

Abstract
A structure-activity relationship study for a 2-chloroanilide derivative of pyrazolo[1,5-a]pyridine revealed that increased EphB3 kinase inhibitory activity could be accomplished by retaining the 2-chloroanilide and introducing a phenyl or small electron donating substituents to the 5-position of the pyrazolo[1,5-a]pyridine. In addition, replacement of the pyrazolo[1,5-a]pyridine with imidazo[1,2-a]pyridine was well tolerated and resulted in enhanced mouse liver microsome stability. The structure-activity relationship for EphB3 inhibition of both heterocyclic series was similar. Kinase inhibitory activity was also demonstrated for representative analogs in cell culture. An analog (32, LDN-211904) was also profiled for inhibitory activity against a panel of two hundred and eighty eight kinases and found to be quite selective for tyrosine kinases. Overall, these studies provide useful molecular probes for examining the in vitro, cellular and potentially in vivo kinase-dependent function of EphB3 receptor.
Erythropoietin-producing hepatocellular carcinoma (Eph) receptors are highly conserved transmembrane proteins composed of multiple domains that participate in an array of complex cell signaling pathways. Sixteen Eph receptors have been identified in vertebrates. They can be divided into two major classes (EphA and EphB) based on sequence similarity in the extracellular domain and binding characteristics. Mammals, including humans, have fourteen Eph receptors (EphA1 – EphA8, EphA10, EphB1 – EphB4 and EphB6).1
The Eph receptors interact with cell surface ligands called Eph receptor interacting proteins (ephrins).2 Currently, nine ephrins are known and are divided into two major classes (ephrin A1 – 6 and ephrin B1 – 3). Humans have all but ephrin A6. Following binding of the Eph receptors to the ephrin ligands, which requires cell-cell interactions, propagation of signaling occurs bi-directionally into both the Eph receptor and the ephrin presenting cells.3 The signaling events resulting from these interactions are important in both neural development4 and during adulthood. For example, the Eph receptors together with ephrins participate in axon guidance by providing repulsive cues during axonal neurogenesis.
The EphB3 receptor subtype is expressed during embryonic development and in discrete areas of the adult brain, including the cerebellum and hippocampus. It co-localizes to brain regions with high levels of ephrin B ligand expression.5 EphB3 receptor expression also increases following central nervous system injury. However, it remains unclear if EphB3 is inhibitory to axonal regeneration or beneficial for axonal repair. For example, following adult optic nerve injury, EphB3 receptor appears and coincides with retinal ganglion cell axon sprouting and remodeling.6 However, after spinal cord injury EphB3 expression increases and appears to contribute to restricted axonal regeneration and sprouting.7 Increased EphB3 receptor expression has also been documented in pancreatic cancer cell lines,8 squamous cell carcinoma,9a and rhabdomyosarcoma.9b
In addition to ligand binding domains, the Eph receptors have an intracellular tyrosine kinase domain, although EphA10 and EphB6 lack essential amino acid residues to enable catalysis. The Eph receptor’s kinase activity is required for some, but not all, of the signal transduction pathways involving Eph receptors.10
Engagement of the ephrin ligands with the Eph receptors initially results in receptor dimerization followed by autophosphorylation of tyrosine residues in the juxtamembrane region of the receptor, which is located between the transmembrane and the kinase domains. These phosphorylation events result in kinase activation by dissociation of the juxtamembrane segment from the kinase domain.11 Once fully active, the kinase domain then can bind and phosphorylate intracellular adaptor molecules perpetuating signaling.
Ligands that target different binding components of Eph receptors could serve as useful molecular probes to help elucidate the cellular biology and in vivo physiology of Eph receptors.12 These ligands could also be used to selectively modulate Eph receptor’s kinase-dependent and independent functions.13 Utilizing a recently developed high throughput screen (HTS) for EphB3 kinase activity,14 the pyrazolo[1,5-a]pyridine derivative 1 was discovered as a moderately potent inhibitor (IC50 ~ 1 µM). Herein, we describe the results of a structure-activity relationship study to optimize EphB3 kinase inhibition and to increase mouse liver microsome stability. In addition, kinase inhibitory activity is also demonstrated in cell culture and a select analog is profiled against a broad panel of kinases.
The synthesis of many pyrazolo[1,5-a]pyridine derivatives was accomplished according to Scheme 1 (Method A). Pyrazolo[1,5-a]pyridine-3-carboxylic acid, 2, was converted to the corresponding acyl chloride and then treated with amines to give 3. Derivatives incorporating other heterocycles in place of the pyrazolo[1,5-a]pyridine were prepared in a similar manner, unless otherwise noted. The amide could be reduced with LiAlH4 to give amine 4.
Scheme 1.

(Method A). Reagents and conditions: (a) (C=O)2Cl2, DCM, cat. DMF, 0 °C to rt, 2 h; (b) R1R2NH, DCM, DIPEA, rt, 18 h; (c) LiAlH4, THF,Δ, 2 h.
Carboxylic acid 2 was also allowed to react with (PhO)2P(O)N3 to generated the corresponding acyl azide, which upon heating in the presence of benzyl alcohol underwent a Curtius rearrangement followed by alcohol addition to the intermediate isocyanate to produce 5 (Scheme 2, Method B).15 Deprotection of the benzyl carbamate in the presence of hydrogen (1 atm) and 10% Pd/C yielded amine 6, which upon treatment of 2-chlorobenzoyl chloride gave 7.
Scheme 2.

(Method B). (a) (PhO)2P(O)N3, THF, DIPEA, rt, 16 h; (b) BnOH, Δ, 12 h, 80% for two steps; (c) H2 (1 atm), 10% Pd/C, MeOH/EtOAc (1:1), 45 min; (d) 2-Cl-PhC(O)Cl, DCM, DIPEA, rt, 16 h, 36% over two steps.
The synthesis of substituted pyrazolo[1,5-a]pyridine and derivatives that incorporate additional nitrogen atoms into the pyrazolo[1,5-a]pyridine is illustrated in Scheme 3 (Method C). Heterocycles 8 were converted to the N-amino derivatives 9 with mesitylSO3NH2.16 Cycloaddition with methyl propiolate in the presence of potassium carbonate gave 10 in low yield.17 Hydrolysis of the esters yielded 11. Conversion of the carboxylic acids to the corresponding acyl chlorides with thionyl chloride followed by treatment with 2-chloroaniline in pyridine gave 12.
Scheme 3.

(Method C). (a) mesitylSO3NH2, DCM, 0 °C to rt, 30 min, 81%; (b) HC≡CCO2Me, K2CO3, DMF, 50 °C, 72 h, 14%; (c) LiOH, MeOH/H2O, rt, 2 h, then 1N HCl, 85–90%; (d) SOCl2, 80 °C, 1 h; (e) 2-ClPhNH2, pyridine, rt, 16 h.
The synthesis of 5-amino substituted pyrazolo[1,5-a]pyridine derivatives is outlined in Scheme 4 (Method D). 4-Chloropyridinium hydrochloride, 13, was treated with an amine to generate 14, via a nucleophilic aromatic substitution. Conversion of 14 to the N-aminopyridine 15 with 2,4-(NO2)2PhONH2 followed by cycloaddition with N-(2-chlorophenyl)-2-propynamide18 produced 16.
Scheme 4.

(Method D). (a) HNR1R2, H2O, MW, 130 °C, 30 min, 90%; (b) 2,4-(NO2)2PhONH2, CH3CN,Δ, 16 h, 81 %; (c) HC≡CC(O)NH-2-ClPh, K2CO3, DMF, 50 °C, 72 h.
The synthesis of imidazo[1,2-a]pyridine derivatives was accomplished according to Scheme 5 (Method E). 2-Amino-5-bromopyridine, 17, was coupled with phenyl boronic acid to give 18. 5-Bromo-2-nitropyridine, 19, was treated with piperidine to give 20, which was subsequently hydrogenated in the presence of 10% Pd/C to yield 21. The 2-aminopyridines 18 or 21 were heated at reflux in toluene with N,N-dimethylformamide dimethyl acetal (DMF-DMA) and then treated with 2-bromo-N-arylacetamides (prepared by coupling anilines with 2-bromoacetyl chloride) to give 22.19 β-Ketoamide 23 was chlorinated with sulfuryl chloride yielding 24, which was used without purification. Heating 24 in the presence of 17 and sodium bicarbonate gave the 2-methyl imidazo[1,2-a]pyridine derivative 25.
Scheme 5.

(Method E). (a) PhB(OH)2, Pd(PPh3)4, Na2CO3, CH3CN, H2O, 90 °C, 16 h, 98%; (b) Piperidine, DMSO, n-Bu4NI, K2CO3, 95 °C, 16 h, 84%; (c) H2 (1 atm), 10% Pd/C, EtOH/MeOH, rt, 16 h, 100%; (d) Me2NCH(OMe)2, toluene, Δ, 6 h then BrCH2C(O)NH-2-R-Ph (where R = Cl or OMe), MeOH, Δ, 16 h, 17 – 45%; e) SO2Cl2, 0 °C to rt; f) 17, toluene, NaHCO3, 120 °C, 3 d, 10%.
The synthesis of several other 5-substituted imidazo[1,2-a]pyridine derivatives was accomplished according to the method outlined in Scheme 6 (Method F). A Pd-mediated coupling of boronic ester 26 with 17 followed by hydrogenation of the alkene yielded 27. Amine 17 was also converted to intermediate 28,20 which was not isolated, but subjected to halogen metal exchange with n-BuLi followed by addition of N-Boc-4-piperidone to give 29 after an acidic work-up. Both 27 and 29 were converted to 30 and 31, respectively, utilizing the procedure described in Method E.19 Finally, removal of the carbamate protecting groups from 30 and 31 with 30% TFA in dichloromethane produced amines 32 and 33, respectively.
Scheme 6.

(Method F). (a) 17, Pd(PPh3)4, Na2CO3, CH3CN, H2O, 90 °C, 6 h; (b) 10% Pd/C, H2 (1 atm), EtOH, 16 h, 67% for two steps; (c) n-BuLi (2 equiv), (Me2SiClCH2)2, THF, − 78 °C, 1 h; (d) n-BuLi (1 equiv), N-Boc-4-piperidone, − 78 °C to rt, 18 h, 58%; e) DMF-DMA, 85 °C, toluene 8 h, then BrCH2C(O)NH-2-Cl-Ph, MeOH, 85 °C, 18 h, 26 – 30%; f) 30% TFA in DCM, 1 h, rt, 82%.
Evaluation of EphB3 kinase inhibitory activity for the various compounds was accessed by measuring 33P incorporation into the BTK-peptide (Cell Signaling) in the presence of varying test compound concentrations. Progress curves for production of phospho-BTK were linear for at least 30 min, allowing the calculation of observed velocities. IC50 values were then determined using a two parameter fit.14
The 2-chloro substituent on the aniline ring was necessary for kinase inhibitory activity (Table 1). Removal of this group, replacing it with other electron withdrawing or donating substituents, or transposing it to the 3- or 4-positions were all detrimental. In addition, methylation of the amide (43), insertion of a methylene to give a benzylamine derivative (44), inverting the amide functional group (7) or reduction of the amide to an amine (45) all resulted in diminished EphB3 kinase inhibitory activity.
Table 1.
IC50 determinations for inhibiting EphB3 phosphorylation of BTK-peptide
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | X | Y | R1 | R2 | Method | IC50 (µM) |
| 1 | C=O | NH | 2-Cl-Ph | H | --- | 1.0 |
| 34 | C=O | NH | Ph | H | A | NA |
| 35 | C=O | NH | 2-F-Ph | H | A | ~ 20 |
| 36 | C=O | NH | 3-Cl-Ph | H | A | NA |
| 37 | C=O | NH | 4-Cl-Ph | H | A | NA |
| 38 | C=O | NH | 2,3-Cl2-Ph | H | A | ~20 |
| 39 | C=O | NH | 2-OMe-Ph | H | A | NA |
| 40 | C=O | NH | 2-CF3-Ph | H | A | > 20 |
| 41 | C=O | NH | 2-CN-Ph | H | A | > 20 |
| 42 | C=O | NH | 2-MeSO2Ph | H | A | NA |
| 43 | C=O | NMe | 2-Cl-Ph | H | A | NA |
| 44 | C=O | NH | CH2-2-Cl-Ph | H | A | NA |
| 7 | NH | C=O | 2-Cl-Ph | H | B | > 20 |
| 45 | CH2 | NH | 2-Cl-Ph | H | A | > 20 |
| 46 | C=O | NH | 2-Cl-Ph | 4-Me | C | ~ 15 |
| 47 | C=O | NH | 2-Cl-Ph | 5-Me | C | 0.26 |
| 48 | C=O | NH | 2-Cl-Ph | 6-Me | C | ~ 15 |
| 49 | C=O | NH | 2-Cl-Ph | 7-Me | C | NA |
| 50 | C=O | NH | 2-Cl-Ph | 4-Cl | C | ~ 20 |
| 51 | C=O | NH | 2-Cl-Ph | 5-Cl | C | 2.0 |
| 52 | C=O | NH | 2-Cl-Ph | 6-Cl | C | > 20 |
| 53 | C=O | NH | 2-Cl-Ph | 5-OMe | C | 0.19 |
| 54 | C=O | NH | 2-Cl-Ph | 5-Ph | C | 0.066 |
| 55 | C=O | NH | 2-Cl-Ph | 5-NMe2 | C | 0.077 |
| 56 | C=O | NH | 2-Cl-Ph | 5-Pyrr | D | 0.20 |
| 57 | C=O | NH | 2-Cl-Ph | 5-Morph | D | 0.24 |
| 58 | C=O | NH | 2-Cl-Ph | 5-Pip | D | 0.063 |
| 59 | C=O | NH | 2-Cl-Ph | 5-OBn | C | ~ 20 |
NA: Not active at 20 µM; Pyrr = N-pyrrolidinyl; Morph = N-morpholinyl; Pip = N-piperidinyl
Interestingly, addition of a methyl substituent to the 4-, 6-, or 7-positions of the pyrazolo[1,5-a]pyridine was also detrimental. However, placement of the methyl substituent in the 5-position (47) resulted in increased activity. Similarly, introduction of a chlorine to the 5-position (51) was also tolerated, but not at the 4- or 6-positions. Based on these results, other substituents were examined at the 5-position of the pyrazolo[1,5-a]pyridine. Introduction of a phenyl (54), dimethylamino (55) or N-piperidinyl (58) further increased activity resulting in IC50 values < 100 nM. However, a larger benzyloxy (59) substituent was not tolerated at the 5-position.
Various replacements of the pyrazolo[1,5-a]pyridine were next examined (Table 2). Most of the heterocycles, including indoles (60 and 61), a quinoline (62), a 1,2-benzisoxazole (63), an indazole (64) and pyrazolo[1,5-a]pyridines incorporating an additional nitrogen atom into the heterocycle (65 – 68) did not inhibit EphB3 kinase activity at concentrations < 15 µM. However, replacement of the pyrazolo[1,5-a]pyridine with an imidazo[1,2-a]pyridine (69) retained activity with an IC50 = 0.46 µM.
Table 2.
IC50 determinations for inhibiting EphB3 phosphorylation of BTK-peptide.
 | |||
|---|---|---|---|
| Cmpd | R | Method | IC50 (µM) |
| 60 | 3-indolyl | a | NA |
| 61 | N-methyl-3indolyl | A | NA |
| 62 | 4-quinolinyl | A | NA |
| 63 | 3-(1,2-benzisoxazolyl) | C | NA |
| 64 | 3-(1H-indazolyl) | Cb | > 20 |
| 65 | R1 | C | > 20 |
| 66 | R2 | Cb | NA |
| 67 | R3 | C | ~ 15 |
| 68 | R4 | C | NA |
| 69 | R5 | Cb | 0.46 |
Prepared by EDCI mediated coupling of indole-3-carboxylic acid with 2-chloroaniline
Method C, steps d and e; NA: Not active at 20µM.
Several of the SAR findings established for the pyrazolo[1,5-a]pyridine derivatives were next examined for the imidazo[1,2-a]pyridine series (Table 3). Introduction of substituents, such as bromo (25), phenyl (70) and N-piperidinyl (71), at the 5-position again resulted in a significant increase in EphB3 inhibitory activity with IC50 values < 100 nM, whereas replacing the 2-chloroanilide with a 2-methoxyanilide (72) or introduction of a methyl substituent at the 2-position of the imidazo[1,2-a]pyridine (73) abolished inhibitory activity. The 4-piperidinyl analog (32) retained significant EphB3 inhibitory activity, while also offering greater aqueous solubility due to the presence of a basic amine. However, the inhibitory activity was eroded with further introduction of a hydroxyl group onto the 4-piperidinyl ring (33).
Table 3.
IC50 determinations for inhibiting EphB3 phosphorylation of BTK-peptide.
 | |||||
|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | Method | IC50 (µM) |
| 25 | Cl | H | Br | E | 0.173 |
| 70 | Cl | H | Ph | E | ~ 0.060a |
| 71 | Cl | H | Pip | E | 0.087 |
| 72 | OMe | H | Ph | E | > 20 |
| 73 | Cl | Me | Ph | E | > 20 |
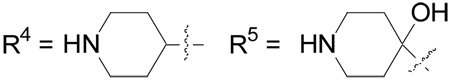
| 32b | Cl | H | R4 | F | 0.079 |
| 33 | Cl | H | R5 | F | 0.228 |
Pip = N-piperidinyl
An accurate IC50 value was difficult to obtain due to poor compound solubility at higher concentrations
boxylate salt.
Next, several derivatives were assessed for in vitro metabolic stability in pooled mouse liver microsomes.14, 21 The results of these studies are shown in Table 4. Both pyrazolo[1,5-a]pyridine and imidazo[1,2-a]pyridine analogs either without substitution or with an amino group attached through a N-C bond at the 5-position of the heterocycle (1, 58, 69 and 71) demonstrated poor stability (t1/2 ≤ 20 min). However, introduction of a substituent through a C-C bond at the 5-position (54 and 70) resulted in increased stability, with 32 demonstrating the best stability with a t1/2 of 348 min and a CLint of 4 µL/min/mg protein.
Table 4.
Mouse liver microsome stability half-life and clearance values.
| Compound | t1/2 (min) | CLint (µL/min/mg protein) |
|---|---|---|
| 1 | 5.3 | 261 ± 18 |
| 32a | 348 | 4 ± 1.4 |
| 54 | 27 | 51 ± 2.1 |
| 58 | 8.3 | 167 ± 4.2 |
| 69 | 3.8 | 361 ± 64 |
| 70 | 53 | 26 ± 0.80 |
| 71 | 20 | 71 ± 2.0 |
oxylate salt
A subset of analogs was also evaluated for inhibition of EphB3 receptor autophosphorylation in HEK293 cells following stimulation with ephrinB3 (Figure 2).14 Several compounds (34, 35, 37, 61, and 72) that were inactive or weakly active in the in vitro biochemical kinase assay were found to be inactive or weakly active in this cell-based assay. In contrast, derivatives (1, 32, 33, 58, and 71) that were active in the biochemical assay again demonstrated potent activity in cells.
Figure 2.

Inhibition assay of EphB3-induced autophosphorylation in HEK293 cells. D = DMSO, EB3 = ephrinB3, [Compound] = 10 µM, N = 3. Note: 32 was the oxylate salt.
Finally, 32 was profiled for functional inhibitory activity against a panel of two hundred and eighty eight kinases at 5 µM.22 The results demonstrated that this compound was quite selective for tyrosine kinases (Table S1 and Figure S1).14 The only noted exceptions were the three serine/threonine kinases p38α, p38β and Qik. In addition, the compound only showed moderate selectivity among the tyrosine kinases and little selectivity verses other EphA and EphB subtypes, except for EphA6 and EphA7.
In conclusion, a structure-activity relationship study of the pyrazolo[1,5-a]pyridine 1, identified as an EphB3 kinase inhibitor utilizing a recently developed HTS assay, revealed that increased inhibitory activity could be accomplished by retaining the 2-chloroanilide and introducing a phenyl or small electron donating substituent to the 5-position of the pyrazolo[1,5-a]pyridine. In addition, replacement of the pyrazolo[1,5-a]pyridine with an imidazo[1,2-a]pyridine was well tolerated and the resulting structure-activity relationship of both heterocyclic series was similar. However, the imidazo[1,2-a]pyridines offered enhanced in vitro metabolic stability as assessed in mouse liver microsomes. In particular, 32 (LDN-211904) was a potent EphB3 inhibitor, displaying excellent liver microsome stability and had enhanced aqueous solubility due to incorporation of a more basic secondary amine. EphB3 inhibitory activity was also demonstrated for representative analogs in cell culture and a correlation with in vitro biochemical activity was demonstrated. Finally, 32 was profiled for inhibitory activity against a panel of kinases and found to be quite selective for tyrosine kinases. These studies provide useful molecular probes for examining the in vitro and cellular kinase-dependent function of EphB3. Continued optimization of this compound series to further increase selectivity could afford additional analogs useful for exploring the in vivo pharmacology of EphB3 kinase inhibition in various animal disease models where EphB3 receptor expression occurs, either endogenously or as a result of injury or disease.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at ().
Figure 1.

EphB3 inhibitor identified by HTS. Also shown is the numbering system for pyrazolo[1,5-a]pyridines.
Acknowledgments
The authors thank the Harvard NeuroDiscovery Center for financial support. This work was also supported in part by an NIH grant (U24 NS049339). We also would like to thank Dr. Bing-Cheng Wang (Case Western Reserve University School of Medicine, Cleveland, OH) for the HEK293 cells expressing wild-type or kinase-inactive EphB3.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Pasquale EB. Nat. Rev. Mol. Cell Biol. 2005;6:462. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- 2.Kullander K, Klein R. Nat. Rev. Mol. Cell. Biol. 2002;3:475. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 3.(a) Pasquale EB. Cell. 2008;133:38. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]; (b) Himanen JP, Saha N, Nikolov DB. Curr. Opin. Cell. Biol. 2007;19:534. doi: 10.1016/j.ceb.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Aoto J, Chen L. Brain Res. 2007;1184:72. doi: 10.1016/j.brainres.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holder N, Cooke J, Brennan C. Eur. J. Neurosci. 1998;10:405. doi: 10.1046/j.1460-9568.1998.00032.x. [DOI] [PubMed] [Google Scholar]
- 5.Willson CA, Foster RD, Onifer SM, Whittemore SR, Miranda JD. J. Mol. Hist. 2006;37:369. doi: 10.1007/s10735-006-9067-0. [DOI] [PubMed] [Google Scholar]
- 6.Liu X, Hawkes E, Ishimaru T, Tran T, Sretavan DW. J. Neurosci. 2006;26:3087. doi: 10.1523/JNEUROSCI.4797-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Willson CA, Miranda JD, Foster RD, Onifer SM, Whittemore SR. Cell. Transplant. 2003;12:279. doi: 10.3727/000000003108746830. [DOI] [PubMed] [Google Scholar]; (b) Miranda JD, White LA, Marcillo AE, Willson CA, Jagid J, Whittemore SR. Exp. Neurol. 1999;156:218. doi: 10.1006/exnr.1998.7012. [DOI] [PubMed] [Google Scholar]
- 8.Farivar RS, Gardner-Thorpe J, Ito H, Arshad H, Zinner MJ, Ashley SW, Whang EE. J. Surg. Res. 2003;115:219. doi: 10.1016/s0022-4804(03)00246-4. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kang JU, Koo SH, Kwon KC, Park JW, Kim JM. BMC Cancer. 2009;9:237. doi: 10.1186/1471-2407-9-237. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Berardi AC, Marsilio S, Rofani C, Salvucci O, Altavista P, Perla FM, Diomedi-Camassei F, Uccini S, Kokai G, Landuzzi L, McDowell HP, Dominici C. Anticancer Res. 2008;28:763. [PubMed] [Google Scholar]
- 10.(a) Miao H, Strebhardt K, Pasquale EB, Shen T-L, Guan J-L, Wang B. J. Biol. Chem. 2005;280:923. doi: 10.1074/jbc.M411383200. [DOI] [PubMed] [Google Scholar]; (b) Kullander K, Mather NK, Diella F, Dottori M, Boyd AW, Klein R. Neuron. 2001;29:73. doi: 10.1016/s0896-6273(01)00181-7. [DOI] [PubMed] [Google Scholar]
- 11.Binns KL, Taylor PP, Sicheri F, Pawson T, Holland SJ. Mol. Cell Biol. 2000;20:4791. doi: 10.1128/mcb.20.13.4791-4805.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For an example of small molecules inhibiting ephrin binding to the EphA2 and EphA4 receptors see: Noberini R, Koolpe M, Peddibhotla S, Dahl R, Su Y, Cosford NDP, Roth GP, Pasquale EB. J. Biol. Chem. 2008;283:29461. doi: 10.1074/jbc.M804103200.
- 13.For other Eph receptor kinase inhibitors see: Choi Y, Syeda F, Walker JR, Finerty PJ, Jr, Cuerrier D, Wojciechowski A, Liu Q, Dhe-Paganon S, Gray NS. Bioorg. Med. Chem. Lett. 2009;19:4467. doi: 10.1016/j.bmcl.2009.05.029.Bardelle C, Cross D, Davenport S, Kettle JG, Ko EJ, Leach AG, Mortlock A, Read J, Roberts NJ, Robins P, Williams EJ. Bioorg. Med. Chem. Lett. 2008;18:2776. doi: 10.1016/j.bmcl.2008.04.015.Kolb P, Kipouros CB, Huang D, Caflisch A. Proteins. 2008;73:11. doi: 10.1002/prot.22028.Miyazaki Y, Nakano M, Sato H, Truesdale AT, Stuart JD, Nartey EN, Hightower KE, Kane-Carson L. Bioorg. Med. Chem. Lett. 2007;17:250. doi: 10.1016/j.bmcl.2006.09.050.Caligiuri M, Molz L, Liu Q, Kaplan F, Xu JP, Majeti JZ, Ramos-Kelsey R, Murthi K, Lievens S, Tavernier J, Kley N. Chem. Biol. 2006;13:711. doi: 10.1016/j.chembiol.2006.05.008.
- 14.See supplementary data for details.
- 15.Ninomiya K, Shioiri T, Yamada S. Tetrahedron. 1974;30:2009. [Google Scholar]
- 16.Tamura Y, Minamikawa J, Miki Y, Matsugashita S, Ikeda M. Tetrahedron Lett. 1972;40:4133. [Google Scholar]
- 17.Tamura Y, Sumida Y, Miki Y, Ikeda M. J. Chem. Soc., Perkin Trans. 1975;1:406. [Google Scholar]
- 18.Coppola GM, Damon RE. Syn. Comm. 1993;23:2003. [Google Scholar]
- 19.Georgescu F, Georgescu E, Draghici C, Iuhas PC, Filip PI. Rev. Roumaine Chim. 2005;50:349. Chem. Abstr. 2005, 144, 350628. [Google Scholar]
- 20.Mayer A, Leumann CJ. Eur. J. Org. Chem. 2007:4038. [Google Scholar]
- 21.Baranczewski P, Stańczak A, Sundberg K, Svensson R, Wallin A, Jannson J, Garberg P, Postlind H. Pharmacol. Rep. 2006;58:453. [PubMed] [Google Scholar]
- 22.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd R, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. Nat. Biotechnol. 2008;26:127. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data associated with this article can be found, in the online version, at ().