

Abstract
Salmonella enterica encodes two virulence-related type III secretion systems in Salmonella pathogenicity islands 1 and 2, respectively. These systems mediate the translocation of protein effectors into the eukaryotic host cell, where they alter cell signaling and manipulate host cell functions. However, the precise role of most effectors remains unknown. Using a genetic screen, we identified the small, reduction/oxidation-regulatory protein thioredoxin as a mammalian binding partner of the Salmonella effector SlrP. The interaction was confirmed by affinity chromatography and coimmunoprecipitation. In vitro, SlrP was able to mediate ubiquitination of ubiquitin and thioredoxin. A Cys residue conserved in other effectors of the same family that also possess E3 ubiquitin ligase activity was essential for this catalytic function. Stable expression of SlrP in HeLa cells resulted in a significant decrease of thioredoxin activity and in an increase of cell death. The physiological significance of these results was strengthened by the finding that Salmonella was able to trigger cell death and inhibit thioredoxin activity in HeLa cells several hours post-infection. This study assigns a functional role to the Salmonella effector SlrP as a binding partner and an E3 ubiquitin ligase for mammalian thioredoxin.
Protein secretion is a basic function in all groups of bacteria. Many secretion systems have been found in Gram-negative bacteria, from the relatively simple type I secretion systems to the complex type III or type IV machines or the recently described type VI systems (reviewed in Refs. 1 and 2). Many pathogenic or symbiotic Gram-negative bacteria rely on type III secretion systems (T3SS)2 for their interaction with host organisms. The T3SS is a protein export pathway that spans the inner membrane, periplasmic space, outer membrane, and host cell membrane. These complex structures are related to flagella and consist of at least 20 different subunits that enable the bacteria to translocate substrates into the cytosol of the eukaryotic host cell (reviewed in Ref. 3). These systems have also been referred to as injectisomes or molecular needles (4).
Proteins secreted and translocated into eukaryotic cells through T3SS are called “effectors.” These effectors have the ability to suppress host defense signaling. Effectors of plant pathogens may target salicylic acid- and abscisic acid-dependent defenses, host vesicle trafficking, or interfere with host RNA metabolism. Effectors from animal pathogens modify the cytoskeleton to facilitate bacterial entry, modulate Rho GTPases and NF-κB, inhibit the host inflammatory response, elicit death of immune cells, and disrupt both adaptative and innate immune responses (reviewed in Ref. 5).
Salmonella enterica produces two distinct T3SS essential for virulence that are encoded by genes located in Salmonella pathogenicity islands 1 and 2 (SPI-1 and SPI-2), respectively. The SPI-1 T3SS secretes at least 16 proteins: AvrA, GogB, SipA, SipB, SipC, SipD, SlrP, SopA, SopB/SigD, SopD, SopE, SopE2, SptP, SspH1, SteA, and SteB (6–8). Six of them have been shown to regulate actin cytoskeleton dynamics (reviewed in Ref. 9). 19 SPI-2 T3SS effectors have been identified: GogB, PipB, PipB2, SifA, SifB, SopD2, SseF, SlrP, SseG, SseI/SrfH, SseJ, SseK1, SseK2, SseL, SspH1, SspH2, SteA, SteB, and SteC. However, the biochemical functions of most of them remain unknown (reviewed in Ref. 10).
The conventional paradigm, supported by in vivo and in vitro studies, is that the SPI-1-encoded T3SS is required for the invasion of M cells and cultured epithelial cells (11, 12) as well as for the inflammatory response of the intestinal cells, and that the SPI-2-encoded T3SS is essential for replication and survival within macrophages and the progression of a systemic infection (13). Recent evidence suggests that the boundaries between SPI-1 and SPI-2 function are not sharply defined: some SPI-1 effectors are detected hours or days after infection and SPI-2-encoded genes may be expressed before penetration of the intestinal epithelium. In addition, as can be noticed comparing the lists of effectors above, some effectors, including SlrP, can be secreted by both T3SS.
SlrP (for Salmonella leucine-rich repeat protein) was identified as a S. enterica serovar Typhimurium host range factor by signature-tagged mutagenesis (14). A mutant in this gene has no difference in virulence with the wild-type strain when infecting calves but is 6-fold attenuated for mouse virulence after oral infection. This gene is located in a 2.9-kb DNA region with features of horizontal acquisition and has similarity to yopM from Yersinia spp. and ipaH from Shigella flexneri. The predicted protein contains 10 copies of a leucine-rich repeat signature, a protein motif frequently involved in protein-protein interactions. Other members of the leucine-rich repeat family in Salmonella are the effectors SspH1 and SspH2, which share 39 and 38% amino acid identity with SlrP, respectively. Similarity in the amino-terminal region of these three proteins helped to define a translocation signal that was also found in four other T3SS effectors: SifA, SifB, SseI, and SseJ (15). Although SlrP can be delivered into mammalian cells by both T3SS, its expression seems to be induced by RtsA, one of the main regulators of SPI-1, independently of HilA or InvF (16).
Although the function of SlrP was completely unknown, the presence of leucine-rich repeats in this protein suggested that it may bind eukaryotic proteins during infection. In addition, recent reports have shown an enzymatic activity, E3 ubiquitin ligase, for effectors of the same family (17, 18).
In this work we demonstrate that SlrP interacts with mammalian thioredoxin-1 (Trx). We also show that SlrP is an E3 ubiquitin ligase that can use Trx as a substrate. Our results support a model in which interaction of SlrP with Trx leads to a decrease in thioredoxin activity and triggers host cell death.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Yeast Strains, and Plasmids
Plasmids and microbial strains used in this study are listed and described in Table 1.
TABLE 1.
Bacterial strains, yeast strains, and plasmids used in this study
| Strain/plasmid | Relevant characteristics | Source/Ref. |
|---|---|---|
| E. coli | ||
| DH5α | supE44 Δ lacU169 (Ø80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 61 |
| BL21(DE3) | F−ompT gal dcm lon hsdSB (rB− mB−; E. coli B strain), with DE3, a λ prophage carrying the T7 RNA pol gene | Stratagene |
| S. enterica | ||
| 14028 | Wild-type | ATCC |
| SV5193 | 14028 slrP ::3×FLAG, Kmr | This study |
| S. cerevisiae | ||
| AH109 | MATa, trp1–901, leu2–3, 112, ura3–52, his3–200, gal4Δ, gal80 Δ, LYS2 :: GAL1UAS-GAL1TATA-HIS3, GAL2UAS-GAL2TATA-ADE2 URA3 :: MEL1UAS-MEL1TATA-LacZ MEL1 | Holtza |
| Y187 | MATα, ura3–52, his3–200, ade2–101, trp1–901, leu2–3, 112, gal4Δ, gal80 Δ, met−, URA3 ::GAL1UAS-GAL1TATA-LacZ MEL1 | 62 |
| Plasmids | ||
| p cDNA3 | Transient or stable transfection vector, Ampr | Invitrogen |
| pCS2+HA3 | Transient transfection vector, Ampr | F. Romero |
| pGADT7 | GAL4-(768–881) AD, LEU2, Ampr | Clontech |
| pGBKT7 | GAL4-(1–147) DB, TRP1, Kmr | Clontech |
| pGBKT7–53 | DB control plasmid | Clontech |
| pGBT10 | GAL4-(1–147) DB, TRP1, Ampr | Clontech |
| pGEX-4T-1 | GST fusion vector, Ampr | GE Healthcare |
| pGEX-4T-3 | GST fusion vector, Ampr | GE Healthcare |
| pIZ1623 | pGEX-4T-3-SlrP | This study |
| pIZ1627 | pGBT10-SlrP | This study |
| pIZ1680 | pGADT7-SlrP | This study |
| pIZ1699 | pGADT7-s3 | This study |
| pIZ1700 | pGADT7-s5 | This study |
| pIZ1702 | pGADT7-s14 | This study |
| pIZ1712 | pGEX-4T-1-Trx | This study |
| pIZ1720 | pCS2-SlrP-3×FLAG | This study |
| pIZ1721 | pCS2–3×HA-Trx | This study |
| pIZ1725 | p cDNA3-SlrP-3×FLAG | This study |
| pIZ1727 | pGEX-4T-1-Trx(C32S/C35S) | This study |
| pIZ1757 | pGEX-4T-3-SlrP(C546A) | This study |
| pIZ1780 | p cDNA3-SlrP(C546A)-3×FLAG | This study |
a A. Holtz, unpublished data.
Chromosomal Gene Epitope Tagging
Addition of the 3× FLAG epitope tag at the 3′ end of srlP was carried out as described (19) using primers SlrP-P1Flag and SlrP-P2Flag (Table 2).
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotide/use | Sequence 5′-3′ (restriction sites are underlined) |
|---|---|
| Epitope tagging of SlrP | |
| slrpflag5′ | GAAAAAAGAGGTGAGCTCGCTCATGAGCGCCTACTGGCGAGACTACAAAGACCATGACGG |
| slrpflag3′ | TAAACAGGGCTCTCTCCCTCTTCTGATAAACTGCGTTCAGCATATGAATATCCTCCTTAG |
| Construction of pIZ1623 | |
| slpgst5′ | GTCAGAATTCTATGTTTAATATTACTAATATACAATC |
| slrpgst3′ | TATAGTCGACTTCTGATAAACTGCGTTCAG |
| Construction of pIZ1635 | |
| slrpkt7 | GTCAGAATTCATGTTTAATATTACTAATATACAATC |
| slrpgst3′ | As above |
| Construction of pIZ1720 | |
| slrpbam5′ | CTGAGGATCCACCATGTTTAATATTACTAATATACAATC |
| slrpflagxba3′ | CTGATCTAGATTACTATTTATCGTCGTCATC |
| Mutagenesis of slrP | |
| slrpC546Adir | GATGAATGCAACGATAAGCGCTGAAGATCGGGTCACAC |
| slrpC546Arev | GTGTGACCCGATCTTCAGCGCTTATCGTTGCATTCATC |
| slrP sequencing | |
| slrpnrev | CTAGCAGTCACGCATCCGTTGTAC |
| slrp1701 | GGTTCTGCCTGAAACACTTC |
| slrp1830rev | ACGGACCAGGTTATTGCGAG |
| slrpc | GATGCTGAAAGAGGCGCCTTTG |
| Construction of pIZ1725 | |
| slrPpcdna5′ | GTCAGAATTCGCCGCCACCATGTTTAATATTACTAATATACAATC |
| slrPflagxba3′ | As above |
| 2H clones sequencing | |
| T7 | TAATACGACTCACTATAGGG |
| Construction of pIZ1712 | |
| trx5′ | GTCAGAATTCATGGTGAAGCAGATCGAGAG |
| trx3′ | CTGACTCGAGATAGCCAATGGCTGGTTATG |
| Mutagenesis of trx | |
| trxcsdir | CTCAGCCACGTGGTCTGGGCCCTCCAAAATGATCAAGCC |
| trxcsrev | GGCTTGATCATTTTGGAGGGCCCAGACCACGTGGCTGAG |
| Construction of pIZ1721 | |
| trx5′ | As above |
| trxxba3′ | CTGATCTAGAATAGCCAATGGCTGGTTATG |
DNA Amplification with the Polymerase Chain Reaction and Oligonucleotides
Amplification reactions were carried out in a PerkinElmer Gene-Amp PCR System 2400 (PerkinElmer Life Sciences). The final reaction volumes were 50 μl, and the final concentration of MgCl2 was 1.5 mm. Reagents were used at the following concentrations: dNTPs, 200 μm; primers, 1 μm; and Taq polymerase (ExpandTM High Fidelity PCR System, Roche Diagnostics SL), 1 unit per reaction. The thermal program included the following steps: (i) initial denaturation, 2 min at 94 °C; (ii) 25 cycles of denaturation (94 °C, 30 s), annealing (55 °C, 30 s), and extension (72 °C, 1 to 3 min); and (iii) final incubation at 72 °C for 7 min, to complete extension. Primers are listed in Table 2.
DNA Sequencing and Sequence Analysis
cDNA from positive clones obtained in the two-hybrid screen, PCR constructs, and mutations were sequenced with an automated DNA sequencer (Sistemas Genómicos, Valencia, Spain). Sequence analysis was performed with molecular biology algorithms from the National Center for Biotechnology Information (NCBI) and the European Bioinformatics Institute (EBI).
Bacterial Culture
The standard culture medium for S. enterica and Escherichia coli was LB broth. Solid LB contained a final concentration of 1.5% agar. Antibiotics were used at the following concentrations for kanamycin (Km), 50 μg/ml, and ampicillin, 100 μg/ml.
Yeast Two-hybrid Methods
Plasmids were introduced into Saccharomyces cerevisiae strains using the lithium acetate procedure, as previously described (20). A human HeLa Matchmaker cDNA library (Clontech) pretransformed in S. cerevisiae strain Y187 was screened using yeast mating, according to the manufacturer's instructions. Briefly, S. cerevisiae strain AH109 transformed with pIZ1627 was grown at 30 °C overnight with shaking in yeast drop-out medium lacking tryptophan. The culture was concentrated, mixed with 1 ml of the library in 45 ml of 2× YPDA, containing 2% yeast extract, 4% peptone, 4% glucose, and 0.006% adenine hemisulfate, and incubated at 30 °C overnight with gentle swirling. The mating mixture was plated in SD medium lacking leucine, tryptophan, and histidine (Clontech). Plates were incubated at 30 °C for 8 days and then colonies were patched on the same medium and replica-plated on medium lacking tryptophan, leucine, histidine, and adenine and in medium lacking leucine and tryptophan and supplemented with 5-bromo-4-chloro-3-indolyl-α-d-galactopyranoside (X-α-gal), to check for the expression of the ADE2 and MEL1 reporters, respectively. Positive clones were rescued, tested for specificity using empty pGBT10, and sequenced with primer T7.
Cell Culture, Lysis, and Transfection
HeLa cells were cultured in Dulbecco's modified essential medium supplemented with 10% fetal calf serum, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin were included in the culture media. All cells were maintained in a 5% CO2 humidified atmosphere at 37 °C. For cell lysis, 2 × 107 to 108 cells/ml were incubated at 4 °C in Nonidet P-40 buffer (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 10% glycerol, 1% Nonidet P-40, 1% aprotinin, 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin, and 1 μg/ml leupeptin) or in Triton X-100 buffer (25 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10% glycerol, 0.5% Triton X-100, 2.5 mm EDTA, 1% aprotinin, 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin, and 1 μg/ml leupeptin) for 20 min. The extract was centrifuged at 20,000 × g for 20 min and the supernatant was stored at −80 °C. For transient transfection assays, 2–5 × 106 HeLa cells/assay were resuspended in 200 μl of 15 mm HEPES-buffered serum-containing medium, mixed with 50 μl of 210 mm NaCl containing 5–10 μg of plasmid DNA, and electroporated using a BTX Electrocell Manipulator 600 set at 240 volts, 720 ohm, and 950 microfarads. Cells were processed 24 h after electroporation. For stable transfection, HeLa cells were electroporated with pcDNA3 or pcDNA3 derivatives and selection was started 24 h after electroporation in medium containing 0.5 mg/ml G418.
GST Fusion Proteins, Electrophoresis and Immunoblot
Expression of the GST fusion proteins was induced by the addition of 1 mm isopropyl β-d-thiogalactoside to bacteria containing pGEX-4T-1, pGEX-4T-3, or their derivatives and the fusion proteins were isolated from bacterial lysates by affinity chromatography with glutathione-agarose beads (Sigma). For lysis, bacteria were sonicated in Nonidet P-40 buffer. For some binding experiments, lysates from S. enterica strain SV5193 were incubated for 2 h with fusion protein bound to glutathione-coupled agarose beads. The precipitates were washed six times in Nonidet P-40 buffer followed by SDS-PAGE. The gel was blotted onto a nitrocellulose membrane and probed with anti-FLAG M2 monoclonal antibodies (1:10,000; Sigma). Goat anti-mouse horseradish peroxidase-conjugated antibodies (Bio-Rad) were used as secondary antibodies. Detection was via chemiluminescence procedures (Pierce).
Oxidation of GST-Trx with H2O2
For some binding experiments GST-Trx was treated with an oxidizing reagent according to a previous report (21). Briefly, GST-Trx bound to glutathione-agarose beads was treated with 1 mm H2O2 in Nonidet P-40 buffer for 15 min and then washed 6 times with buffer before incubation with bacterial lysates. As a control to revert oxidation, a part of the H2O2-treated fusion protein was incubated with buffer containing 100 mm DTT for 15 min after washing and before incubation with lysate.
Coimmunoprecipitation Experiments
Lysates from HeLa cells cotransfected with plasmids pIZ1720 and pIZ1721, expressing SlrP-3×FLAG and 3×HA-Trx, respectively, were incubated with protein A/G plus-agarose beads (Santa Cruz Biotechnology) for 1 h at 4 °C. After centrifugation the supernatants were incubated for 2 h with monoclonal anti-FLAG antibodies or preimmune serum, overnight with protein A/G plus-agarose beads, and then centrifuged. The beads were washed five times in Nonidet P-40 buffer. Proteins were eluted and dissolved into Laemmli sample buffer (50 mm Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 0.0005% bromphenol blue) containing 5% β-mercaptoethanol, incubated at 95 °C for 5 min, and subjected to SDS-PAGE. Proteins were transferred to a nitrocellulose filter and probed with anti-FLAG (Sigma) and anti-HA-peroxidase (clone 3F10, Roche).
Mutagenesis
Mutants Cys546 to Ala (C546A) in SlrP and double mutant Cys32 and Cys35 to Ser (C32S/C35S) in Trx were made in the pGEX or pcDNA3 vectors using the “QuikChange® II Site-directed Mutagenesis Kit” (Stratagene). Primers used are listed in Table 2.
In Vitro Ubiquitination Assays
Ubiquitination reactions were performed in a 40-μl mixture containing buffer A (25 mm Tris-HCl, pH 7.5, 50 mm NaCl, 5 mm ATP, 10 mm MgCl2, 0.1 mm DTT), 2 μg of HA-tagged ubiquitin (Boston Biochem), 0.5 μg of human recombinant E1 (Boston Biochem), and 2 μg of E2 (human recombinant UbcH5b from Boston Biochem) in the presence or absence of 1 μg of GST-SlrP, GST-SlrP(C546A), or GST-Trx. Reactions were incubated at 37 °C for 1 h and stopped by the addition of an equal volume of Laemmli sample buffer containing 100 mm DTT. Reaction mixtures were separated by SDS-PAGE, transferred onto a nitrocellulose membrane, and probed with specific antibodies. Some reaction mixtures were immunoprecipitated with anti-Trx antibodies (Santa Cruz Biotechnology) and then submitted to immunoblot analysis.
Trx-reducing Assay
Trx activity assay was performed in 96-well plates with an end point insulin assay (22, 23). For the reactions, cell lysates with 40 μg of total protein were mixed with 3.8 μl of 1.7 mm Trx reductase from rat liver (Sigma), 20 μl of a 1:12 mixture of solution N (40 mg/ml β-NADPH) and solution M (210 mm HEPES, pH 7.6, 790 μm insulin, 20 mm EDTA) and TE (50 mm Tris-HCl, pH 7.6, 20 mm EDTA) to reach a total volume of 50 μl. The reaction solutions without Trx reductase were used as the control, which was subtracted. After the reaction was performed at 37 °C for 30 min, 200 μl of 1 mm 5,5′-dithiobis(nitrobenzoic acid) in 6 m guanidine hydrochloride solution was added to stop the reaction and the absorbance was read at 412 nm.
Cell Death Assays
The percentage of HeLa cell death was determined by measuring lactate dehydrogenase (LDH) release in confluent cultures using the CytoTox 96 nonradioactive cytotoxicity assay kit (Promega), following instructions provided by the manufacturer. Percentage cytotoxicity was calculated as 100 × (experimental LDH release − spontaneous LDH release)/(maximum LDH release − spontaneous LDH release), where spontaneous LDH release was the level detected in a non-confluent HeLa cell culture.
Infections of HeLa Cells with Salmonella
Two days before the infection, 250,000 HeLa cells/well were seeded in 6-well plates (Nunc, Roskilde, Denmark) and grown at 37 °C, 5% CO2 in media without antibiotics. Bacteria were grown overnight in LB broth with 0.3 m NaCl at 37 °C without shaking, and were added to HeLa cells to reach a multiplicity of infection of 50 bacteria/eukaryotic cell. Sixty minutes after the infection, cells were washed twice with phosphate-buffered saline and incubated in fresh Dulbecco's modified essential medium containing 100 μg/ml gentamicin. Sixty minutes later the concentration of gentamicin was lowered to 16 μg/ml and the incubation was continued for an additional 6 or 14 h.
Statistical Analysis
Each Trx activity or percent cell death is the mean of at least three independent experiments. Student's t test was used to compare different strains or conditions. p values of 0.05 or less are considered significant.
RESULTS
A Yeast Two-hybrid Screen Identifies Mammalian Thioredoxin as a Partner for Salmonella SlrP
The initial hypothesis for this work was that SlrP interacts with one or more host proteins after translocation by a T3SS. To identify potential mammalian partners, SlrP from S. enterica serovar Typhimurium strain 14028 was fused to the DNA binding domain (DB) of the yeast Gal4 transcriptional activator and used as bait in the yeast two-hybrid system. This fusion was coexpressed in yeast with a HeLa cDNA library fused to the activation domain (AD) of Gal4. Plasmid pGBT10 (24) was used as a DB vector and 2 × 106 clones were screened. Fifteen colonies were able to grow in synthetic medium lacking histidine, used to select for the interactions. Most of these positive clones also expressed the additional reporter genes ADE2 and MEL1. The candidate AD plasmids were subsequently tested for reporter gene activation with another construct (pGBKT7-p53) and with the empty DB vector. Sequencing of the specific clones revealed that three of them, termed s3, s5, and s14, carried the complete coding sequence for human Trx, although in the wrong reading frame, which could be explained through the production of frameshifted polypeptides (25). To verify the interactions, the complete open reading frame present in clone s3 was cloned in-frame in pGADT7 and pGBT10 and tested for interaction with SlrP expressed in fusion with Gal4 DB from pGBT10 or with Gal4 AD from pGADT7, respectively. As seen in Fig. 1, the interaction detected with the original clones was confirmed with the in-frame construction, as well as the switched vectors.
FIGURE 1.

Salmonella SlrP interacts with human Trx in the yeast two-hybrid system. Diploids were obtained by conjugation between strain AH109, containing derivatives of pGBT10, and strain Y187, containing derivatives of pGADT7, as indicated. The interaction between the two hybrid proteins is shown by growth in the absence of histidine. Empty vectors (pGBT10 or pGADT7) were used as negative controls. DB, fusion with the DNA-binding domain of Gal4. AD, fusion with the activation domain of Gal4. s3, s5, and s14 are three different clones containing the coding sequence of trx obtained in the two-hybrid screen with SlrP as a bait.
Copurification and Coimmunoprecipitation Confirm the Interaction between Trx and SlrP
The result obtained with the two-hybrid system was confirmed by two independent approaches. For the first approach, DNA encoding a 3×FLAG epitope was introduced at the 3′ end of slrP in the chromosome of S. enterica serovar Typhimurium strain 14028 to generate strain SV5193. This strain was then transformed with plasmid pGEX4T-1, expressing GST alone, or with plasmid pIZ1712, a derivative of pGEX4T-1 coding for a GST-Trx fusion protein. GST and GST-Trx were purified from bacterial lysates using glutathione-agarose beads and analyzed by immunoblot with anti-FLAG monoclonal antibodies. As seen in Fig. 2A, SlrP-3×FLAG copurified with GST-Trx but not with GST, showing the specificity of the interaction. Because this interaction can only take place in vivo in the host cell, we decided to investigate if both proteins could coimmunoprecipitate from mammalian cell lysates. HeLa cells were cotransfected with two derivatives of plasmid pCS2, one expressing SlrP-3×FLAG and the other expressing 3×HA-Trx. Lysates were prepared, SlrP-3×FLAG was immunoprecipitated with anti-FLAG antibodies, and the presence of 3×HA-Trx in the immunoprecipitation complex was investigated by immunoblot with anti-HA antibodies (Fig. 2B). Trx was clearly coimmunoprecipitated with SlrP. On the other hand Trx was not detected when using a preimmune serum instead of the anti-FLAG antibodies, showing again the relevance and specificity of this interaction.
FIGURE 2.

Trx interacts with SlrP in bacteria and host cells. A, Trx co-purifies with SlrP from Salmonella lysates. Lysates from derivatives of strain SV5193 (14028 slrP::3xFLAG) expressing GST or the GST-Trx fusion protein from plasmid pGEX4T-1 were prepared in Nonidet P-40 lysis buffer. GST proteins were isolated by affinity chromatography with glutathione-agarose beads. After washing, proteins eluted in sample buffer were resolved by SDS-PAGE, blotted on nitrocellulose filters, and developed with monoclonal anti-FLAG. Aliquots of the original lysates (1/18 the amount used in the interaction experiments) were also included. B, coimmunoprecipitation of SlrP and human Trx. HeLa cells were transiently co-transfected with 5 μg of two derivatives of plasmid pCS2, one expressing SlrP-3×FLAG, and the other 3×HA-Trx. Nonidet P-40 lysates from 9 × 106 transfected cells were subjected to immunoprecipitation (IP) with preimmune serum (PI) or anti-FLAG monoclonal antibodies and, after washing, resolved by SDS-PAGE, transferred to nitrocellulose membranes, and developed with the indicated antibodies: monoclonal anti-FLAG for the upper part of the membrane, monoclonal anti-HA for the lower part. Lysates from 5 × 105 transfected cells are included in the blot. IgG, IgG heavy chain W, Western blot.
Hydrogen Peroxide Inhibits the Binding of SlrP to Trx in Vitro
Trx contains a conserved active site with two cysteines that are essential for its redox activity. We decided to investigate if this site was necessary for interaction with SlrP as it is known for other Trx partners (26). GST, GST-Trx, and GST-Trx(C32S/C35S), a Cys32 and Cys35 mutant, were purified from E. coli BL21(DE3). GST proteins bound to glutathione-agarose beads were incubated with lysates of strain SV5193, expressing SlrP-3×FLAG. After washing, the complexes were analyzed by immunoblot with anti-FLAG antibodies. Fig. 3A shows that both the wild-type and the mutant forms of Trx are able to interact with SlrP, suggesting that the integrity of the active site is not necessary for this interaction. Next we investigated if the redox status of Trx could be relevant for the interaction. For this purpose, GST-Trx bound to glutathione-agarose beads was treated with H2O2 before the in vitro interaction assay. As seen in Fig. 3B, this oxidative treatment significantly reduced the interaction. Importantly, this effect was reverted by reduction with DTT.
FIGURE 3.
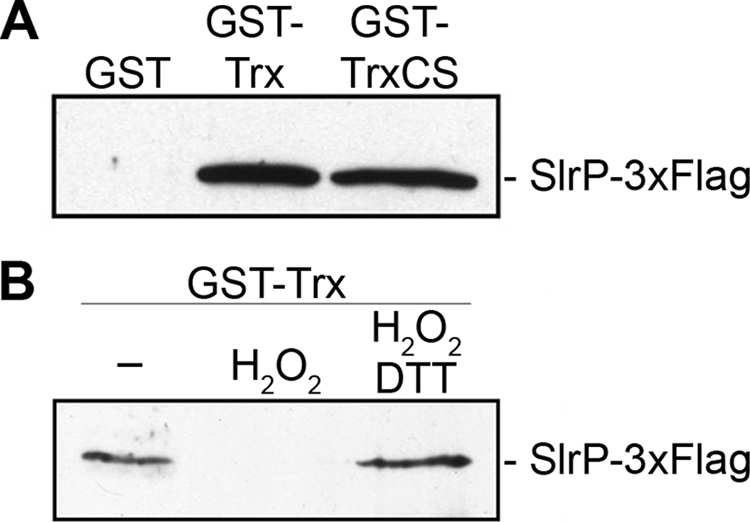
Hydrogen peroxide inhibits the binding of SlrP to Trx in vitro. A, SlrP binds to the mutated form of Trx(C32S/C35S). GST, GST-Trx, and GST-Trx(C32S/C35S) (GST-TrxCS) were produced in E. coli BL21(DE3), purified with glutathione-agarose beads, and incubated with Nonidet P-40 lysates of S. enterica serovar Typhimurium strain SV5193, expressing SlrP-3×FLAG. The precipitates were washed and submitted to immunoblot with monoclonal anti-FLAG. B, oxidation inhibits the interaction between SlrP and Trx. GST-Trx produced in E. coli BL21(DE3) and immobilized on glutathione-agarose beads was incubated with Nonidet P-40 buffer or with buffer supplemented with 1 mm H2O2 for 15 min at room temperature. After extensive washing in the same buffer, the proteins were submitted to a binding assay as above. As a control to revert oxidation, part of the H2O2-treated fusion protein was incubated with buffer containing 100 mm DTT for 15 min after washing and before incubation with lysates.
SlrP Is an E3 Ubiquitin Ligase for Mammalian Trx
Recent reports demonstrated that IpaH9.8, a Shigella type III secretion effector, and the Salmonella effectors SspH1 and SspH2, are E3 ubiquitin ligases (17, 18) and that the isolated carboxyl-terminal domain of SlrP also has this activity (17). We decided to test if the entire SlrP protein also had E3 ubiquitin ligase activity on Trx. In vitro reactions were performed in the presence of HA-ubiquitin, E1, the E2 enzyme UbcH5B, and GST-SlrP. Anti-HA immunoblot revealed the presence of several bands that appeared only when GST-SlrP was included in the reactions. These bands corresponded in size to oligomers of HA-ubiquitin (Fig. 4A, third lane), indicating that SlrP can direct the polyubiquitination of ubiquitin. Because Trx interacts with SlrP, we used GST-Trx as a possible substrate in ubiquitin reactions. The fourth lane in Fig. 4A shows additional bands detected with anti-HA. Importantly, the immunoblot with anti-Trx (Fig. 4B) revealed in addition to GST-Trx, some larger species that formed only in the presence of GST-SlrP, suggesting that SlrP is an E3 ubiquitin ligase for human Trx. To confirm this hypothesis immunoprecipitation with anti-Trx antibodies was carried out after the in vitro ubiquitination reactions and the immunoprecipitates were submitted to immunoblot with anti-HA. The results in Fig. 4C show that Trx is ubiquitinated only in the presence of SlrP. A Cys residue that is present in all members of the IpaH effector family has been shown to be important for the in vitro ubiquitination activity (18). Indeed, when Cys546 was replaced by Ala in SlrP, the ubiquitination activity was no longer observed (Fig. 4D).
FIGURE 4.

SlrP is an E3 ubiquitin ligase for Trx. A, SlrP ubiquitinates ubiquitin. Reactions performed in the presence of HA-ubiquitin (E1), UbcH5B (E2), GST-SlrP (SlrP), and GST-Trx (Trx), as indicated, were submitted to 15% SDS-PAGE and immunoblot analysis using anti-HA monoclonal antibodies. B, SlrP ubiquitinates Trx. The same blot was reincubated with anti-Trx polyclonal antibodies. C, the immunoprecipitation (IP) experiment confirms that SlrP ubiquitinates Trx. Reactions performed in the presence of HA-ubiquitin (E1), UbcH5B (E2), GST-SlrP (SlrP), and GST-Trx (Trx), as indicated, were submitted to immunoprecipitation with anti-Trx antibodies. The precipitates were resolved in 10% SDS-PAGE and analyzed by immunoblot with anti-HA antibodies. D, SlrP mutated in Cys546 does not possess E3 ubiquitin ligase activity. Reactions performed in the presence of HA-ubiquitin (E1), UbcH5B (E2), and GST-SlrP (SlrP), or GST-SlrPC546A (C546A), as indicated, were resolved by 10% SDS-PAGE and submitted to immunoblot analysis using anti-HA monoclonal antibodies. Molecular mass markers, in kDa, are indicated on the left of each panel.
Expression of SlrP in HeLa Cells Decreases the Reducing Activity of Trx and Triggers Cell Death
To investigate the physiological consequences of the interaction of SlrP with Trx in vivo, we established a stable slrP-3×FLAG-transfected HeLa cell line. The expression of tagged SlrP was confirmed by Western blot (Fig. 5A). The reducing activity of Trx was measured with the insulin reduction assay (see “Experimental Procedures”). In the initial experiments, with growing cells, no effect of SlrP was observed (data not shown). When the activity was measured in confluent cultures, a significant reduction of Trx activity was detected in the SlrP expressing cell (Fig. 5B). The role of ubiquitination in this phenotype was evaluated by establishing a HeLa cell line that stably produces the C546A mutant form of tagged SlrP. The level of expression of the mutant version of this protein was similar to the level of expression of the wild-type version (Fig. 5A); however, the Trx activity in this cell line was not significantly different from the activity in HeLa transfected with empty vector (Fig. 5B). The cytosolic mammalian Trx has numerous functions in defense against oxidative stress, control of growth, and apoptosis (27). Therefore we decided to compare spontaneous cell death in confluent cultures of slrP and vector-transfected cells. Fig. 5C shows a significant increase in cytotoxicity in SlrP expressing cells. Interestingly, the level of cell death in SlrP(C546A) expressing cells was intermediate between vector-transfected and wild-type slrP-transfected cells. Taken together, these results suggest that the presence of SlrP in the host cell decreases Trx activity and renders cells more prone to cell death and that these effects are, at least partially, dependent on the E3 ubiquitin ligase activity of SlrP.
FIGURE 5.
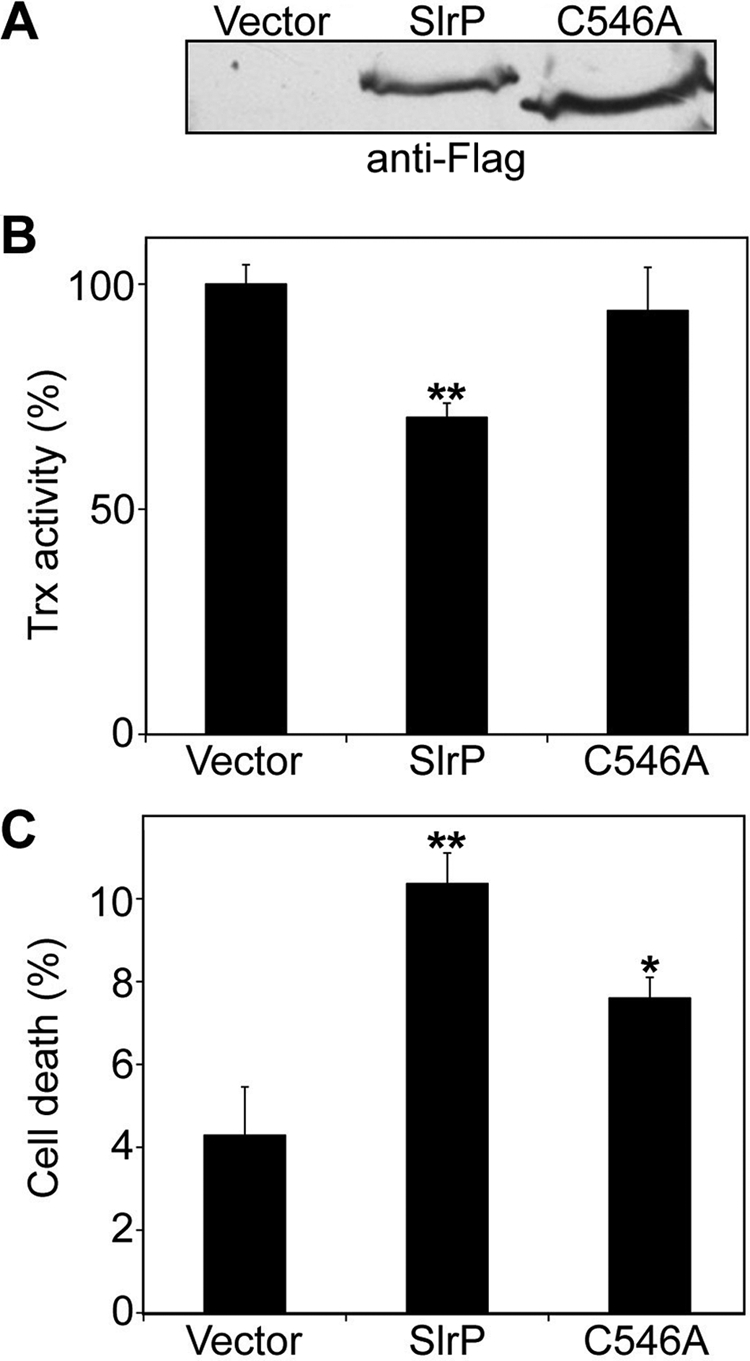
Expression of SlrP in HeLa cells decreases Trx activity and triggers cell death. A, immunoblot analysis using anti-FLAG monoclonal antibodies of lysates from HeLa cells stably transfected with pcDNA3 (vector), pcDNA3-SlrP-3×FLAG (SlrP), or pcDNA3-SlrP(C546A)-3×FLAG (C546A). B, effects of SlrP on HeLa Trx-reducing activity. The insulin disulfide reduction assay was performed in lysates obtained from confluent cultures as described under “Experimental Procedures.” Activities for HeLa cells transfected with pcDNA3-SlrP-3×FLAG and pcDNA3-SlrP(C546A)-3×FLAG are shown relative to the control (HeLa cells transfected with pcDNA3 vector), which is assigned as 100%. The results are the mean ± S.D. of three samples. The asterisks indicate significance from vector-transfected cells at p < 0.01. C, effects of SlrP on cell death. The HeLa transfectants were cultured until confluence and cell death were determined by LDH release. Values are the means of three separate experiments with an error bar representing S.D. The asterisks indicate significance from vector-transfected cells (*, p < 0.05; **, p < 0.01). The differences between SlrP and SlrP(C546A) expressing cells are also statistically significant (p < 0.01).
Salmonella Infection Decreases Trx Activity and Triggers Cell Death in HeLa Cultures
The effects of SlrP on HeLa cells prompted us to investigate if Salmonella infections could have a similar impact on Trx activity. HeLa cells were infected with S. enterica serovar Typhimurium strain 14028 and Trx activity was measured 8 and 16 h post-infection. As shown in Fig. 6A, lysates from infected cells had reduced Trx activity compared with lysates from non-infected cells. In addition, a high percentage of cell death was detected in cultures of infected HeLa cells (Fig. 6B).
FIGURE 6.
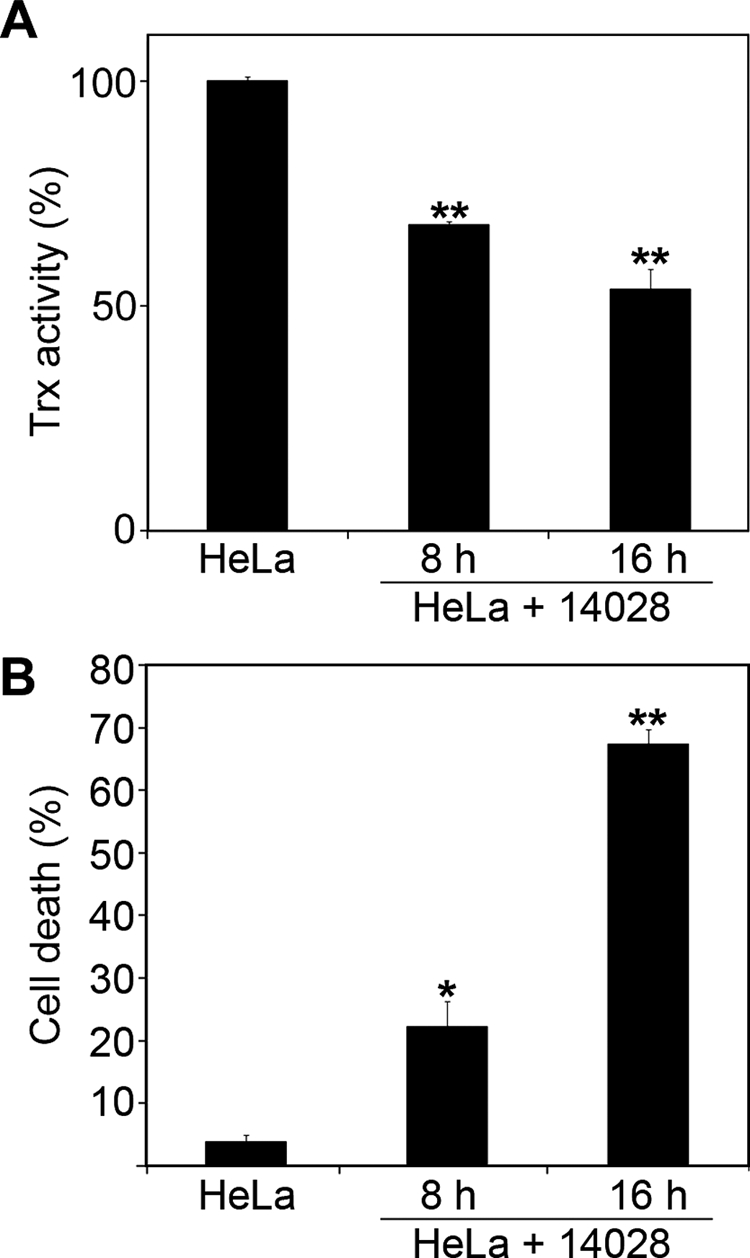
Salmonella infection decreases Trx activity and triggers delayed cell death in HeLa human epithelial cells. A, decrease of Trx activity in HeLa cells in response to Salmonella infection. Confluent cultures of HeLa cells were infected with S. enterica serovar Typhimurium strain 14028 at a multiplicity of infection of 50:1. 8 or 16 h post-infection lysates were prepared and Trx activities were measured as described under “Experimental Procedures.” Activities for HeLa cells infected with Salmonella are shown relative to the control (non-infected cells), which is assigned as 100%. The results are the mean ± S.D. of three samples. The asterisks indicate significance from non-infected cells at p < 0.01. B, death of HeLa cells in response to infection with wild-type S. enterica serovar Typhimurium 14028. Cytotoxicity of Salmonella on HeLa cells was measured 8 and 16 h post-infection with an LDH release method and compared with the level of spontaneous cell death in non-infected cells. The results are the mean ± S.D. of three samples. The asterisks indicate significance from non-infected cells (*, p < 0.05; **, p < 0.01).
DISCUSSION
More than 30 effectors are already known for the two T3SS involved in Salmonella virulence and considerable effort is being made to elucidate their functions. However, the number of effectors in this and other bacterial pathogens is constantly growing and the role of most of them in the interaction with the host remains unknown. In this study we have been able both to find a partner and to demonstrate a biochemical activity for SlrP, a Salmonella effector whose function was previously unknown.
The presence of a leucine-rich repeat domain in SlrP, a motif frequently involved in protein-protein interactions, prompted us to look for mammalian partners for this protein. A two-hybrid screen detected Trx as a partner for SlrP (see Fig. 1). The relevance of the SlrP-Trx interaction was confirmed by affinity chromatography and coimmunoprecipitation (see Fig. 2).
Trx plays a variety of redox-related roles that are conserved from E. coli to humans. It is a small, ubiquitous, multifunctional protein containing a redox-active disulfide/dithiol within its active site sequence, -Cys-Gly-Pro-Cys-. It functions together with NADPH and Trx reductase as a protein disulfide-reducing system (28). Trx was originally identified in 1964 (63) in E. coli as a hydrogen donor for ribonucleotide reductase required for DNA synthesis. In mammals there are two main isoforms of Trx, one predominantly cytosolic (Trx1) and the other of mitochondrial localization (Trx2). The importance of the Trx system is emphasized by the fact that knock-out mice lacking either of these Trx die early during embryogenesis (29, 30). The isoform found to interact with SlrP in our two-hybrid screen was Trx1. In contrast, no interaction was observed with Trx2 (data not shown). Trx1 plays an important role in various redox-regulated signaling events and is known to participate in the control of numerous physiological processes including growth, cell survival/apoptosis, development, differentiation, and proliferation (31, 32). In response to various stress conditions, Trx1 can be translocated from the cytosol to the nucleus and increase the activity of certain transcription factors, such as NF-κB, AP-1, and p53 (33) or can be secreted and have immunomodulatory functions, with co-cytokine or chemokine activities (34–36). In regard to cell survival control, there are several ways through which a functional Trx system can oppose apoptosis (reviewed in Ref. 37). One example is the inhibition of ASK-1 by the binding of reduced Trx to this kinase. Another example is the support of peroxiredoxin activity and other antioxidant functions that counteract the triggering of apoptosis through oxidative stress. It is interesting to note that the levels of Trx1 are higher in confluent cultures than in sparse cultures of HeLa cells (38). Other stress response proteins have been shown to increase as cells reach confluence including MnSOD (39) and NADH cytochrome b5 reductase (40), but the increase of Trx1 seems particularly relevant to the decrease of reactive oxygen species that is also observed in confluent cells (38, 40). These previous data could give a supportive explanation for our observation that decreased Trx activity in confluent cultures of SlrP expressing cells is correlated with increased cell death (see Fig. 5).
Conversion of Cys32 and Cys35 in the active redox site of Trx to Ser, which prevents interaction with ASK-1 (41), does not inhibit interaction with SlrP (Fig. 3A). However, oxidation of Trx abolishes both associations (see Ref. 41 and Fig. 3B). To understand this result it should be taken into account that cysteines in the redox active site are not the only residues in Trx that are affected by oxidizing conditions. In fact oxidization leads to Trx dimerization through a disulfide bond between Cys73 in both monomers (42, 43). The residues necessary for interaction with SlrP could be buried in the dimeric form of Trx.
In this study we have also demonstrated that SlrP is an E3 ubiquitin ligase that can mediate ubiquitination of its host partner Trx (Fig. 4). This post-translational modification consists of the covalent attachment of ubiquitin onto a target protein through a three-step enzymatic cascade involving the activity of an ubiquitin-activating enzyme (E1), an ubiquitin-conjugating enzyme (E2), and an ubiquitin ligase (E3), which controls the specificity of the reaction by recruiting the target protein (44). Ubiquitin is conjugated to the target protein by the formation of an isopeptide bond between the carboxyl terminus of ubiquitin and the ϵ-amino group of a lysine residue on the target protein. Ubiquitin itself has seven lysines; one of these lysines can be further conjugated by another ubiquitin in a highly processive manner to form a polyubiquitin chain (45). Although protein degradation by the 26 S proteasome is the best characterized function of ubiquitination, other regulatory functions of ubiquitin have been uncovered. For instance, monoubiquitination does not normally target a protein for degradation by the proteasome, but it can be a signal for DNA repair, transcriptional regulation, and transport of membrane proteins (46). Polyubiquitin chains can also have functions independent of proteolysis. For example, Lys63-linked polyubiquitin chains have been shown to mediate protein kinase activation, DNA repair, and vesicle trafficking. And although the classical view is that ubiquitin chains linked by Lys48 target substrates to the proteasome for degradation, recent studies also reveal some novel non- proteolytic functions for Lys48-linked polyubiquitin chains (reviewed in Ref. 47). An example of these non-proteolytic functions is regulation of the activity of Met4 in S. cerevisiae (48). Lys48 polyubiquitination represses activity of this transcription factor without affecting its stability.
There is a growing number of T3SS and T4SS effectors from animal and plant bacterial pathogens that exploit the host ubiquitin system (reviewed in Ref. 49). Some effectors use the ubiquitin system to ensure their own degradation or modification by ubiquitination, including the Salmonella SPI-1 T3SS effectors SopA, SopB, and SopE. Other effectors, such as YopJ from Yersinia pseudotuberculosis, interfere with the ubiquitination levels of host proteins important for the immune response. Finally, there are effectors that mimic host E3 ubiquitin ligases. Examples are AvrPtoB from Pseudomonas syringae pv. potato and SopA from S. enterica serovar Typhimurium. The first belongs to the RING-type and the second to the HECT-type of E3 ubiquitin ligases.
In addition to these examples, it has been recently shown that Shigella effector IpaH9.8 and Salmonella effectors SspH1 and SspH2, belonging to the leucine-rich repeat family of effectors, are E3 ubiquitin ligases (17, 18). They constitute a novel class of E3 that do not share sequence or structural similarity with the main groups of E3 ubiquitin ligases, the HECT type and the RING type (17, 50, 51). They contain a novel E3 ligase domain called novel E3 ligase (NEL) that is also present in SlrP (17, 18). We now confirm that full-length SlrP has E3 ubiquitin ligase activity and that the Cys546 residue, conserved in all family members, is necessary for this activity. Therefore SlrP is a member of this class of E3 ubiquitin ligases.
IpaH9.8 is able to ubiquitinate the yeast protein Ste7 in vitro and production of IpaH9.8 in S. cerevisiae results in the proteasome-dependent disappearance of Ste7 (18), which is consistent with the well known role of polyubiquitination in targeting substrates for degradation. Expression of SlrP in HeLa cells does not appear to trigger Trx degradation (data not shown), suggesting that ubiquitination in this case is performing a proteasome-independent function. A non-proteolytic function of ubiquitination would also be a plausible explanation for the known data about the interaction between SspH1 and its host target PKN1. SspH1 is a Salmonella T3SS effector belonging to the IpaH family that acts as E3 ubiquitin ligase for the mammalian serine/threonine kinase PKN1 (18). However, the current view is that the interaction between SspH1 and PKN1 in mammalian cells leads to activation of the kinase, which in turn inhibits the NF-κB signaling pathway (52). This view would not be consistent with an SspH1-mediated degradation of PKN1.
Our results with SlrP and Trx support a model in which ubiquitination of Trx by SlrP would lead, through a proteolytic or a non-proteolytic pathway, to a decrease in the global Trx activity of the cell, which in turn could increase susceptibility of the cell to apoptosis under certain circumstances. It is interesting to note, however, that the C546A mutant form of SlrP is able to trigger cell death, although to a level significantly lower than the level induced by wild-type SlrP. This result suggests that SlrP may have some roles that are independent of its E3 ubiquitin ligase activity. To understand the physiological significance of these results, we investigated the effects of Salmonella infection on HeLa cells. Interestingly, we were able to demonstrate that infection with S. enterica serovar Typhimurium strain 14028 triggered death of HeLa cells several hours after bacterial entry, and that this cytotoxicity was accompanied by a reduction in Trx activity (Fig. 6).
What is the role of host cell death in Salmonella infections? Rapid and delayed forms of Salmonella-induced host cell death have been more extensively studied in macrophages (reviewed in Ref. 53). Salmonella does not induce rapid cell death in epithelial cell lines HeLa, Caco-2, and Henle-407 (54). However, intestinal epithelial cells HT-29 undergo apoptosis 12–18 h after bacterial entry (55). In addition, colonization of HeLa epithelial cells for 24 h by S. enterica serovar Typhimurium PhoPc strain initiates apoptosis in 9–16% of the cell population (56). Other bacterial pathogens also trigger apoptosis of intestinal epithelial cells (57). Programmed cell death by apoptosis or pyroptosis (53) could be seen as forms of host protection that would contribute to fight the infection through the elimination of infected tissues. However, Salmonella factors are contributing to modulate these host responses. In fact, rapid pyroptosis and delayed apoptosis in macrophages are mediated by the SPI-1 effector SipB (53, 58), and could be a way to remove host effector cells and to weaken the host immune response. Early apoptosis of epithelial cells, which could be detrimental both for Salmonella and the host, is prevented by the SPI-1 effector SopB/SigD (59). Late apoptosis, in contrast, could be necessary to escape from the host epithelial cell, infect new cells, and facilitate dissemination. The SPI-2 T3SS, the spv genes and the SPI-1 effector AvrA are involved in this delayed epithelial cell apoptosis (56, 60). Our results suggest that SlrP could be another effector contributing to host cell death. In summary, the identification of mammalian Trx as a target for SlrP and the demonstration of SlrP as an E3 ubiquitin ligase for Trx are relevant contributions to the understanding of the function of this Salmonella T3SS effector.
Acknowledgments
We are grateful to F. Romero for kindly providing some plasmids and J. Casadesús and F. Romero for helpful discussions and critical reading of the manuscript.
This work was supported by Spanish Ministry of Science and Innovation and the European Regional Development Fund Grant SAF2007-60738 and Grant P08-CVI-03487 from Consejería de Innovación, Ciencia y Empresa, of the Junta de Andalucía.
- T3SS
- type III secretion system
- Trx
- thioredoxin
- SPI
- Salmonella pathogenicity island
- Km
- kanamycin
- LDH
- lactate dehydrogenase
- DB
- DNA binding domain
- AD
- activation domain
- DTT
- dithiothreitol
- E1
- ubiquitin-activating enzyme
- E2
- ubiquitin-conjugating enzyme
- E3
- ubiquitin ligase
- HA
- hemagglutinin
- GST
- glutathione S-transferase.
REFERENCES
- 1.Filloux A., Hachani A., Bleves S. (2008) Microbiology 154, 1570–1583 [DOI] [PubMed] [Google Scholar]
- 2.Gerlach R. G., Hensel M. (2007) Int. J. Med. Microbiol. 297, 401–415 [DOI] [PubMed] [Google Scholar]
- 3.Ghosh P. (2004) Microbiol. Mol. Biol. Rev. 68, 771–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cornelis G. R. (2006) Nat. Rev. Microbiol. 4, 811–825 [DOI] [PubMed] [Google Scholar]
- 5.Stavrinides J., McCann H. C., Guttman D. S. (2008) Cell. Microbiol. 10, 285–292 [DOI] [PubMed] [Google Scholar]
- 6.Coombes B. K., Wickham M. E., Brown N. F., Lemire S., Bossi L., Hsiao W. W., Brinkman F. S., Finlay B. B. (2005) J. Mol. Biol. 348, 817–830 [DOI] [PubMed] [Google Scholar]
- 7.Galán J. E. (1996) Curr. Top. Microbiol. Immunol. 209, 43–60 [DOI] [PubMed] [Google Scholar]
- 8.Geddes K., Worley M., Niemann G., Heffron F. (2005) Infect. Immun. 73, 6260–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ly K. T., Casanova J. E. (2007) Cell. Microbiol. 9, 2103–2111 [DOI] [PubMed] [Google Scholar]
- 10.Ramsden A. E., Holden D. W., Mota L. J. (2007) Trends Microbiol. 15, 516–524 [DOI] [PubMed] [Google Scholar]
- 11.Jones B. D., Ghori N., Falkow S. (1994) J. Exp. Med. 180, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Penheiter K. L., Mathur N., Giles D., Fahlen T., Jones B. D. (1997) Mol. Microbiol. 24, 697–709 [DOI] [PubMed] [Google Scholar]
- 13.Kuhle V., Hensel M. (2004) Cell. Mol. Life Sci. 61, 2812–2826 [DOI] [PubMed] [Google Scholar]
- 14.Tsolis R. M., Townsend S. M., Miao E. A., Miller S. I., Ficht T. A., Adams L. G., Bäumler A. J. (1999) Infect. Immun. 67, 6385–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao E. A., Miller S. I. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 7539–7544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellermeier C. D., Slauch J. M. (2003) J. Bacteriol. 185, 5096–5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quezada C. M., Hicks S. W., Galán J. E., Stebbins C. E. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 4864–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohde J. R., Breitkreutz A., Chenal A., Sansonetti P. J., Parsot C. (2007) Cell Host Microbe 1, 77–83 [DOI] [PubMed] [Google Scholar]
- 19.Uzzau S., Figueroa-Bossi N., Rubino S., Bossi L. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 15264–15269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherman F., Fink G. R., Hicks J. B. (1986) Methods in Yeast Genetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 21.Nishiyama A., Matsui M., Iwata S., Hirota K., Masutani H., Nakamura H., Takagi Y., Sono H., Gon Y., Yodoi J. (1999) J. Biol. Chem. 274, 21645–21650 [DOI] [PubMed] [Google Scholar]
- 22.Arnér E. S., Zhong L., Holmgren A. (1999) Methods Enzymol. 300, 226–239 [DOI] [PubMed] [Google Scholar]
- 23.Luthman M., Holmgren A. (1982) Biochemistry 21, 6628–6633 [DOI] [PubMed] [Google Scholar]
- 24.Chardin P., Camonis J. H., Gale N. W., van Aelst L., Schlessinger J., Wigler M. H., Bar-Sagi D. (1993) Science 260, 1338–1343 [DOI] [PubMed] [Google Scholar]
- 25.Fromont-Racine M., Rain J. C., Legrain P. (1997) Nat. Genet. 16, 277–282 [DOI] [PubMed] [Google Scholar]
- 26.Junn E., Han S. H., Im J. Y., Yang Y., Cho E. W., Um H. D., Kim D. K., Lee K. W., Han P. L., Rhee S. G., Choi I. (2000) J. Immunol. 164, 6287–6295 [DOI] [PubMed] [Google Scholar]
- 27.Arnér E. S., Holmgren A. (2000) Eur. J. Biochem. 267, 6102–6109 [DOI] [PubMed] [Google Scholar]
- 28.Holmgren A. (1985) Annu. Rev. Biochem. 54, 237–271 [DOI] [PubMed] [Google Scholar]
- 29.Matsui M., Oshima M., Oshima H., Takaku K., Maruyama T., Yodoi J., Taketo M. M. (1996) Dev. Biol. 178, 179–185 [DOI] [PubMed] [Google Scholar]
- 30.Nonn L., Williams R. R., Erickson R. P., Powis G. (2003) Mol. Cell. Biol. 23, 916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nordberg J., Arnér E. S. (2001) Free Radic. Biol. Med. 31, 1287–1312 [DOI] [PubMed] [Google Scholar]
- 32.Powis G., Montfort W. R. (2001) Annu. Rev. Biophys. Biomol. Struct. 30, 421–455 [DOI] [PubMed] [Google Scholar]
- 33.Hirota K., Murata M., Sachi Y., Nakamura H., Takeuchi J., Mori K., Yodoi J. (1999) J. Biol. Chem. 274, 27891–27897 [DOI] [PubMed] [Google Scholar]
- 34.Bertini R., Howard O. M., Dong H. F., Oppenheim J. J., Bizzarri C., Sergi R., Caselli G., Pagliei S., Romines B., Wilshire J. A., Mengozzi M., Nakamura H., Yodoi J., Pekkari K., Gurunath R., Holmgren A., Herzenberg L. A., Ghezzi P. (1999) J. Exp. Med. 189, 1783–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bizzarri C., Holmgren A., Pekkari K., Chang G., Colotta F., Ghezzi P., Bertini R. (2005) Antioxid. Redox Signal. 7, 1189–1194 [DOI] [PubMed] [Google Scholar]
- 36.Pekkari K., Goodarzi M. T., Scheynius A., Holmgren A., Avila-Cariño J. (2005) Blood 105, 1598–1605 [DOI] [PubMed] [Google Scholar]
- 37.Arnér E. S., Holmgren A. (2006) Semin. Cancer Biol. 16, 420–426 [DOI] [PubMed] [Google Scholar]
- 38.Spielberger J. C., Moody A. D., Watson W. H. (2008) J. Cell. Biochem. 104, 1879–1889 [DOI] [PubMed] [Google Scholar]
- 39.Oberley T. D., Schultz J. L., Li N., Oberley L. W. (1995) Free Radic. Biol. Med. 19, 53–65 [DOI] [PubMed] [Google Scholar]
- 40.Bello R. I., Alcaín F. J., Gómez-Díaz C., López-Lluch G., Navas P., Villalba J. M. (2003) J. Bioenerg. Biomembr. 35, 169–179 [DOI] [PubMed] [Google Scholar]
- 41.Saitoh M., Nishitoh H., Fujii M., Takeda K., Tobiume K., Sawada Y., Kawabata M., Miyazono K., Ichijo H. (1998) EMBO J. 17, 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andersen J. F., Sanders D. A., Gasdaska J. R., Weichsel A., Powis G., Montfort W. R. (1997) Biochemistry 36, 13979–13988 [DOI] [PubMed] [Google Scholar]
- 43.Weichsel A., Gasdaska J. R., Powis G., Montfort W. R. (1996) Structure 4, 735–751 [DOI] [PubMed] [Google Scholar]
- 44.Pickart C. M. (2001) Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 45.Pickart C. M. (2000) Trends Biochem. Sci. 25, 544–548 [DOI] [PubMed] [Google Scholar]
- 46.Johnson E. S. (2002) Nat. Cell. Biol. 4, E295–E298 [DOI] [PubMed] [Google Scholar]
- 47.Li W., Ye Y. (2008) Cell Mol. Life Sci. 65, 2397–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flick K., Ouni I., Wohlschlegel J. A., Capati C., McDonald W. H., Yates J. R., Kaiser P. (2004) Nat. Cell. Biol. 6, 634–641 [DOI] [PubMed] [Google Scholar]
- 49.Angot A., Vergunst A., Genin S., Peeters N. (2007) PLoS Pathog. 3, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singer A. U., Rohde J. R., Lam R., Skarina T., Kagan O., Dileo R., Chirgadze N. Y., Cuff M. E., Joachimiak A., Tyers M., Sansonetti P. J., Parsot C., Savchenko A. (2008) Nat. Struct. Mol. Biol. 15, 1293–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Y., Li H., Hu L., Wang J., Zhou Y., Pang Z., Liu L., Shao F. (2008) Nat. Struct. Mol. Biol. 15, 1302–1308 [DOI] [PubMed] [Google Scholar]
- 52.Haraga A., Miller S. I. (2006) Cell. Microbiol. 8, 837–846 [DOI] [PubMed] [Google Scholar]
- 53.Fink S. L., Cookson B. T. (2007) Cell. Microbiol. 9, 2562–2570 [DOI] [PubMed] [Google Scholar]
- 54.Lundberg U., Vinatzer U., Berdnik D., von Gabain A., Baccarini M. (1999) J. Bacteriol. 181, 3433–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim J. M., Eckmann L., Savidge T. C., Lowe D. C., Witthöft T., Kagnoff M. F. (1998) J. Clin. Invest. 102, 1815–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collier-Hyams L. S., Zeng H., Sun J., Tomlinson A. D., Bao Z. Q., Chen H., Madara J. L., Orth K., Neish A. S. (2002) J. Immunol. 169, 2846–2850 [DOI] [PubMed] [Google Scholar]
- 57.Philpott D. J., Girardin S. E., Sansonetti P. J. (2001) Curr. Opin. Immunol. 13, 410–416 [DOI] [PubMed] [Google Scholar]
- 58.Knodler L. A., Finlay B. B. (2001) Microbes Infect. 3, 1321–1326 [DOI] [PubMed] [Google Scholar]
- 59.Knodler L. A., Finlay B. B., Steele-Mortimer O. (2005) J. Biol. Chem. 280, 9058–9064 [DOI] [PubMed] [Google Scholar]
- 60.Paesold G., Guiney D. G., Eckmann L., Kagnoff M. F. (2002) Cell. Microbiol. 4, 771–781 [DOI] [PubMed] [Google Scholar]
- 61.Hanahan D. (1983) J. Mol. Biol. 166, 557–580 [DOI] [PubMed] [Google Scholar]
- 62.Harper J. W., Adami G. R., Wei N., Keyomarsi K., Elledge S. J. (1993) Cell 75, 805–816 [DOI] [PubMed] [Google Scholar]
- 63.Laurent T. C., Moore E. C., Reichard P. (1964) J. Biol. Chem. 239, 3436–3444 [PubMed] [Google Scholar]