

Abstract
The actin cytoskeleton is a dynamic network required for intracellular transport, signal transduction, movement, attachment to the extracellular matrix, cellular stiffness and cell shape. Cell shape and the actin cytoskeletal configuration are linked to chondrocyte phenotype with regard to gene expression and matrix synthesis. Historically, the chondrocyte actin cytoskeleton has been studied after formaldehyde fixation - precluding real-time measurements of actin dynamics, or in monolayer cultured cells. Here we characterize the actin cytoskeleton of living low-passage human chondrocytes grown in three-dimensional culture using a stably expressed actin-GFP construct. GFP-actin expression does not substantially alter the production of endogenous actin at the protein level. GFP-actin incorporates into all actin structures stained by fluorescent phalloidin, and does not affect the actin cytoskeleton as seen by fluorescence microscopy. GFP-actin expression does not significantly change the chondrocyte cytosolic stiffness. GFP-actin does not alter the gene expression response to cytokines and growth factors such as IL-1β and TGF-β. Finally, GFP-actin does not alter production of extracellular matrix as measured by radiosulfate incorporation. Having established that GFP-actin does not measurably affect the chondrocyte phenotype, we tested the hypothesis that IL-1β and TGF-β differentially alter the actin cytoskeleton using time-lapse microscopy. TGF-β increases actin extensions and lamellar ruffling indicative of Rac/CDC42 activation, while IL-1β causes cellular contraction indicative of RhoA activation. The ability to visualize GFP-actin in living chondrocytes in 3D culture without disrupting the organization or function of the cytoskeleton is an advance in chondrocyte cell biology and provides a powerful tool for future studies in actin-dependent chondrocyte differentiation and mechanotransduction pathways.
Introduction
The relationship between cell shape and chondrocyte phenotype is well established (1). Cell shape influences chondrogenic differentiation of mesenchymal precursor cells, as shown in experimental models using cultured embryonic limb bud cells. These cells undergo a shape-dependent differentiation into chondrocytes, and experimental manipulations which alter cell shape can either prevent or promote this chondrogenic differentiation (2-4). Cell shape also influences the maintenance of the chondrocyte phenotype and extracellular matrix production in differentiated chondrocytes (5). In monolayer culture, the expression of cartilage markers and the production of cartilage matrix by articular chondrocytes is reduced. The cartilage phenotype can be reestablished if the cells are subsequently placed in 3D culture where they maintain a rounded shape (6, 7).
The actin cytoskeleton is a major determinant of chondrocyte shape. The disruption of the actin cytoskeleton in cells grown as monolayers causes a rounding of the cells. In 3D culture, overall chondrocyte actin expression is reduced, the actin cytoskeleton assumes a cortical arrangement, and many actin features prominent in monolayer culture such as stress fibrils, focal adhesions, lamellipodia and filopodia, are no longer apparent. The link between the disruption of the actin cytoskeleton and chondrocyte phenotype is observed both in mesenchymal precursors from limb buds (8), and in adult articular chondrocytes (9). Microtubules appear to contribute very little to chondrocyte shape, and agents that disrupt microtubules do not affect either cell shape or differentiation (8).
The response of chondrocytes to anabolic and catabolic factors important for cartilage homeostasis is influenced both by cell shape and the actin cytoskeleton. Disruption of the actin cytoskeleton in monolayer cultured chondrocytes inhibits the anabolic effect of bone morphogenetic proteins (10) and affects the response to catabolic IL-1β (11). The actin cytoskeleton is intimately involved in sensing mechanical forces applied to chondrocytes, which in moderate amounts can act as an anabolic stimulus to promote the expression of cartilage markers and the synthesis of extracellular matrix components (12). A requirement for the actin cytoskeleton in mechanotransduction is explained by the tensegrity theory (13, 14), and virtually all models of mechanotransduction involve a major role for the actin cytoskeleton (see examples in (15) and (16).
The actin cytoskeleton is rapidly remodeled during the application of forces, which alters the mechanical properties of the cells (17). The rapid dynamic remodeling of the actin cytoskeleton was described in many culture systems including vascular endothelial cells under shear forces (18), chondrocytes under hydrostatic pressure (19), chondrocytes under cyclic osmotic pressure (20), and agarose-embedded chondrocytes responding to either static (21) or cyclic (22) strain. In addition to cell shape, the ability to rapidly remodel the actin cytoskeleton is critically important in determining the proper cellular response. This was highlighted by the recent observation that reagents which either stabilize or destabilize actin filaments promote chondrocyte differentiation in monolayer culture (23), yet actin destabilization in 3D cultures can have opposite effects (24).
A limitation in current techniques to visualize the actin cytoskeleton is that they require fixation, permeablization, and staining of F-actin. All of these steps introduce artifacts (25), and because they do not allow real-time visualization of chondrocyte actin dynamics in live cells, the current technique is inherently incapable of measuring actin remodeling kinetics. In this report we describe a method for live-cell imaging of the actin cytoskeleton using lentiviral GFP-actin transduction. We characterized the transduced cells with regard to overall actin expression, cytoskeletal appearance, responses to anabolic and catabolic stimulus, cartilage matrix production, and cell biomechanical properties.
Materials and Methods
Cell Source
Primary human articular chondrocytes were obtained from the femoral and tibial condyles as previously described(7). Chondrocytes were expanded in monolayer culture until nearly confluent, then passaged up to three times. Media was changed every 2-3 days. Representative results are shown from chondrocytes isolated from up to four individual donors. Within each experiment, all comparisons were done on cells from the same donor at the same passage.
Culture of Chondrocytes in 3D
Chondrocytes were released from monolayer culture with trypsin, centrifuged and resuspended at 10 × 106 cells/ml, mixed with an equal volume of 6% low gelling point agarose at 42°C, then immediately pipetted into a custom casting chamber exactly 2 mm thick. The agarose was gelled at 4°C for 30 minutes, and 6mm discs made with a dermal punch. The final cell density was 5 × 106 cells/ml in 3% agarose. Cells were cultured in DMEM supplemented with 10% fetal calf serum (FCS), antibiotics, and 25 mM ascorbic acid.
GFP-actin Transduction
The coding region of human beta-actin was PCR-amplified from a cDNA plasmid (Open Biosystems, Huntsville AL) and cloned into the peGFP-C2 plasmid (Clontech, Mountainview, CA) to yield a construct coding for the GFP-actin fusion protein. This was sub-cloned into the pENTR/SD/DTOPO Gateway entry vector, then into the pLenti4/TO/V5-DEST Gateway destination vector. As a control, eGFP was cloned into the pLenti4/TO/V5-DEST vector. These constructs were used with ViraPower T-Rex reagents to generate virus according to the manufacturer's instructions (Invitrogen, Carlsbad CA). Briefly, replication-defective but infectious Lentivirus was produced in 293FT cells, concentrated two-fold by ultracentrifugation, and stored at -80°C until ready for use. To transduce chondrocytes, 2 × 105 freshly trypsinized chondrocytes were seeded into 6-well plates and allowed to attach for 4-6 hours. Viral stock was used in the presence of hexadimethryne bromide to infect chondrocytes overnight. Infection rates were approximately 90%-95% by counting GFP-positive cells.
Cell Deformation Measurements
To determine whether GFP-actin expression or lentiviral transduction alters cell stiffness, the deformation of chondrocytes subjected to 15% strain was measured. Chondrocytes were cultured in 3% agarose and tested 1 day after gelling, before the establishment of a detectable extracellular matrix that might interfere with such measurements. Cells were labeled with 1 μg/ml CalceinAM (Invitrogen) for 60 minutes to evenly label the cytoplasm. A custom-designed loading chamber on the stage of a Zeiss LSM-510 confocal microscope was used to apply precisely controlled compression in the X axis. A 63× water-immersion objective was used to obtain a 1μm z-slice through the largest area of the cell. Cellular deformation was estimated using the ratio of the X and Y diameters of the cells (17, 26). A total of 30 uncompressed cells and 30 compressed cells from each of the control, GFP, and GFP-actin groups were measured. X to Y ratios were compared between groups using oneway ANOVA with Tukey's HSD post-hoc test.
Cell Stiffness Calculations
To determine whether the observed cellular deformation corresponded with previously published measurements of chondrocyte cytosolic stiffness, we applied a finite element analysis model basically as described (17).
ImmunoBlotting
To determine whether GFP-actin expression or lentiviral transduction alters endogenous actin production, immunoblotting was used to compare actin levels in transduced and untransduced cells. Four days after viral infection, chondrocytes were re-plated onto 6-well plates at subconfluent density for two days and cultured in monolayer in DMEM supplemented with 10% FCS. Cells were washed with PBS then lysed with RIPA buffer (50 mM Tris-HCl pH 7.4, 1%NP-40, 0.25% sodium-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1mM PMSF, with protease and phosphatase inhibitor cocktails). Cell debris was removed by centrifugation. Ten μg of cell lysate was subjected to SDS-PAGE on 4-20% gradient gels (Invitrogen), and transferred to Immobilon-FL membranes (Millipore, Billerica, MA). Actin levels were probed with a rabbit polyclonal anti-pan-actin antibody (catalog number AAN01-A, Cytoskeleton, Denver CO) followed by qDot655-conjugated secondary antibody (Invitrogen). To verify even protein loading, an equal amount of cell extract was probed using an anti-GAPDH antibody followed by HRP-conjugated secondary antibody and SuperSignal West Dura substrate (Pierce, Rockford IL). Images were captured using a 16-bit digital camera on a BioRad XRS gel documentation system with UV-transillumination (actin) or 2 × 2 binning (GAPDH) and quantified with Quantity One software (Bio-Rad, Hercules CA).
Expression Analysis by RNA Isolation and Quantitative RT-PCR
To determine whether GFP-actin or lentiviral transduction alter the response to growth factors and cytokine stimulation, quantitative RT-PCR was used to compare gene expression responses to TGF-β and IL-1β. Cells were seeded into 6-well plates at 80% confluency and allowed to attach overnight. Cells were serum-starved for 24 hours in DMEM with 0.1% FCS, and then treated for 6 hours with 5 ng/ml TGF-β or IL-1β. Total RNA was isolated using RNEasy reagents (Qiagen, Valencia CA). Quantitative RT-PCR was performed using TaqMan EZ-rTtH RT-PCR reagents and Assays-on-Demand primer/probe sets (Applied Biosystems, Foster City CA) to detect GAPDH, VEGF, IL-6, and iNOS expression. RT-PCR was performed in triplicate for each RNA, and student's t-test used to assess statistical significance. Expression levels were normalized to GAPDH using the ΔCt method. Induction of IL-6 and iNOS mRNA by IL-1β treatment in transduced cells was compared to the induction of IL-6 and iNOS mRNA by IL-1β in control cells. Similarly, the induction of VEGF by TGFβ in transduced cells was compared to that in control cells.
Sulfate Incorporation
To determine whether GFP-actin expression or lentiviral transduction alters extracellular matrix production in 3D-cultured chondrocytes, sulfate incorporation assays were used to compare proteoglycan production in transduced and control cells. Chondrocytes were embedded in agarose overnight then cultured 24 hours in the presence of 20 μCi/ml 35S-labeled H2SO4. After washing in HBSS, gels were digested overnight with 100 μg/ml proteinase K at 55°C, unincorporated isotope removed by size exclusion chromatography on PD-10 columns (GE Healthcare, Piscataway NJ), and the matrix-incorporated radioactivity counted in a scintillation counter as counts per minute (CPM). DNA was measured using the PicoGreen DNA Quantification kit (Molecular Probes, Eugene OR). Data is presented as incorporated CPM/ng DNA. Four gels were analyzed for each condition, and CPM/ng DNA values were compared using oneway ANOVA with Tukey's HSD to test for differences between groups.
Time-lapse Confocal Microscopy
To determine whether anabolic and catabolic stimuli alter the chondrocyte actin cytoskeleton, GFP-actin expressing chondrocytes were seeded onto glass-bottom cell culture dishes and repeatedly imaged on the confocal microscope. Images were taken every 10 to 60 seconds for up to 2 hours to establish a baseline morphology for the actin cytoskeleton. After 2 hours, 20ng/ml TGF-β or 5ng/ml IL-1β were added to the cell cultures, and images continued to be collected every 10 to 60 seconds for up to 6 hours.
Results
Cells from the same donor were split into three groups and were either left untransfected (control), or infected with lentivirus coding either for GFP or for the GFP-actin fusion protein, with otherwise identical culture conditions. To test the effect of viral transduction and GFP-actin expression on endogenous actin, we used a pan-actin antibody to detect all actin isoforms produced in the chondrocytes. Actin production at the protein level was comparable in all groups (Fig. 1), indicating that GFP-actin does not greatly alter the endogenous actin level.
Figure 1.
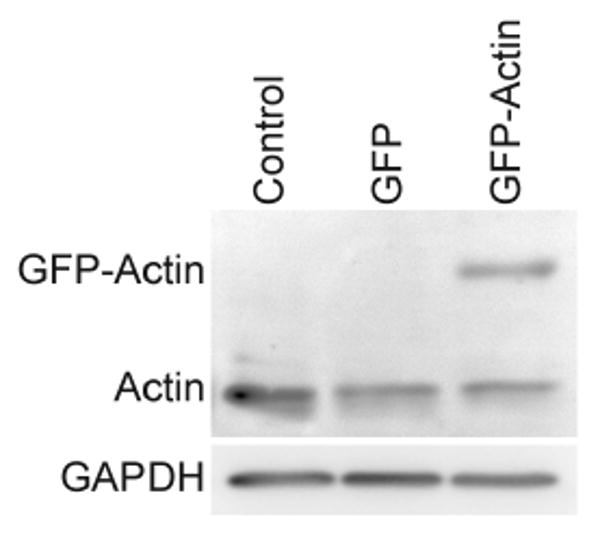
Comparison of actin protein levels in GFP and GFP-Actin transduced chondrocytes versus control cells. Similar amounts of endogenous actin were detected in all cell extracts by western blot analysis. A duplicate blot probed with anti-GAPDH shows even protein loading.
Next we wanted to examine the cytoskeleton in chondrocytes in control and transduced cells expressing GFP or GFP-actin. The cytoskeleton of fixed cells was essentially similar in all 3 groups; neither GFP nor GFP-actin transduction caused missing or extra features compared to control cells (Fig. 2, top). In monolayer culture, the chondrocyte sizes ranged from 60 to 120 μm, and were no more than 1 μm thick in all areas except the nucleus which was slightly thicker. The prominent phalloidin-stained features of monolayer cells were thick stress fibrils running the length of the cells, with even stronger staining along the long edges of the cells. Other features included smaller actin projections of 1 μm to 3 μm from the sides of the cells. Transduction of GFP and GFP-actin did not alter the presence or distribution of these features. GFP was localized evenly throughout the cytosol. GFP-actin was localized in all of the phalloidin-stained features of the monolayer chondrocytes. Additional GFP-actin was observed throughout the cytosol that was not identified by the phalloidin stain. No GFP-actin was detected in the nucleus.
Figure 2.
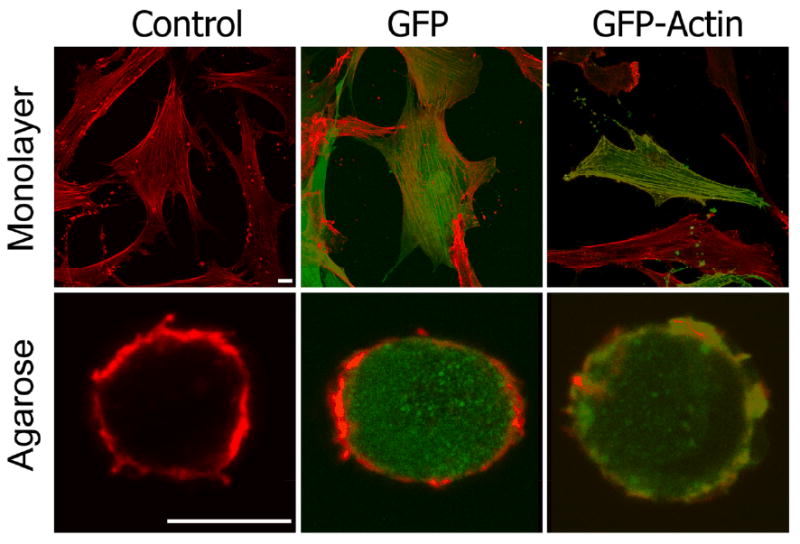
Comparison of the actin cytoskeleton in monolayer chondrocytes and after 1 week of 3D culture in agarose (red = phalloidin; green = GFP and GFP-actin; white bar = 10 μm). GFP-actin appears in all phalloidin stained actin structures both in monolayer and 3D culture. GFP and GFP-actin did not appear to alter the actin cytoskeleton.
In 3D agarose culture, the chondrocyte cytoskeleton is drastically different from that in monolayer culture. Consistent with published observations (27), 3D chondrocytes were spherical and appeared much smaller, ranging in diameter from 8μm to 12μm (Fig. 2, bottom). The standard actin features of monolayer cells such as stress fibrils, focal adhesions, filipodia, and lamellipodia were not identifiable. Instead, there was a cortical mesh of much smaller interlaced actin filaments. Isolated punctate spots were observed throughout the cytosol. The only actin feature observed in 3D that may be homologous to the monolayer culture were the smaller actin projections of 1μm to 3μm from the sides of the cells. Transduction of GFP and GFP-actin did not alter the presence or distribution of these features. GFP was localized throughout the cytosol of the chondrocytes. GFP-actin was incorporated into all phalloidin-stained actin filaments. As in the monolayer culture system, we also found GFP-actin in areas that were not stained by phalloidin.
The actin cytoskeleton is thought to influence the stiffness of the chondrocytes, which in turn may influence the chondrocyte response to mechanical stimuli. To determine whether cell stiffness was affected by transduction of GFP or GFP-actin, we measured the deformation of live agarose-embedded chondrocytes before and after 15% bulk strain was applied to the agarose. We found that the expression of GFP or GFP-actin did not alter the cellular deformation (Fig. 3), and from this result we concluded that the cellular stiffness is not measurably altered.
Figure 3.
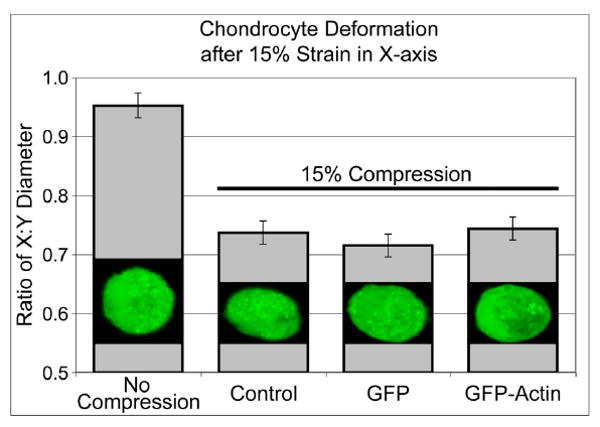
Cell deformation estimated by the ratio of the cell diameter in the X to Y direction before and after 15% strain in the X axis. No significant difference in cellular deformation was observed between control and GFP or GFP-actin transduced cells by oneway ANOVA with Tukey's HSD (p=0.6). 30 cells were measured in each condition, and a typical cell is shown. Error bars represent a pooled estimate of error variance.
The chondrocyte is entirely responsible for the secretion and maintenance of the cartilage matrix. To determine whether GFP or GFP-actin transduction altered the synthesis of extracellular matrix, we measured proteoglycan synthesis by the sulfate incorporation in 3D cultured chondrocytes. We found no significant effect of GFP or GFP-actin transduction on matrix production relative to that in untransduced control cells (Fig. 4).
Figure 4.
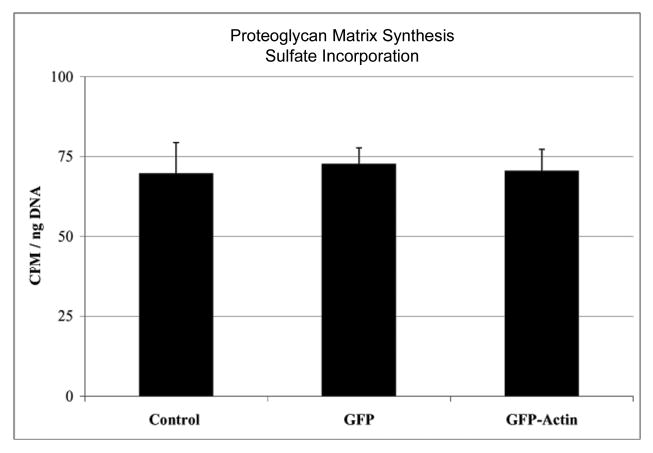
Glycosaminoglycan production measured by radiosulfate incorporation. Agarose-embedded chondrocytes were cultured for 24 hours with 20 μCi/ml H235SO4, and radioactive glycosaminoglycan normalized to DNA content. No significant differences in matrix production was observed between control, GFP, and GFP-actin transduced chondrocytes by one-way ANOVA with Tukey's HSD (p>0.84). Four gels were measured for each condition from a typical experiment. Error bars represent standard deviation.
The actin cytoskeleton is involved in signal transduction of both anabolic and catabolic stimuli, we therefore determined whether these systems were affected by GFP or GFP-actin transduction in chondrocytes. We measured the chondrocyte response to TGF-β and IL-1β, representing the major anabolic and catabolic factors in cartilage, respectively. TGF-β causes a 4- to 10-fold induction of VEGF, and IL-1β is a potent inducer of iNOS and IL-6. To compare the change in expression among different donors, we expressed the induction of these mRNAs in GFP- and GFP-actin-transduced cells as a percentage of the induction in control cells from the same donor. We found that transduction with GFP or GFP-actin caused no substantial differences of the cellular responses to IL-1β or TGF-β treatment (Fig. 5).
Figure 5.
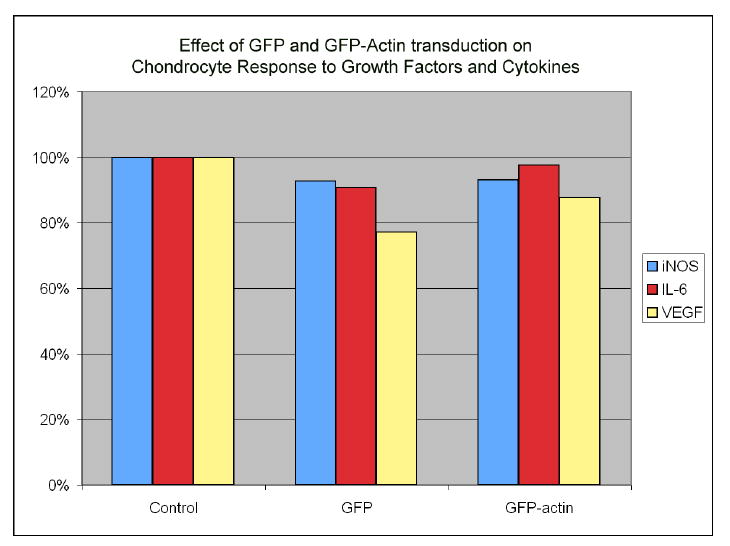
Comparison of chondrocyte response to growth factors and cytokines in transduced versus control cells. IL-1 response was measured by induction of iNOS and IL-6 expression, and TGF-β response was measured by induction of VEGF expression. Transduction with GFP and GFP-actin did not substantially alter the cellular response to TGF-β or IL-1β treatment.
Having established that the chondrocyte phenotype is not adversely affected by GFP or GFP-actin expression in these five important parameters, we then used live-cell imaging of the GFP-actin cytoskeleton to examine the response to anabolic and catabolic stimuli. Treatment with the catabolic IL-1β cytokine reproducibly decreased cellular area by 15% (Fig 6). Treatment with the anabolic TGF-β consistent increased lamellipodial extensions from the periphery of the chondrocytes, which was often also accompanied by a decrease in filopodia (Fig 7).
Figure 6.
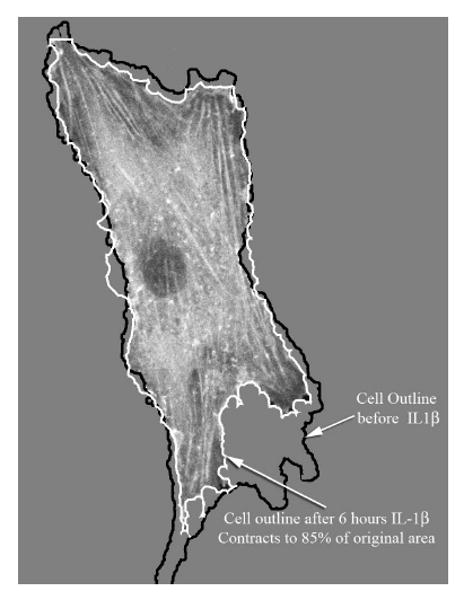
Time-lapse confocal microscopy of 6 hours of IL-1β treatment. Cellular area was measured before and after 6 hours or IL-1β treatment. A typical cell is shown, demonstrating the 15% reduction in cell area. Actin-based cellular contraction is often mediated by Rho GTPases.
Figure 7.
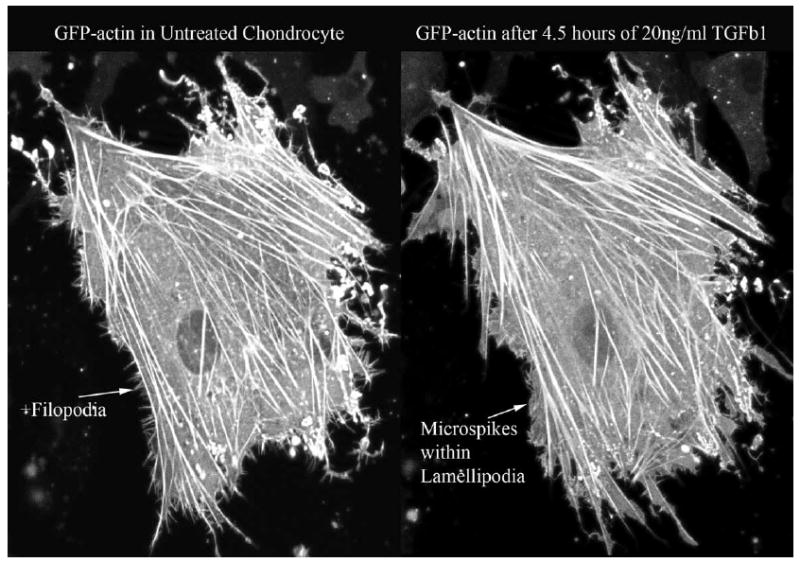
Time lapse confocal microscopy of 4.5 hours of TGF-β treatment. A typical cell shown, demonstrating a decrease in filopodia and an increase in the lamellipodia actin extensions from the cell periphery. Actin filopodia and lamellipodia are usually caused by Cdc42 and Rac GTPases, respectively.
Discussion
Our objective was to generate an experimental system to visualize the dynamic reorganization of the actin cytoskeleton of chondrocytes in a native 3D culture system. To this end, we chose lentiviral introduction of a GFP-actin fusion protein. This system efficiently infects chondrocytes and provides long-term stable protein production. Transduced chondrocytes were cultured in 3D conditions for at least four weeks with continuous production of the GFP-actin, permitting experiments that require the synthesis and assembly of extracellular matrix.
Chimeric GFP-β-actin retains the functions of endogenous β-actin in a majority of non-chondrocyte systems in which actin functions have been studied. GFP-actin incorporated into all cellular actin structures including lamellipodia, filopodia, focal contacts and stress fibers (28), as well as more specialized structures such as podosomes in osteoclasts (29). In most cases, GFP-actin did not interfere with cell growth (28, 30, 31), although it slightly impairment cytokinesis in quickly growing Dictyostelium discoideum (32). GFP-actin dynamically incorporates into rapidly remodeled actin (28, 33), and GFP-actin responded to actin destabilizing cytochalasin-B treatment similarly to normal actin (30). In vitro assays showed that low amounts of GFP-actin had little effect on either filament polymerization or movement along a surface of heavy meromyosin (32). However, Westphal, Feng and others observed that GFP-actin function is somewhat compromised, causing filament instability (32) and altering the initial dynamics of integrin-based cell spreading (34). Despite this, mice expressing GFP-actin from a profilin promoter are viable, implying that there are no drastic effects of GFP-actin on overall development (33).
While virtually all phalloidin-stained actin was also positive for GFP-actin, we observed additional GFP-actin in areas that were phalloidin-negative. Phalloidin only binds with F-actin. Actin binding proteins including cofilin, actin depolymerizing factor (ADF) and others, can alter the configuration of actin filaments to prevent phalloidin binding (35, 36). Also, actin sequestering proteins bind monomeric actin to prevent filament formation while maintaining an available pool of G-actin. These proteins include the twinfilins, beta-thymosins, ADFs/cofilins, Srv2/CAPs, profilins, and others (37-39). We expect that the phalloidin-negative GFP-actin was either bound by actin-binding proteins that prevent phalloidin binding, or present as monomers ready for incorporation into filaments.
Since the actin cytoskeleton of 3D cultured chondrocytes differs substantially from the cytoskeleton in other cell types, we felt it necessary to characterize the effect of GFP-actin transduction on the chondrocyte phenotype with regard to several parameters important in chondrocyte biology. We verified that neither the viral infection, the production of a recombinant protein, or the production of GFP-actin altered the nature of the chondrocytes or disrupted the actin cytoskeleton. GFP-actin was incorporated into all actin structures visible after phalloidin staining of formaldehyde-fixed cells. GFP-actin caused no substantial differences in endogenous actin levels, actin cytoskeletal structures, cytosolic stiffness, extracellular matrix production, or cellular responses to anabolic and catabolic stimuli.
Having established that these important parameters of chondrocytes are not adversely affected by the GFP-actin expression, we used live-cell imaging to evaluate how the actin cytoskeleton responds to anabolic and catabolic growth factors and cytokines. We found that catabolic IL-1β treatment causes a cellular contraction of approximately 15% within 6 hours. Contractile forces generated through the actin cytoskeleton generally involve Rho GTPase activation, Rho Kinase, and stress fibers in other cell types, and it is likely but unproven that a similar pathway is activated by IL-1 β treatment in the chondrocytes. We also found that TGF- β treatment caused a decrease in filopodia and an increase in lamellipodia on the periphery of the chondrocytes within 4.5 hours. In other cell types, filopodia and lamellipodia are indicative of Cdc42 and Rac GTPase activity, and again it is likely but unproven that similar pathways are activated in the chondrocytes. The 3D equivalent of stress fibers, filopodia and lamellipodia have not yet been described in detail for any cell type. The molecular tools and techniques presented in this manuscript will enable us to identify the 3D equivalents of these actin structures.
In summary, we demonstrate that lentivirally transduced GFP-actin has no significant impact on several important aspects of chondrocyte biology. The ability to visualize the dynamic reorganization of the actin cytoskeleton in living chondrocytes in a three-dimensional culture without altering the nature of the chondrocyte, and without disrupting the organization and function of the cytoskeleton, is an advancement in the field of chondrocyte cell biology. This provides a powerful tool for the study of cell biomechanics and for studies in actin-dependent signaling pathways present in the chondrocyte, including responses to growth factors, cytokines, and mechanotransduction pathways.
Acknowledgments
This work was funded through NIH grant AG07996 (MKL) and a generous donation from Donald P. and Darlene V. Shiley (DDL). The authors have no professional or financial affiliations that may be perceived to bias the presented results. We are grateful to Peter C. Chen, PhD for expert technical assistance and for his help in data analysis and interpretation.
References
- 1.Daniels K, Solursh M. J Cell Sci. 1991;100(Pt 2):249–254. doi: 10.1242/jcs.100.2.249. [DOI] [PubMed] [Google Scholar]
- 2.Archer CW, Rooney P, Wolpert L. Cell differentiation. 1982;11:245–251. doi: 10.1016/0045-6039(82)90072-0. [DOI] [PubMed] [Google Scholar]
- 3.Levitt D, Dorfman A. Proceedings of the National Academy of Sciences of the United States of America. 1972;69:1253–1257. doi: 10.1073/pnas.69.5.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solursh M, Linsenmayer TF, Jensen KL. Dev Biol. 1982;94:259–264. doi: 10.1016/0012-1606(82)90090-2. [DOI] [PubMed] [Google Scholar]
- 5.Glowacki J, Trepman E, Folkman J. Proc Soc Exp Biol Med. 1983;172:93–98. doi: 10.3181/00379727-172-41533. [DOI] [PubMed] [Google Scholar]
- 6.Benya PD, Shaffer JD. Cell. 1982;30:215–224. doi: 10.1016/0092-8674(82)90027-7. [DOI] [PubMed] [Google Scholar]
- 7.Haudenschild DR, McPherson JM, Tubo R, Binette F. Anat Rec. 2001;263:91–98. doi: 10.1002/ar.1079. [DOI] [PubMed] [Google Scholar]
- 8.Zanetti NC, Solursh M. J Cell Biol. 1984;99:115–123. doi: 10.1083/jcb.99.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newman P, Watt FM. Exp Cell Res. 1988;178:199–210. doi: 10.1016/0014-4827(88)90391-6. [DOI] [PubMed] [Google Scholar]
- 10.Vinall RL, Lo SH, Reddi AH. Exp Cell Res. 2002;272:32–44. doi: 10.1006/excr.2001.5395. [DOI] [PubMed] [Google Scholar]
- 11.Tew SR, Hardingham TE. J Biol Chem. 2006 doi: 10.1074/jbc.M604322200. [DOI] [PubMed] [Google Scholar]
- 12.Kim YJ, Sah RL, Grodzinsky AJ, Plaas AH, Sandy JD. Arch Biochem Biophys. 1994;311:1–12. doi: 10.1006/abbi.1994.1201. [DOI] [PubMed] [Google Scholar]
- 13.Ingber DE. J Cell Sci. 1993;104(Pt 3):613–627. doi: 10.1242/jcs.104.3.613. [DOI] [PubMed] [Google Scholar]
- 14.Ingber DE, Dike L, Hansen L, Karp S, Liley H, Maniotis A, McNamee H, Mooney D, Plopper G, Sims J, et al. Int Rev Cytol. 1994;150:173–224. doi: 10.1016/s0074-7696(08)61542-9. [DOI] [PubMed] [Google Scholar]
- 15.Salter DM, Millward-Sadler SJ, Nuki G, Wright MO. Clin Orthop Relat Res. 2001:S49–60. doi: 10.1097/00003086-200110001-00006. [DOI] [PubMed] [Google Scholar]
- 16.Pritchard S, Guilak F. Ann Biomed Eng. 2004;32:103–111. doi: 10.1023/b:abme.0000007795.69001.35. [DOI] [PubMed] [Google Scholar]
- 17.Freeman PM, Natarajan RN, Kimura JH, Andriacchi TP. J Orthop Res. 1994;12:311–320. doi: 10.1002/jor.1100120303. [DOI] [PubMed] [Google Scholar]
- 18.Franke RP, Grafe M, Schnittler H, Seiffge D, Mittermayer C, Drenckhahn D. Nature. 1984;307:648–649. doi: 10.1038/307648a0. [DOI] [PubMed] [Google Scholar]
- 19.Parkkinen JJ, Lammi MJ, Inkinen R, Jortikka M, Tammi M, Virtanen I, Helminen HJ. J Orthop Res. 1995;13:495–502. doi: 10.1002/jor.1100130404. [DOI] [PubMed] [Google Scholar]
- 20.Chao PH, West AC, Hung CT. Am J Physiol Cell Physiol. 2006;291:C718–725. doi: 10.1152/ajpcell.00127.2005. [DOI] [PubMed] [Google Scholar]
- 21.Lee DA, Knight MM, Bolton JF, Idowu BD, Kayser MV, Bader DL. J Biomech. 2000;33:81–95. doi: 10.1016/s0021-9290(99)00160-8. [DOI] [PubMed] [Google Scholar]
- 22.Knight MM, Toyoda T, Lee DA, Bader DL. J Biomech. 2006;39:1547–1551. doi: 10.1016/j.jbiomech.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Woods A, Wang G, Beier F. J Biol Chem. 2005;280:11626–11634. doi: 10.1074/jbc.M409158200. [DOI] [PubMed] [Google Scholar]
- 24.Woods A, Beier F. J Biol Chem. 2006;281:13134–13140. doi: 10.1074/jbc.M509433200. [DOI] [PubMed] [Google Scholar]
- 25.Arcangeletti C, Sutterlin R, Aebi U, De Conto F, Missorini S, Chezzi C, Scherrer K. J Struct Biol. 1997;119:35–58. doi: 10.1006/jsbi.1997.3871. [DOI] [PubMed] [Google Scholar]
- 26.Knight MM, Ghori SA, Lee DA, Bader DL. Med Eng Phys. 1998;20:684–688. doi: 10.1016/s1350-4533(98)00080-0. [DOI] [PubMed] [Google Scholar]
- 27.Langelier E, Suetterlin R, Hoemann CD, Aebi U, Buschmann MD. J Histochem Cytochem. 2000;48:1307–1320. doi: 10.1177/002215540004801002. [DOI] [PubMed] [Google Scholar]
- 28.Choidas A, Jungbluth A, Sechi A, Murphy J, Ullrich A, Marriott G. Eur J Cell Biol. 1998;77:81–90. doi: 10.1016/S0171-9335(98)80075-7. [DOI] [PubMed] [Google Scholar]
- 29.Destaing O, Saltel F, Geminard JC, Jurdic P, Bard F. Mol Biol Cell. 2003;14:407–416. doi: 10.1091/mbc.E02-07-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ballestrem C, Wehrle-Haller B, Imhof BA. J Cell Sci. 1998;111(Pt 12):1649–1658. doi: 10.1242/jcs.111.12.1649. [DOI] [PubMed] [Google Scholar]
- 31.Doyle T, Botstein D. Proc Natl Acad Sci U S A. 1996;93:3886–3891. doi: 10.1073/pnas.93.9.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westphal M, Jungbluth A, Heidecker M, Muhlbauer B, Heizer C, Schwartz JM, Marriott G, Gerisch G. Curr Biol. 1997;7:176–183. doi: 10.1016/s0960-9822(97)70088-5. [DOI] [PubMed] [Google Scholar]
- 33.Gurniak CB, Witke W. Eur J Cell Biol. 2006 doi: 10.1016/j.ejcb.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Feng Z, Ning Chen W, Vee Sin Lee P, Liao K, Chan V. Biomaterials. 2005;26:5348–5358. doi: 10.1016/j.biomaterials.2005.01.069. [DOI] [PubMed] [Google Scholar]
- 35.Bamburg JR. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 36.Hawkins M, Pope B, Maciver SK, Weeds AG. Biochemistry. 1993;32:9985–9993. doi: 10.1021/bi00089a014. [DOI] [PubMed] [Google Scholar]
- 37.Safer D, Golla R, Nachmias VT. Proc Natl Acad Sci U S A. 1990;87:2536–2540. doi: 10.1073/pnas.87.7.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huff T, Rosorius O, Otto AM, Muller CS, Ballweber E, Hannappel E, Mannherz HG. J Cell Sci. 2004;117:5333–5341. doi: 10.1242/jcs.01404. [DOI] [PubMed] [Google Scholar]
- 39.Paavilainen VO, Bertling E, Falck S, Lappalainen P. Trends Cell Biol. 2004;14:386–394. doi: 10.1016/j.tcb.2004.05.002. [DOI] [PubMed] [Google Scholar]