were scraped from the plate, and the lysates were centrifuged for 5 min at 4 °C and 13,000 rpm (16,000 × g) in a tabletop centrifuge. Protein concentration in cell lysate supernatants was determined with the BCA assay (Pierce).
SDS-PAGE and Western Blotting
Proteins were resolved by 12% SDS-PAGE and transferred onto nitrocellulose membranes. The following primary antibodies were used: rabbit anti-EDA1 serum AL166 (11), mouse monoclonal antibody anti-P IκBα mouse (Cell Signaling, Beverly, MA), polyclonal rabbit antibody anti-IκBα (Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal antibody anti-α-tubulin (Sigma), M2 mouse monoclonal antibody anti-Flag (Sigma), and horseradish peroxidase-coupled antibody anti-human IgG (Jackson ImmunoResearch). Blots were revealed with ECL (GE Healthcare).
Purification of Flag-tagged Proteins
Various Flag-tagged EDA1 constructs were purified from supernatants of transiently transfected 293T cells using anti-Flag M2-agarose (Sigma). Flag-EDA1 were eluted with citrate-NaOH, pH 3 and neutralized with Tris-HCl, pH 9. Buffer was exchanged for PBS, and the protein was stored at −70 °C until use.
Purification of Fc-tagged Proteins
Fc-PreScission-EDA1-E245, Fc-EDA-A238, and hEDAR-PreScission-Fc proteins were purified from culture supernatants of stably transfected CHO cell clones by protein A affinity chromatography (15).
Cytotoxicity Assays
The cytotoxicity assay using EDAR:Fas Jurkat cells was performed as described for FasL on Jurkat cells (2).
Prescission Protease Digestion of Proteins
Proteins were digested in PBS, 1 mm EDTA with 40 units/ml of GST-Prescission protease (GE Healthcare). Digestions were performed either in solution or on protein A-Sepharose beads for 24 h at 4 °C on a rotating wheel. Digestion efficiency was checked by Western blotting with anti-EDA1 antibodies.
Gel Filtration Chromatography and Enzyme-linked Immunosorbant Assay
100 μg of Fc-PreScission-EDA1-E245 (cleaved or not with PreScission protease), were applied onto a Superdex 200 column (GE Healthcare) and eluted in PBS at 0.5 ml/min. Fractions of 250 μl were collected, of which 10 μl were analyzed by Western blotting. Various Flag-EDA and Flag-FasL constructs in Opti-MEM supernatants were analyzed similarly. In this case, 2, 5, 10, or 50 μl of the 500 μl fractions were used to detect the ligands by ELISA, essentially as described previously (3, 11). Briefly, EDAR-Fc or Fas-Fc (Alexis, Lausen, Switzerland) were coated at 1 μg/ml to capture the ligands, and biotinylated anti-Flag M2 antibody (Sigma) followed by horse radish peroxidase-coupled streptavidin were used for revelation. The Superdex 200 column was calibrated with the following standard proteins: thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), bovine serum albumin (67 kDa) ovalbumin (43 kDa), chymotrypsinogen A (25 kDa), and ribonuclease A (13.7 kDa).
Flow Cytometry Analysis
HaCat cells were detached with PBS, 1 mm EDTA, and washed once with FACS buffer (PBS, 1% FCS). Staining was performed in 96-well plates with 0.5 × 106 to 1 × 106 cells/well, using 10 μl of Fc-PS-EDA1-E245 Opti-MEM supernatant and 40 μl of FACS buffer, for 20 min at 4 °C. In some instances, FACS buffer was replaced by 20-fold concentrated Opti-MEM supernatant containing the soluble receptors EDAR-COMP-Flag or BCMA-COMP-Flag. After washing with FACS buffer, Fc-EDA1 was revealed with phycoerythrin (PE)-coupled anti-human Fc antibodies (Southern Biotech, Birmingham, AL).
293T cells co-transfected with EDAR-GPI (or XEDAR-GPI control) and EGFP, and Jurkat cells transfected with syndecan-1, syndecan-2, or glypican-1 expression constructs, were stained with various Fc-ligands or Flag-ligands as described previously (14).
Intraperitoneal Injections in Newborn Tabby Mice
Tabby mice (Jackson Laboratories, Bar Harbor, ME) were handled according to institutional and Swiss Federal Veterinary Office guidelines, with the authorization of the Office vétérinaire cantonal du canton de Vaud.
Pups were labeled by puncture of a footpad with a 30-gauge needle dipped in Aramis tattoo ink (Braintree Scientific, Braintree, MA). Intraperitoneal injections of EDA1 proteins were performed within 24 h after birth with a maximal volume of 15 μl using 0.5 ml of U-100 insulin syringe (Becton Dickinson, Franklin Lakes, NJ). When two injections were required, Fc-EDA1 and EDAR-Fc were each administered in 7.5 μl. Examination and photography of tail hairs were performed 3–4 weeks postinjection.
Genotyping
TNF−/− mice (17) were kindly provided by Dr. Fabienne Tacchini-Cottier. The following pairs of oligonucleotides were used for genotyping: TNF−/−: 5′-AGGGCTGTGGGACCTAAATGTC-3′ and 5′-TTTGAAGCGTGCGAGAATGC-3′. TNF+/+: 5′-AGGGCTGTGGGACCTAAATGTC-3′ and 5′-TTTGAGTTCTTGGAGGAAGTGGC-3′.
RESULTS
HaCat Cells Express EDAR and Activate NF-κB in Response to EDA1 Stimulation
HaCat cells have been shown previously to express messenger RNA for EDAR (18). We have used a fusion protein, in which the receptor-binding portion of EDA1 was fused to the Fc portion of human immunoglobulin G1 (Fc-EDA1) (12), as a probe to detect surface expression of EDAR in HaCat cells (Fig. 2A). As expected for a specific interaction, this staining was dose-dependent (supplemental Fig. S1) and could be competed by preincubation of Fc-EDA1 with a soluble form of recombinant EDAR, but not by preincubation with an irrelevant receptor (Fig. 2A). We conclude that HaCat cells express endogenous EDAR at the cell surface.
FIGURE 2.

EDA1 activates NF-κB in HaCat keratinocytes. Panel A, HaCat keratinocytes were stained with Fc-EDA1-E245 (EDA1), in the presence or absence of EDAR-COMP-Flag (sol. EDAR) or BCMA-COMP-Flag (sol. BCMA), and bound Fc-EDA1 was monitored by FACS. Panel B, HaCat cells were stimulated with 200 ng/ml of Fc-TNF (TNF) or 200 ng/ml of Fc-EDA1-E245 (EDA1), for the indicated period of time. Cell extracts were analyzed by Western blotting for the presence of total IκBα (IκBα), phosphorylated IκBα (P-IκBα), or the loading control tubulin.
To determine whether EDA1 or TNF could activate NF-κB signaling in HaCat cells, the phosphorylation and degradation of the NF-κB inhibitor IκBα (19) was assessed following EDAR or TNF receptor engagement. HaCat cells were starved in serum-free medium to down-regulate spontaneous NF-κB activation, and subsequently stimulated with TNF as a positive control for NF-κB activation. TNF induced robust phosphorylation of IκBα within 5 min. This event was followed by the degradation of IκBα, as observed after 15 and 30 min, and its resynthesis after 1 and 4 h (Fig. 2B). Discrete upper bands of phospho-IκBα detected at 5 min most probably represent ubiquitin derivatives of IκBα that are targeted to degradation (19). Compared with untreated control cells, biologically active Fc-EDA1 (12) also induced some IκBα phosphorylation, although the response was weaker and slower than with TNF, and the disappearance of IκBα was at best partial at later time points (Fig. 2B). These results indicate that HaCat cell can respond to biologically active EDA1.
EDAR Does Not Signal Cell Death in HaCat Cells and Does Not Rescue Putative TNF-induced Cell Death during Embryonic Development
Mouse embryos with impaired NF-κB signaling suffer from massive liver damage and die at around gestational day 15, but can be rescued by TNF or TNF-R1 inactivation, indicating that TNF is the cause of liver damage (20, 21). When NF-κB is compromised, TNF-R1 signals apoptosis through a death domain-dependent secondary signaling complex (22). As EDAR resembles TNF-R1 not only by its ability to activate NF-κB, but also by the presence of an intracellular death domain, we wondered whether EDA1 could negatively affect viability of HaCat cells with compromised NF-κB signaling. HaCat cells expressing a super-repressor of NF-κB (non-degradable IκBα) died as expected in response to TNF, but remained completely resistant to EDA1, suggesting that EDAR does not transmit death signals similar to TNF-R1 (supplemental Fig. S2A).
Nevertheless, cells involved in early placode development could be killed in a TNF-dependent manner in the absence of protective, EDAR-mediated NF-κB signals, as observed for fetal liver cells in response to TNF (20, 21). In this scenario, EDA deficiency should be rescued by concomitant inactivation of TNF. This was however experimentally not the case, as EDA-deficient mice expressing TNF or not both displayed identical phenotype of ectodermal dysplasia (supplemental Fig. S2B).
The TNF Homology Domain of EDA1 Is a Poor NF-κB Agonist Compared with Longer Forms of EDA1
Although the THD of EDA1 (EDA-E245) is both required and sufficient to bind EDAR (11) (supplemental Fig. S3B), high concentration of this ligand was required to induce only a weak NF-κB signal in HaCat cells (Fig. 3A). In contrast, a longer form of EDA1 starting at the furin cleavage site (EDA1-S160) and comprising the collagen domain in addition to the THD was estimated to be at least 100-fold more potent than EDA1-E245 in this NF-κB assay (Fig. 3A and supplemental Fig. S3A). The same was true for naturally processed EDA1 (Fig. 3B). Of note, EDA1-S160 constructs with in-frame deletions in the collagen domain (Δ185–196 and Δ218–223) were as efficient as the wild-type protein in this assay (Fig. 3, A and B). These results indicate that the presence of the collagen domain increases the biological activity of EDA1 about 100-fold, regardless of whether or not it contains short in-frame deletions.
FIGURE 3.

The collagen domain confers signaling capacity to the TNF domain of EDA1. HaCat cells were stimulated for 20 min with various EDA1 constructs, and levels of phospho-IκBα, IκBα and tubulin were visualized by Western blotting. Panel A, cells were stimulated with the indicated EDA1 proteins, which were obtained from Fc-PreScission-EDA1 that had been cleaved with PreScission protease (see supplemental Fig. S3). Panel B, cells were stimulated with supernatants of 293T cells transfected with full-length EDA1, Flag-tagged soluble EDA1, and deletion mutants thereof (see supplemental Fig. S3). The stimulation was performed in the presence or absence of anti-Flag antibody. Anti-Flag alone had no effect in this assay (data not shown). Panel C, cells were stimulated with Fc-EDA1-E245 or Fc-PreScission-EDA1-E245 that had been treated or not with PreScission protease (see supplemental Fig. S4).
Cross-linking Antibodies Can Functionally Replace the Collagen Domain of EDA
Results presented above indicate that the collagen domain of EDA1 is important for the activation of NF-κB in HaCat cells, despite the fact that it is not required for receptor binding. This suggests that the collagen domain may activate EDA1 by oligomerization of THD trimers, but alternative, non-mutually exclusive hypotheses such as recruitment of interaction partners or engagement of co-receptors via the collagen domain could also account for these observations.
Like EDA1-E245, Flag-EDA1-E245 failed to activate NF-κB in HaCat cells. However, Flag-EDA1-E245 could activate NF-κB when cross-linked with anti-Flag antibodies (Fig. 3B and supplemental Fig. S3B). Flag-tagged forms of EDA1-S160, with or without in-frame deletions in the collagen domain, were constitutively active and, as expected, remained active in the presence of cross-linking antibodies (Fig. 3B).
The Fc Portion of IgG1 Can Functionally Replace the Collagen Domain of EDA
EDA1 constructs were generated in which the collagen domain was replaced with the Fc portion of human IgG1, with or without insertion of a PreScission viral protease site between the Fc and THD domains. Fusion of the dimeric Fc fragment to trimeric EDA1 is predicted to yield a hexameric protein containing 3 Fc moieties and 2 EDA1 trimers moieties, or even higher oligomers (supplemental Fig. S3A, scheme). Fc-EDA1-E245 indeed eluted with high apparent molecular weight by size exclusion chromatography, consistent with an oligomeric form of the protein (supplemental Fig. S4B), whereas treatment of the protease-sensitive protein with PreScission protease resulted in a quantitative release of EDA1-E245 that eluted at the position expected for a trimer (supplemental Fig. S4, A and C).
Uncleaved Fc-EDA1 activated NF-κB in HaCat cells regardless of the presence or absence of the protease site (Fig. 3C). This biological effect was lost by physical separation of the Fc domain from the EDA1 domain by cleavage with PreScission protease, but, as expected, the same treatment did not affect the protease-resistant Fc-EDA1 (Fig. 3C).
Taken together, these data are consistent with the notion that the collagen domain renders EDA1 active by oligomerization. This function can be mimicked by other oligomerization means, such as Flag plus anti-Flag or fusion to the Fc portion of IgG1.
The Collagen Domain Potentiates the Ability of EDA1 to Deliver Oligomerization-dependent Signals in an Engineered Cell Death Assay by More than Three Orders of Magnitude
The HaCat assay provided qualitative rather than quantitative information on EDA1 activity and unfortunately lacked robustness due to the relatively weak NF-κB activation. We therefore thought to adapt a well-characterized FasL-dependent cell death assay to the purpose of measuring the functional effect of EDA1 oligomerization. Thus, Fas-deficient Jurkat T cells were transfected with a chimeric receptor consisting of the extracellular domain of EDAR fused to the transmembrane and intracellular domains of Fas, and clones undergoing apoptosis in an EDA1-sensitive manner were selected.
Purified Flag-EDA1-S160, with or without in-frame deletions in the collagen domain, killed Jurkat EDAR:Fas cells with an IC50 of about 1 ng/ml, and was further activated only about 2-fold in the presence of anti-Flag antibodies (Fig. 4 and supplemental Fig. S5). Flag-EDA1-E245 displayed no activity by itself in this assay, but this latency was overcome in the presence of a cross-linking anti-Flag antibody that increased the activity about 1000-fold (Fig. 4). Interestingly, a 3-day preincubation at 50 °C did not affect the activity of Flag-EDA1-S160 but decreased the activity of mutants with in-frame deletions in the collagen domain roughly 10-fold. Inline with the results obtained for NF-κB activation in HaCat cells, we conclude that the collagen domain of EDA1 fulfills a prominent role to stimulate the signaling ability of EDA1 trimers by oligomerization. Although deletions in the collagen domain have no or little impact on the activity of the protein, they decreased heat stability of the protein. Taken together, these in vitro results demonstrate that the collagen domain is a positive regulator of EDA signals.
FIGURE 4.

The collagen domain of EDA1 is required for signaling through an oligomerization-dependent reporter pathway. Jurkat T cell clone stably expressing a chimeric receptor consisting of the extracellular domain of EDAR fused to the transmembrane and intracellular domains of Fas was treated for 16 h with varying amounts of the indicated Flag-EDA1 (see supplemental Fig. S5), in the presence (black symbols) or absence (white symbols) of anti-Flag antibody. Flag-EDA1 was either used directly (squares) or preincubated for 3 days at 50 °C prior to the assay (circles). Cell viability was monitored with the PMS/MTS assay.
EDA Oligomerizes via Its Collagen Domain
To determine the oligomerization status of EDA, its size was estimated by gel permeation chromatography (Fig. 5 and Table 1). The TNF domain only (EDA1-E245) eluted at a size 3.2× that of the monomer, in line with the trimeric structure determined by crystallography (23). EDA1-S160, EDA1-S160 with mutation KKKGKK → SASGAS in front of the collagen domain and ACRP-EDA1-E245 (with the collagen domain of ACRP30) had apparent multiplicities of 10.4, 14.4, and 16.4, respectively, suggesting that they are bigger than trimers. However, size exclusion chromatography overestimates the size of asymmetric proteins, such as a protein containing an elongated collagen domain. Constructs of another TNF family ligand, FasL, were therefore analyzed in parallel. The TNF homology domain of FasL taken alone eluted as expected as a timer. FasL preceded by the collagen domain of EDA1 and ACRP-FasL eluted with apparent multiplicities of 9.7 and 12.7. As ACRP-FasL was previously characterized as a hexamer by electron microscopy (3), it can be concluded that the presence of a collagen domain roughly doubles the molecular weight determined by size exclusion chromatography. When taking into account this correction factor, estimated multiplicities of EDA1-S160 and EDA1-S160 KKKGKK → SASGAS are about 5.2 and 7.2. This is in any case bigger than a trimer and re-enforces the assumption that EDA with a collagen domain (from EDA or ACRP) forms multimers, probably hexamers.
FIGURE 5.
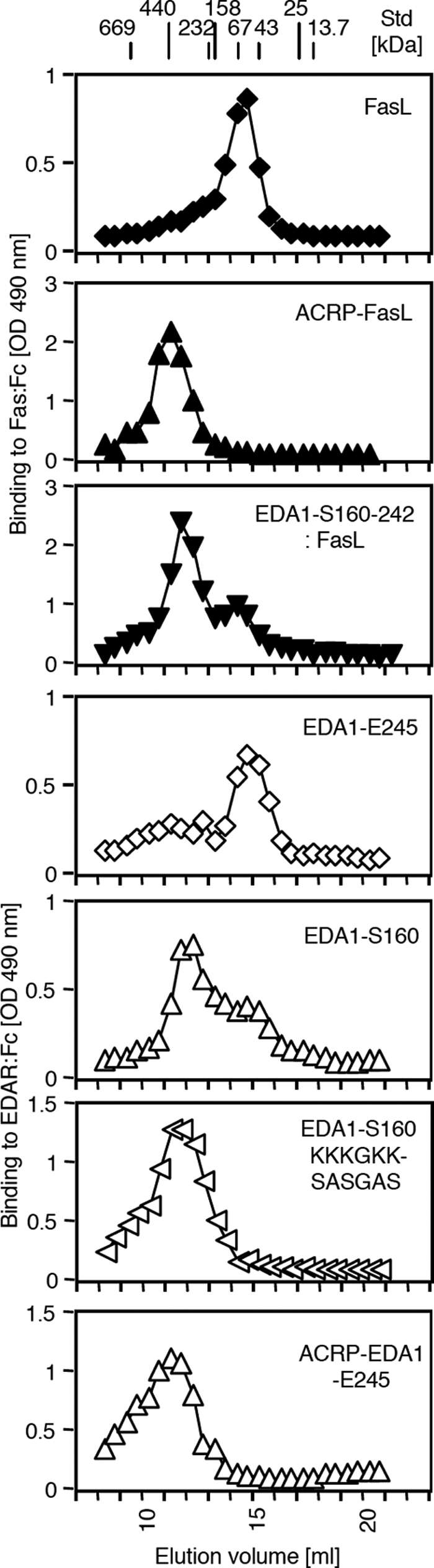
Size exclusion chromatography of EDA1 and FasL with or without collagen domains. The indicated Flag-tagged proteins were fractionated by size exclusion chromatography on a Superdex 200 column. Active ligands were detected in the eluted fractions by means of a receptor-binding ELISA. The elution position of molecular mass markers (in kDa) is indicated at the top of the elution profile. The analysis of the data is shown in Table 1.
TABLE 1.
Sizes of EDA1 and FasL with or without collagen domains by size exclusion chromatograpy
| Ligand | Elution volume | Apparent size | Theoretical monomer molecular massa | Number of globular subunitsb | Estimated number of subunitsc |
|---|---|---|---|---|---|
| ml | kDa | KDa | |||
| FasL | 14.6 | 78 | 25.7 | 3.0 | 3.0 |
| ACRP-FasL | 11.25 | 447 | 35.1 | 12.7 | 6.4 |
| EDA1-S160–242:FasL | 11.85 | 327 | 33.7 | 9.7 | 4.8 |
| EDA1-E245 | 15 | 63 | 19.7 | 3.2 | 3.2 |
| EDA1-S160 | 12 | 302 | 29.1 | 10.4 | 5.2 |
| EDA1-S160 KKKGKK → SASGAS | 11.5 | 392 | 27.3 | 14.4 | 7.2 |
| ACRP-EDA1-E245 | 11.25 | 447 | 27.3 | 16.4 | 8.2 |
a Theoretical molecular mass of the mature polypeptide + 7.5 kDa (for three N-linked glycans of FasL) or + 1.3 kDa (for one N-linked glycan on half of the EDA1).
b Apparent size divided by the theoretical monomer molecular mass.
c Number of globular subunits divided by 2 if the molecule contains a collagen domain.
Less than 4-h Exposure to Active EDA1 Is Sufficient to Induce Tail Hair Formation in Newborn, EDA-deficient Tabby Mice
Tabby mice have no hair on the tail, a defect that can be corrected by ip administration of Fc-EDA1-E245 in newborn mice (12). As expected, tail hair formation induced by a dose of 2 μg (2 mg/kg) Fc-EDA1-E245 was abrogated by co-injection of an excess (37.5 μg) of the decoy receptor EDAR-Fc. Interestingly, delayed administration of EDAR-Fc for up to 1 h prevented tail hair induction, but failed to do so after 3.5 or 18 h (Fig. 6). This demonstrates that less than 4 h exposure to active EDA1 is sufficient to induce tail hair formation and suggests that this experimental system should be relatively insensitive to the half-life of EDA1.
FIGURE 6.

4-h exposure to Fc-EDA1 in newborn Tabby mice is sufficient to induce tail hair formation. Panel A, 1-day-old Tabby mice received ip 2 μg of Fc-EDA1, followed by 37.5 μg of EDAR-Fc at the indicated time point. Pictures of the tail were taken 3–4 weeks later. Panel B, quantification of tail hair induction of the mice shown in Panel A. Mean ± S.D. of three mice per group. Tail hair induction score was determined as described in the legend to Fig. 7.
EDA1 S160 Is Less Active than EDA1 E245 in Vivo, in Contrast to in Vitro Results
From the combined in vitro results, we predicted that EDA-S160 and EDA-E245 would be, respectively, active and inactive when administered in vivo. To test this hypothesis, newborn Tabby mice were treated ip with increasing doses of EDA1-E245 or EDA1-S160, and examined 3–4 weeks later for the presence of hair on the tail. To our surprise, Flag-EDA1-E245 was relatively efficient at inducing development of tail hair in this setting, whereas Flag-EDA1-S160 only induced few tail hairs, even at the highest dose that we could test (6 mg/kg) (Fig. 7 and data not shown). This suggests first that EDAR signaling in vivo is possibly less sensitive to the oligomerization status of the ligand than expected from in vitro result and, second, that Flag-EDA1-S160 may not freely gain access to the receptor to initiate signaling.
FIGURE 7.

Differential effects of the collagen domain and HSPG-binding domain of EDA1 in vitro and in vivo. Panel A, Jurkat EDAR:Fas cells were treated with different Flag-EDA1 at the indicated concentration as described in the legend to Fig. 4. Panel B, EDA-deficient Tabby mice were treated ip at day 1 after birth with the indicated dose of the same Flag-EDA1 proteins used in panel A. Presence of hair on the tails was scored 3–4 weeks later (Score 0: no hair, like Tabby; 0.5: very few hair; 1: sparse hair on ventral side; 2: numerous hair on ventral side; 3: numerous hair on both sides; 4: dense hair on both sides, like wild type). Pictures of the tail of three mice per group treated at the dose of 2.4 mg/kg are shown on the right. The ventral and dorsal sides of the tail are facing left and right, respectively.
EDA1 Contains an HSPG-binding Domain
We noticed the presence of a basic stretch of amino acid residues at the N terminus of the collagen domain, which is conserved in EDA of mammals, birds, frogs, and even fishes (Fig. 8A). We wondered whether this sequence could mediate binding to proteoglycans, as shown previously to be the case for APRIL, another TNF family member (24). Indeed, Flag-EDA1-S160 that contains the basic region, but not Flag-EDA1-E245 that lacks this region (and the entire collagen domain), bound to proteoglycan-positive 293T cells in a heparin-sensitive manner (Fig. 8B). This binding was unlikely to be mediated by the collagen domain itself, because in-frame deletions in the collagen domain did not affect the binding, and because addition of an heterologous collagen domain to the THD of EDA1 (ACRP-EDA1) did not confer binding to 293T cells (Fig. 8B). These observations were confirmed using Fc-EDA1 constructs, all of which bound surface-expressed EDAR, but not the EDA2-specific receptor XEDAR (25). Heparin-sensitive binding of Fc-EDA1-S160 to 293T cells was still observed after complete deletion of the collagen domain (Δ181–234), but was completely abolished by mutation of the conserved lysine residues of the basic stretch (KKKGKK → SASGAS) (Fig. 8C). Moreover, a perfect correlation was observed between the presence of the basic sequence, and the ability of EDA1 to interact with heparin (Fig. 8D). Finally and similarly to APRIL, EDA1-S160 that contains the basic sequence, but not EDA1-E245 in which this region is absent or EDA1-S160 KKKGKK → SASGAS in which the basic site was mutated, interacted specifically with Jurkat cells transfected with syndecan1, syndecan2, or glypican1, but not with mock-transfected cells, in a heparin-inhibitable manner (supplemental Fig. S6). These results indicate that the basic region of EDA1 mediates binding to heparin and proteoglycans.
FIGURE 8.

EDA1 contains a heparan sulfate proteoglycan binding sequence in front of the collagen domain. Panel A, sequence alignment around the mature N terminus of EDA1. Conserved residues are highlighted. The basic sequence responsible for HSPG binding, and the collagen domain are indicated, furin consensus sites are boxed, and the furin cleavage site (of human EDA) is shown with an arrowhead. Species analyzed are Homo sapiens, Bos taurus, Canis familiaris, Mus musculus, Gallus gallus, Xenopus tropicalis, and Gasterosteus aculeatus. Panel B, untransfected, EDAR-negative 293T cells were stained with the indicated Flag-tagged versions of EDA1 in the presence or absence of heparin, and analyzed by FACS. Panel C, 293T cells were transfected with GPI-anchored extracellular domains of EDAR or XEDAR, together with an EGFP tracer, and then stained with the indicated Fc-tagged versions of EDA1, in the presence or absence of heparin. Cells were analyzed by 2-color FACS. Panel D, indicated Flag-tagged EDA1 constructs were pulled down with either EDAR-Fc (upper panel) or with heparin-Sepharose (middle panel), and detected by Western blot with an anti-Flag antibody. Immunoprecipitated EDAR-Fc was revealed with an anti-human IgG antibody (lower panel). All EDA1 constructs bound to EDAR-Fc, but only those with an intact proteoglycan-binding region interacted with heparin.
The HSPG-binding Domain of EDA1 Attenuates Its Activity in Vivo
We wondered whether the poor activity of EDA1-S160 in vivo after ip administration could be explained by its interaction with ubiquitously expressed HSPGs that would sequester EDA1 away from developing skin. We found that EDA1-S160 with a mutated HSPG binding site (KKKGKK → SASGAS) induced numerous tail hair in Tabby mice very similar in density to those of a wild-type mouse. Such a phenotype was never achieved with wild-type EDA1-S160, containing the HSPG-binding site, and was also stronger than that obtained with EDA1-E245. The THD of EDA1 fused to the collagen domain of ACRP (ACRP-EDA1) or to the Fc domain of an immunoglobulin displayed in vivo activities similar to EDA1-S160 with mutated HSPG-binding site (Fig. 7 and supplemental Fig. S5). It is noteworthy that mutation of the HSPG-binding domain reduced both heat stability and activity of EDA1 on Jurkat EDAR:Fas cells, suggesting that this region also contributes to the oligomerization and stability of EDA1 (Figs. 4 and 7). Taken together, these data are inline with the hypothesis that the HSPG-binding site of EDA1-S160 restricts its distribution in the organism.
DISCUSSION
About one in four mutations affecting EDA in XLHED patients destroys the furin-processing site, indicating that EDA must be cleaved in order to exert its activity (11, 26). A similar proportion of patients display point mutations or in-frame deletions in the collagen domain, pointing to its essential but poorly characterized function.
We demonstrate that the collagen domain of EDA functionally acts as a cross-linker of the receptor-binding, trimeric THD domain. This cross-linking function of the collagen domain can be mimicked in several ways: antibody-mediated cross-linking, fusion with the Fc portion of IgG1 or fusion with the collagen domain of ACRP/adiponectin. All of these means rendered the ligand as much as 1000-fold more active than the THD alone in a surrogate assay measuring activation of the oligomerization-dependent Fas pathway. Size estimation of the EDA1 proteins suggests that the collagen domain may serve as a scaffold to bring at least two trimeric TNF homology domains into the same molecule.
Some TNF family ligands such as TNF or TWEAK are active as soluble trimeric ligands that do not require further oligomerization to signal through (at least one of) their receptors. In contrast, membrane-bound ligands such as FasL or CD40L are inactive in a soluble form, unless oligomerized to meet the requirements of their oligomerization-dependent receptors (2, 3, 27, 28). EDA appears be a “mixture” of both cases: although it is released in a soluble form, it can nevertheless activate its oligomerization-dependent receptor because of a built-in oligomerization domain.
Inline with the hypothesis that the signaling pathway downstream of EDAR is oligomerization-dependent, activation of NF-κB in EDAR-positive HaCat cells required the collagen domain of EDA1, and oligomerization of EDA1 was also required for its full activity when administered in newborn Tabby mice. However, although trimeric EDA1 was essentially inactive in in vitro assays, it displayed some activity in vivo, suggesting that the requirement of EDAR for oligomerized EDA1 in the context of hair development induced by a recombinant ligand may not be as stringent as initially thought.
It only takes a few hours of exposure to EDA to induce hair (and sweat gland, data not shown) formation. However, as different ectodermal appendages can develop at distinct time points, agonists with longer half-lives should be able to induce development of a wider spectrum of structures. In this respect, agonist anti-EDAR antibodies may prove to be particularly useful.
A short stretch of basic amino acids, mostly encoded by the 23 bp exon 4 of EDA, precedes the collagen domain. It is striking that this feature, like the furin site and the collagen domain, is evolutionarily conserved between mammals, birds, amphibians, and fishes. We demonstrate here that this sequence is an HSPG-binding site that impairs in vivo activity of recombinant EDA. For exogenously added EDA, this domain probably results in the scavenging of EDA before it reaches its target organ, an effect that can be partially overcome by increasing the dose. For endogenous EDA, HSPG binding probably restricts diffusion and fine tunes its effects. HSPG binding generally allows to establish concentration gradients for proteins such as chemokines that coordinate immune responses (29), or morphogens of the Wnt, TGF-β, Hedgehog, and FGF families of ligands (30–33). We know that neither the HSPG-binding domain nor the collagen domain of EDA1 are absolutely required to signal through EDAR in vivo, as both can be replaced by the Fc portion of an IgG. However, even high dose treatments with recombinant (12) or transgenic EDA (34, 35) failed to rescue subtle phenotypes such as kinks in zigzag hairs, for which the dose and spatio-temporal distribution of EDA1 is likely crucial (36) and could well be regulated by the HSPG-binding domain. If HSPG binding is functionally relevant, one can wonder why it was never found mutated in XLHED patients. First, exon 4 cannot be deleted in-frame. Second, mis-sense mutation of just one of the basic residues is probably not sufficient to completely abolish HSPG binding, and may not translate into a phenotype severe enough to be noticed. Alternatively, decreased binding to HSPG may promote EDA signaling rather than hampering it, by allowing an easier access to the receptor. Interestingly, mutation of the basic site not only abolished proteoglycan binding but also decreased heat stability, suggesting that the basic sequence may also help stabilizing the multimeric structure of EDA1.
In-frame deletions in the collagen domain that deactivate EDA in XLHED patients have apparently no impact on the cross-linking potential of the collagen domain. Apart from the expected slight reduction of MW by SDS-PAGE, these in-frame deletions affected neither expression levels upon transient transfection of the full-length or soluble proteins, nor proteolytic processing, nor gel filtration elution profiles, nor the activity in vitro, nor proteoglycan binding. It did however display a modest effect on heat stability of the purified proteins. It is uncertain whether this partial stability defect is sufficient to explain why patients harboring these mutations are as severely affected as those with non-sense mutations resulting in total loss of EDA1 (11). Endogenous levels of EDA1 are probably limiting, and a decrease of EDA1 protein levels as a result of in-frame deletions in the collagen domain, which could be due to impaired binding to an unidentified cofactor or to impaired stability of the protein, may severely compromise formation of ectodermal appendages. Recombinant proteins lacking this domain are probably active because they saturate the system, thus masking subtle regulatory mechanisms.
These findings on the collagen and HSPG-binding domains of EDA1 are relevant to the choice of recombinant EDA that may potentially be used for the early treatment of XL-HED: so far, Fc-EDA1 remains the best agonist tested in vivo, probably because of a numbers of reasons. Indeed, the Fc favorably impacts the protein half-life in addition to providing the protein with the potential to be carried through maternal Fc-receptors. Also, the Fc provides the necessary cross-linking and alleviates the potentially deleterious effect of the HSPG-binding domain. Finally, Fc-EDA1 is easier to produce than proteins containing the collagen domain of EDA.
Supplementary Material
Acknowledgments
We thank Fabienne Tacchini-Cottier (WHO, Epalinges, Switzerland) for providing TNF−/− mice, Olivier Micheau (University of Dijon) for the gift of Jurkat-JOM2 cells, Riikka Santalahti and Raija Savolainen (Institute of Biotechnology, University of Helsinki) for excellent technical assistance, and Jürg Tschopp for continuous support and helpful discussions.
This work was supported by grants from the Swiss National Science Foundation and the NCCR Molecular Oncology.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6 and Table S1.
- TNF
- tumor necrosis factor
- EDA1
- ectodysplasin A1
- EDAR
- EDA receptor
- XEDAR
- X-linked EDAR
- THD
- TNF homology domain
- HSPG
- heparan sulfate proteoglycan
- DMEM
- Dulbecco's modified Eagle's medium
- PBS
- phosphate-buffered saline
- FACS
- fluorescent-activated cell sorting
- HSPG
- heparan sulfate proteoglycan
- FCS
- fetal calf serum.
REFERENCES
- 1.Bodmer J. L., Schneider P., Tschopp J. (2002) Trends Biochem. Sci 27, 19–26 [DOI] [PubMed] [Google Scholar]
- 2.Schneider P., Holler N., Bodmer J. L., Hahne M., Frei K., Fontana A., Tschopp J. (1998) J. Exp. Med. 187, 1205–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holler N., Tardivel A., Kovacsovics-Bankowski M., Hertig S., Gaide O., Martinon F., Tinel A., Deperthes D., Calderara S., Schulthess T., Engel J., Schneider P., Tschopp J. (2003) Mol. Cell. Biol. 23, 1428–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui C. Y., Schlessinger D. (2006) Cell Cycle 5, 2477–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mikkola M. L. (2008) Cytokine Growth Factor Rev. 19, 219–230 [DOI] [PubMed] [Google Scholar]
- 6.Schmidt-Ullrich R., Aebischer T., Hülsken J., Birchmeier W., Klemm U., Scheidereit C. (2001) Development 128, 3843–3853 [DOI] [PubMed] [Google Scholar]
- 7.Schmidt-Ullrich R., Tobin D. J., Lenhard D., Schneider P., Paus R., Scheidereit C. (2006) Development 133, 1045–1057 [DOI] [PubMed] [Google Scholar]
- 8.Mou C., Jackson B., Schneider P., Overbeek P. A., Headon D. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 9075–9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pummila M., Fliniaux I., Jaatinen R., James M. J., Laurikkala J., Schneider P., Thesleff I., Mikkola M. L. (2007) Development 134, 117–125 [DOI] [PubMed] [Google Scholar]
- 10.Monreal A. W., Zonana J., Ferguson B. (1998) Am. J. Hum. Genet. 63, 380–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider P., Street S. L., Gaide O., Hertig S., Tardivel A., Tschopp J., Runkel L., Alevizopoulos K., Ferguson B. M., Zonana J. (2001) J. Biol. Chem. 276, 18819–18827 [DOI] [PubMed] [Google Scholar]
- 12.Gaide O., Schneider P. (2003) Nat. Med. 9, 614–618 [DOI] [PubMed] [Google Scholar]
- 13.Soneoka Y., Cannon P. M., Ramsdale E. E., Griffiths J. C., Romano G., Kingsman S. M., Kingsman A. J. (1995) Nucleic Acids Res. 23, 628–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bossen C., Ingold K., Tardivel A., Bodmer J. L., Gaide O., Hertig S., Ambrose C., Tschopp J., Schneider P. (2006) J. Biol. Chem. 281, 13964–13971 [DOI] [PubMed] [Google Scholar]
- 15.Schneider P. (2000) Methods Enzymol. 322, 322–345 [DOI] [PubMed] [Google Scholar]
- 16.Holler N., Kataoka T., Bodmer J. L., Romero P., Romero J., Deperthes D., Engel J., Tschopp J., Schneider P. (2000) J. Immunol. Methods 237, 159–173 [DOI] [PubMed] [Google Scholar]
- 17.Marino M. W., Dunn A., Grail D., Inglese M., Noguchi Y., Richards E., Jungbluth A., Wada H., Moore M., Williamson B., Basu S., Old L. J. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 8093–8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Headon D. J., Overbeek P. A. (1999) Nat. Genet. 22, 370–374 [DOI] [PubMed] [Google Scholar]
- 19.Karin M., Ben-Neriah Y. (2000) Annu. Rev. Immunol. 18, 621–663 [DOI] [PubMed] [Google Scholar]
- 20.Alcamo E., Mizgerd J. P., Horwitz B. H., Bronson R., Beg A. A., Scott M., Doerschuk C. M., Hynes R. O., Baltimore D. (2001) J. Immunol. 167, 1592–1600 [DOI] [PubMed] [Google Scholar]
- 21.Beg A. A., Sha W. C., Bronson R. T., Ghosh S., Baltimore D. (1995) Nature 376, 167–170 [DOI] [PubMed] [Google Scholar]
- 22.Micheau O., Tschopp J. (2003) Cell 114, 181–190 [DOI] [PubMed] [Google Scholar]
- 23.Hymowitz S. G., Compaan D. M., Yan M., Wallweber H. J., Dixit V. M., Starovasnik M. A., de Vos A. M. (2003) Structure 11, 1513–1520 [DOI] [PubMed] [Google Scholar]
- 24.Ingold K., Zumsteg A., Tardivel A., Huard B., Steiner Q. G., Cachero T. G., Qiang F., Gorelik L., Kalled S. L., Acha-Orbea H., Rennert P. D., Tschopp J., Schneider P. (2005) J. Exp. Med. 201, 1375–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan M., Wang L. C., Hymowitz S. G., Schilbach S., Lee J., Goddard A., de Vos A. M., Gao W. Q., Dixit V. M. (2000) Science 290, 523–527 [DOI] [PubMed] [Google Scholar]
- 26.Chen Y., Molloy S. S., Thomas L., Gambee J., Bächinger H. P., Ferguson B., Zonana J., Thomas G., Morris N. P. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 7218–7223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haswell L. E., Glennie M. J., Al-Shamkhani A. (2001) Eur. J. Immunol. 31, 3094–3100 [DOI] [PubMed] [Google Scholar]
- 28.Grell M., Douni E., Wajant H., Löhden M., Clauss M., Maxeiner B., Georgopoulos S., Lesslauer W., Kollias G., Pfizenmaier K., Scheurich P. (1995) Cell 83, 793–802 [DOI] [PubMed] [Google Scholar]
- 29.Lortat-Jacob H., Grosdidier A., Imberty A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 1229–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson R. A., Nurcombe V., Cool S. M. (2006) Gene 379, 79–91 [DOI] [PubMed] [Google Scholar]
- 31.Belenkaya T. Y., Han C., Yan D., Opoka R. J., Khodoun M., Liu H., Lin X. (2004) Cell 119, 231–244 [DOI] [PubMed] [Google Scholar]
- 32.Franch-Marro X., Marchand O., Piddini E., Ricardo S., Alexandre C., Vincent J. P. (2005) Development 132, 659–666 [DOI] [PubMed] [Google Scholar]
- 33.Han C., Belenkaya T. Y., Wang B., Lin X. (2004) Development 131, 601–611 [DOI] [PubMed] [Google Scholar]
- 34.Srivastava A. K., Durmowicz M. C., Hartung A. J., Hudson J., Ouzts L. V., Donovan D. M., Cui C. Y., Schlessinger D. (2001) Hum. Mol. Genet 10, 2973–2981 [DOI] [PubMed] [Google Scholar]
- 35.Mustonen T., Pispa J., Mikkola M. L., Pummila M., Kangas A. T., Pakkasjärvi L., Jaatinen R., Thesleff I. (2003) Dev. Biol. 259, 123–136 [DOI] [PubMed] [Google Scholar]
- 36.Hammerschmidt B., Schlake T. (2007) Dev. Biol. 305, 246–261 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.