

Abstract
A critical event in atherogenesis is the interaction of macrophages with subendothelial lipoproteins. Although most studies model this interaction by incubating macrophages with monomeric lipoproteins, macrophages in vivo encounter lipoproteins that are aggregated. The physical features of the lipoproteins require distinctive mechanisms for their uptake. We show that macrophages create an extracellular, acidic, hydrolytic compartment to carry out digestion of aggregated low-density lipoproteins. We demonstrate delivery of lysosomal contents to these specialized compartments and their acidification by vacuolar ATPase, enabling aggregate catabolism by lysosomal acid hydrolases. We observe transient sealing of portions of the compartments, allowing formation of an “extracellular” proton gradient. An increase in free cholesterol is observed in aggregates contained in these compartments. Thus, cholesteryl ester hydrolysis can occur extracellularly in a specialized compartment, a lysosomal synapse, during the interaction of macrophages with aggregated low-density lipoprotein. A detailed understanding of these processes is essential for developing strategies to prevent atherosclerosis.
INTRODUCTION
Recruitment of monocytes into developing lesions and accumulation of nonmotile foam cells are integral to atherosclerotic plaque formation. A critical step in foam cell formation is the uptake of cholesterol from subendothelial lipoproteins. Most in vitro studies have investigated this interaction by incubating macrophages with monomeric lipoproteins, in particular modified lipoproteins (Brown et al., 1979; Goldstein et al., 1979). Such studies, coupled with data from animal models of atherosclerosis (Suzuki et al., 1997; de Winther et al., 1999), have led to adoption of a model in which the uptake of modified low-density lipoprotein (LDL) via various types of scavenger or pattern recognition receptors constitutes the major pathway for foam cell formation in vivo. However, much of the LDL in atherosclerotic plaques is aggregated and avidly bound to the subendothelial matrix (Smith et al., 1976; Tabas, 1999; Boren et al., 2000). For example, only 8% of lesional lipoproteins in human aortic fatty streaks could be released by extraction or by electrophoresis (Smith et al., 1976). The physical nature of these aggregated lipoprotein deposits must be considered because it requires different cellular uptake processes than those used for endocytosis of monomeric lipoproteins.
The specialized cellular processes involved in the interaction of macrophages with matrix-retained and aggregated LDL (agLDL) have been partially characterized. Macrophages internalize agLDL by patocytosis, a nonscavenger receptor-mediated pathway (Sakr et al., 2001; Kruth, 2002). The agLDL is sequestered in deep invaginations at the cell surface, termed surface-connected compartments (SCCs; Zhang et al., 1997). The membrane of the SCC is continuous with the cell surface for prolonged periods of time, during which the rate of hydrolysis of the cholesteryl ester (CE) moiety of agLDL was found to significantly exceed that of the protein moiety (Buton et al., 1999). This is in stark contrast to receptor-mediated endocytosis of acetylated LDL in which the rates of lysosomal hydrolysis of the CE and protein moieties are roughly equivalent (Goldstein et al., 1985). However, the mechanism of CE hydrolysis and the function of the SCC are currently unknown.
One possibility that has been proposed is that CE is transferred to lysosomes, either by a selective CE transport mechanism, similar to that which transfers CE from high-density lipoprotein via scavenger receptor-B1 (SR-B1; Azhar and Reaven, 2002) or by endocytosis of the CE-containing core of the lipoprotein aggregates while leaving the apo proteins outside the cell. An alternative possibility is that the macrophage digests the CE in the SCC during the prolonged contact of the macrophage with the aggregate. There is some prior evidence for partial catabolism of lipoproteins during prolonged extracellular contact. Fluorescence resonance energy transfer between two lipid analogs in the outer shell of β-very low-density lipoprotein was lost while the lipoprotein was still in a specialized SCC (Myers et al., 1993), indicating that the outer layer of phospholipids was catabolized before internalization. Additionally, macrophages and other cells in atherosclerotic lesions secrete sphingomyelinase, which can partially catabolize lipoproteins extracellularly (Hakala et al., 2003). However, this degradation consists of limited hydrolysis that leads to processes such as fusion of the lipoproteins to form agLDL (Devlin et al., 2008).
Hydrolysis of CE from agLDL requires lysosomal acid lipase (LAL; Buton et al., 1999). LAL is released from cultured human macrophages and is present in human atherosclerotic lesions (Schissel et al., 1998). However, because LAL requires an acidic pH for activity (Sheriff et al., 1995), it has been unclear how extracellular LAL could hydrolyze CEs in agLDL. Although it is well known that advanced atherosclerotic plaques are acidic, foam cell formation occurs even at the earliest stages of atherosclerosis, the fatty streak, when the extracellular environment is neutral. Thus, it has not been clear how lysosomal hydrolases could catabolize lipoproteins extracellularly during the initiation of atherosclerosis.
Osteoclasts are tissue-specific macrophage-like cells that create an extracellular, acidic, lytic compartment that degrades bone (Stenbeck, 2002; Jurdic et al., 2006). There have also been reports of an extracellular, acidic environment created by macrophages to degrade extracellular matrix (Baron et al., 1985; Punturieri et al., 2000). Based on such studies, we investigated the possibility that when macrophages interact with agLDL the CE moiety is hydrolyzed extracellularly in SCCs. Herein, we report that SCCs can be acidified and that lysosomal contents are secreted into them. Portions of the SCC are dynamic and change in permeability, thereby allowing the formation of an “extracellular” pH gradient. We also show that some hydrolysis of CEs occurs extracellularly.
We propose that rapid CE hydrolysis and the transfer of free cholesterol (FC) directly from LDL aggregates to the macrophage plays a major role in the conversion of a macrophage into a foam cell. The importance of this novel cholesterol uptake pathway is highlighted by a recent investigation that reported that loss of receptor-mediated lipid uptake via SR-A or CD36 pathways does not prevent foam cell formation in hyperlipidemic mice (Manning-Tobin et al., 2009). Thus, mechanisms of cholesterol uptake that do not require receptor-mediated endocytosis, such as the one described herein, may play an important role in atherogenesis. A detailed understanding of these processes is essential for developing strategies to prevent or reverse foam cell formation and, consequently, atherosclerosis.
MATERIALS AND METHODS
Cells and Cell Culture
Mouse macrophage cell line J774A.1 was obtained from the American Type Culture Collection (Manassas, VA). Cells were maintained in DMEM supplemented with 1% (vol/vol) penicillin/streptomycin, 3.7 g/l sodium bicarbonate, and 10% (vol/vol) fetal bovine serum (FBS). Human monocyte-derived macrophages (hMDMs) were obtained from human peripheral blood mononuclear cells as described previously (Muller and Weigl, 1992). The isolated monocytes were differentiated in vitro by incubation in RPMI containing 10% heat-inactivated FBS and 10 ng/ml macrophage colony-stimulating factor (R&D Systems, Minneapolis, MN) for 5 d. For all live cell imaging experiments, media was changed to DMEM containing 25 mM 4-(2-hydroxyethyl)-1-pipierazine ethane sulfonic acid (HEPES) without phenol red or sodium bicarbonate.
Lipoproteins and Reagents
LDL was isolated from fresh human plasma by preparative ultracentrifugation as previously described (Havel et al., 1955). LDL was vortex aggregated for 10 s (Buton et al., 1999). Acyl-CoA cholesterol acyltransferase (ACAT) inhibitor 58035, originally from Sandoz (East Hanover, NJ), was kindly provided by Dr. Ira Tabas (Columbia University, New York). CypHer 5E Mono N-hydroxysuccinimide ester (CypHer 5E) was purchased from GE Healthcare (Chalfont St. Giles, United Kingdom). AlexaFluor488 (Alexa488), Alexa546, Alexa633, biotin-fluorescein-dextran (10,000 MW), biotin-Alexa546, Alexa555-cholera toxin subunit B (CtB) and Alexa488-CtB were purchased from Invitrogen (Carlsbad, CA). All solvents (acetic acid, chloroform, diethyl ether, dimethyl sulfoxide [DMSO], hexane, and sodium hydroxide), as well as 99% fatty-acid free bovine serum albumin (BSA), bafilomycin A1, biotin, streptavidin, HEPES, sphingomyelinase from Bacillus cereus, paraformaldehyde (PFA), saponin- and streptavidin-coated red fluorescent latex beads (1-μm diameter) were purchased from Sigma-Aldrich (St. Louis, MO).
Delivery of Lysosomal Contents
Lysosome labeling of J774 cells plated on poly-d-lysine–coated, glass-coverslip bottom dishes was accomplished via an 18-h pulse with 2.2 mg/ml biotin-fluorescein-dextran. Cells were chased for 2 h in DMEM and subsequently incubated with coaggregated streptavidin-LDL and Alexa546-LDL (3:1 ratio) for 90 min. Before fixation, cells were incubated with 200 μM biotin for 10 min to bind any unoccupied streptavidin sites before cell permeabilization. Control cells were incubated with agLDL pretreated with biotin. Cells were then fixed with 0.5% PFA for 5 min, washed, and permeabilized with 1% Triton for 30 s. For quantitative measurements, the average fluorescein signal over a 1-μm2 area associated with either nonphagocytosed beads in contact with macrophages (as determined by the Nomarski differential interference contrast [DIC] image) or cell-associated agLDL was measured. Beads touching aggregates were excluded from this analysis as lysosomal exocytosis may be targeted to them because of their proximity to agLDL. The density of streptavidin on both the aggregates and beads was determined by incubating with excess biotin-fluorescein-dextran. The fluorescein signal per unit area was measured on beads and aggregates saturated with biotin-fluorescein-dextran, and average values were used to correct for differences in streptavidin density. Images were acquired with a Zeiss LSM 510 laser scanning confocal microscope (Thornwood, NY) using a 63× 1.4 numeric aperture (NA) objective (axial resolution 1.0 μm).
pH Measurements
J774, primary mouse peritoneal or bone marrow–derived macrophages, or hMDMs were plated on poly-d-lysine–coated glass-coverslip bottom dishes. The cells were incubated for 30 min with CypHer 5E, a pH-sensitive fluorophore, and Alexa488, a pH-insensitive fluorophore, dual-labeled agLDL. The pH values within each pixel were assessed quantitatively by comparison with ratio images obtained in calibration buffers of varying pH. Live cells were imaged on the confocal microscope described above using a 63× 1.4 NA objective. Cell temperature was maintained at 37°C with a heated stage and objective heater. For time-lapse data sets, a 40× 0.8 NA objective was used, and pinholes were opened (so laser power could be lowered to reduce photobleaching), resulting in an axial resolution of 14 μm. This caused an increase in the lowest pH observed when compared with truly confocal images (axial resolution of 1.0 μm) because of axial spatial averaging. Where indicated, Alexa555-CtB (5 μg/ml) or bafilomycin A1 (2 μM) were added to dishes on the microscope stage. Cells were reimaged <1 min after the addition of CtB and 10 min after the addition of bafilomycin A1.
To fabricate the micropatterned arrays of agLDL, a thin layer of parylene c was deposited on 100-mm glass wafers (Precision Glass & Optics, Santa Ana, CA) using the PDS-2010 Labcoater 2 (Specialty Coating Systems, Indianapolis, IN). The wafers were then spin-coated with a photoresist layer and patterned using standard photolithography techniques (Wu et al., 2004). The parylene-coated surfaces were incubated with N-aminopropytrimethoxysilane (Sigma Aldrich; 2% vol/vol in acetone) for 10 min and NHS-biotin was attached to the patterns of amino-derivatized silanes. The surface was incubated with streptavidin- labeled Alexa488-CypHer 5E-LDL for 1 h, treated with 50 mU/ml sphingomyelinase in 5 mM MgCl2 for 30 min to induce aggregation (Devlin et al., 2008), and washed extensively with DMEM. The micropatterned surfaces were incubated with J774 cells for 30 min, and live cell imaging was performed as described above.
All data were analyzed with MetaMorph image analysis software, (Molecular Devices, Downingtown, PA). A binary mask was created using the Alexa488 signal intensity and applied to both channels to remove background noise. Images were convolved with a 7 × 7-pixel Gaussian filter, and ratio images were generated.
Electron Microscopy
J774 cells were incubated with colloidal gold-labeled agLDL for 1 h (Handley et al., 1981). After incubation with agLDL the cells were fixed with a modified Karnovsky's solution containing 2.5% glutaraldehyde, 4% PFA, and 0.02% picric acid; postfixed with 1% osmium tetroxide, 1.5% potassium ferricyanide, enbloc contrasted with uranyl acetate; dehydrated through a graded ethanol series; and embedded in LX112 resin. En face serial sections were cut at 250 nm and picked up on formvar-coated, four-slot copper grids. Sections were further contrasted with uranyl acetate and lead citrate. Images were acquired at the New York Structural Biology Center on a JEM 2100F electron microscope (Jeol, Peabody, MA) operating at 200 kV at a set magnification of ×8000. The tilt range was approximately −60° to +60°, using a nominal 2° interval with cosine adjustment. Tomogram reconstruction was performed using the Protomo software package (Winkler and Taylor, 2006).
CtB Labeling
For surface labeling with fluorescent CtB, J774 macrophages were incubated with Alexa546-agLDL at 37°C for 2 h, labeled with Alexa488-CtB on ice for 3 min, rinsed with ice-cold medium, and fixed with PFA. Images were taken with a 63× objective on the confocal microscope described above (axial resolution 0.8 μm).
Hydrolysis of Cholesteryl-[4-14C]-oleate–containing LDL
Cholesteryl-[4-14C]-oleate (55 μCi/mmol) was purchased from American Radiolabeled Chemicals (Saint Louis, MO). LDL was reconstituted with cholesteryl-[4-14C]-oleate as described previously (Krieger, 1986). J774 cells were plated in six-well poly-d-lysine–coated dishes. AgLDL was delivered to cells in media containing 0.2% fatty-acid free BSA (to prevent nonspecific protein binding), 1% ACAT inhibitor (to prevent reesterification of hydrolyzed [14C]CE), and, where indicated, 2 μM bafilomycin A1 in DMSO. For the control samples, DMSO was added instead of bafilomycin A1. Cells were incubated for 30 min with radiolabeled agLDL, and then the supernatant was gently aspirated to remove any unbound aggregates. Degradation of bound aggregates proceeded for an additional 60 min, and cells were then washed vigorously to remove extracellular but cell-engaged aggregates. The lipids from the aggregates were extracted twice with 2 ml of hexane–isopropenol (3:2 vol/vol) for 30 min, dried under argon, and resuspended in hexane. For each well, the [14C]CE and [14C]FC radioactivity of supernatant/wash was quantified by TLC. Samples were spotted on TLC Whatman Partisil LK5D plates (20 × 20 cm) purchased from VWR International (Batavia, IL) using a TLC Autospotter (Romer Labs, Union, MO). The solvent system used to separate cellular lipids was hexane/diethylether/acetic acid (70/30/1, vol/vol/vol). TLC plates were exposed to phosphorimaging screens (Amersham Biosciences, Piscataway, NJ) for 1 wk at room temperature and then scanned on a STORM 860 Phosphor Imager system (Molecular Dynamics, Sunnyvale, CA). CE hydrolysis is expressed as the percentage of the sum of the signals of FC and CEs. A lactose dehydrogenase assay (Roche Diagnostics, Alameda, CA) was used to ensure that cell detachment during washing was not responsible for the FC seen in the supernatant/wash. No lactose dehydrogenase activity was detected in the released material (data not shown).
RESULTS
Lysosome Exocytosis Is Targeted to SCCs
In order for aggregates to undergo catabolism in SCCs, macrophages must secrete a hydrolase. LAL is a lysosomal hydrolase with an acidic pH optimum that is essential for the intracellular degradation of CEs. Hydrolysis of CEs in agLDL was completely absent during interaction of LAL−/− macrophages with agLDL (Buton et al., 1999). We tested whether lysosomal contents, which include LAL, were delivered to SCCs. Biotin-fluorescein-dextran was incubated with J774 macrophages overnight, leading to endocytosis of the dextran and delivery to lysosomes (Mukherjee et al., 1997). We verified that the cells exhibited punctate fluorescence indicative of efficient lysosome labeling. Cells were then exposed to streptavidin-labeled agLDL for 90 min followed by a 30-s treatment with biotin-Alexa546 to label extracellular aggregates. Next, excess biotin was applied for 10 min to block any remaining free streptavidin on the aggregates. Cells were then fixed and permeabilized to remove the bright lysosomal fluorescence and facilitate identification of the weaker more diffuse SCC fluorescein signal.
When macrophages were in contact with agLDL for 90 min (Figure 1, A–D) there was significant deposition of biotin-fluorescein-dextran onto the agLDL in regions in contact with cells. Consistent with previous studies, the biotin-Alexa546 staining (Figure 1D) clearly indicated that the aggregate was largely extracellular after the 90-min incubation (Buton et al., 1999). Colocalization of the fluorescein and Alexa546 signals demonstrated that lysosomal contents were exocytosed and delivered to SCCs. In control experiments, the retention of biotin-fluorescein-dextran on the agLDL was inhibited if streptavidin-labeled agLDL was pretreated with biotin before exposure to cells (Figure 1, E–G). This indicates that the excess biotin treatment prevented binding of intracellular biotin-fluorescein-dextran to the aggregates upon permeabilization. These data show that lysosomal contents were delivered to SCCs, and the previously internalized biotin-fluorescein-dextran became associated with extracellular aggregates.
Figure 1.

Lysosome exocytosis is targeted to SCCs. (A–D) J774 cells were incubated overnight with 2.2 mg/ml biotin-fluorescein-dextran to deliver the dextran to lysosomes. Cells were then incubated with streptavidin-Alexa633-agLDL for 90 min followed by a 30-s treatment with 200 μM biotin-Alexa546 to label extracellular aggregates contained in SCCs. Next, 200 μM biotin was applied for 10 min to block any remaining free streptavidin. Cells were then fixed and permeabilized to remove unbound biotin-fluorescein-dextran from the lysosomes. Representative images of (A) Nomarski DIC, (B) streptavidin-Alexa633-agLDL, (C) biotin-fluorescein-dextran, and (D) biotin-Alexa546. Colocalization of the fluorescein and Alexa546 signals demonstrated that the lysosomal contents are delivered to SCCs. (E–G) Control cells in which the streptavidin-Alexa633-agLDL was treated with 200 μM biotin before incubation to ensure that excess biotin treatment prevented binding of the cellular biotin-fluorescein-dextran to the aggregates upon permeabilization. Representative images of (E) Nomarski DIC, (F) streptavidin-Alexa633-agLDL, and (G) biotin-fluorescein-dextran. (H–K) Cells loaded with lysosomal biotin-fluorescein-dextran were coincubated with streptavidin-Alexa546-agLDL and streptavidin-coated red fluorescent beads for 90 min followed by incubation with 200 μM biotin for 10 min. Cells were then fixed and permeabilized. Representative images of (H) Nomarski DIC, (I) streptavidin-Alexa546-agLDL and red fluorescent beads, and (J) and biotin-fluorescein-dextran. (K) The average fluorescein signal per unit area associated with nonphagocytosed beads in contact with macrophages (an example is depicted with an arrow in I) or agLDL in SCCs was measured. The average fluorescein signal associated with beads (constitutive exocytosis) and aggregates, corrected for streptavidin density, is shown. Error bars, ±SEM. Scale bars, 25 μm.
To investigate the importance of this pathway, we quantified the number of macrophages that exhibited lysosomal exocytosis after a 90-min incubation with agLDL. More than 80% of macrophages in contact with agLDL demonstrated SCC fluorescein staining. To see if this was a specific response to agLDL, we examined the interaction of streptavidin-coated beads with macrophages (Figure 1, H–K). Biotin-fluorescein-dextran was delivered to J774 macrophage lysosomes by an overnight incubation. Streptavidin-labeled agLDL and streptavidin-coated beads were coincubated with the cells for 90 min. The fluorescein signal associated with either nonphagocytosed beads in contact with macrophages (arrow in Figure 1I) or cell-associated agLDL was measured. Beads touching aggregates were excluded from this analysis as lysosomal exocytosis may be targeted to them because of their proximity to agLDL. Ten cells, with several beads and aggregates per cell, were examined. We found that only small amounts of biotin-fluorescein-dextran became associated with the beads before phagocytosis (Figure 1, I and J). A quantitative comparison of the average amount of fluorescein signal per unit area associated with the beads and the aggregates, that corrects for streptavidin density, shows that agLDL induced roughly fivefold more lysosome secretion than was observed upon macrophage interaction with streptavidin-coated beads (Figure 1K). Thus, the lysosomal exocytosis to SCCs containing agLDL is a specialized response.
Portions of SCCs Are Actively Acidified
In order for LAL or other secreted lysosomal acid hydrolases to function in the SCC, an acidic environment is required. To test whether SCCs are acidified, we labeled LDL with CypHer 5E and Alexa488. The fluorescence of CypHer 5E increases as the pH decreases from 7 to 5 (Beletskii et al., 2005), whereas Alexa488 is pH-independent in this range. Macrophages were incubated with agLDL, and the pH surrounding the aggregate was determined from the ratio of CypHer 5E to Alexa488 fluorescence compared with values obtained in pH calibration buffers. When J774 macrophages interacted with the dual-labeled agLDL, regions of low pH could be seen at the contact sites (Figure 2, A and B). Similar results were obtained using hMDMs (Figure 2, C and D) and murine peritoneal and bone marrow-derived macrophages (data not shown). A wide range of acidic pH values were measured. The most acidic value observed was 5.0. The range of optimal activity for lysosomal acid lipase is reported to be at pH 5.5, with 10% of maximal activity at pH 6.5 (Sheriff et al., 1995). Macrophage lysosomal pH is typically at pH 4.8 (Majumdar et al., 2007). Thus, the measured pH values are sufficient for activation of lysosomal hydrolases. We have observed significant acidification in contact regions of more than 1000 cells under a variety of conditions and over a wide range of aggregate sizes, indicating that the acidification is a robust phenomenon.
Figure 2.

Ratiometric images of acidified SCCs as macrophages interact with agLDL. Cells were incubated for 30 min with agLDL labeled with CypHer 5E, a pH-sensitive fluorophore, and Alexa488, a pH-insensitive fluorophore. The pH values within each pixel were determined by comparison with ratio images obtained in calibration buffers. The color range is scaled for each image to provide the maximum dynamic range. Thus, colors from panel to panel are not comparable. (A and B) Images of J774 cells forming acidic SCCs. In A, an inset of the DIC image with increased contrast shows cellular projections that engulf the aggregate. (C and D) Images of hMDMs forming acidic SCCs. (E) J774 cells. (F) Image acquired 10 min after the addition of 2 μM bafilomycin A1 to the cells shown in E. Bafilomycin A1 abolished the low pH seen in E. (G) J774 cell. (H) Image acquired after the addition of Alexa555-CtB (5 μg/ml) to the cell shown in G. The regions of low pH are surrounded by plasma membrane, demonstrating that the acidic domains are extracellular. (I) J774 macrophages were incubated on a surface micropatterned with agLDL labeled with CypHer 5E and Alexa488. AgLDL attached to the coverslip is exposed to an acidic environment underneath macrophages. (J) Histograms of the pH values of macrophage-associated patterns (red trace) and patterns not in contact with cells (blue trace) in I. Scale bars, 10 μm.
The acidification depended on the activity of the vacuolar (H+)-ATPase (V-ATPase), as the acidic regions were abolished by treatment with the V-ATPase inhibitor, bafilomycin A1 (Figure 2, E and F). V-ATPases on the macrophage plasma membrane serve to translocate protons from the cytoplasm to the extracellular environment (Grinstein et al., 1992). Bafilomycin A1 is a potent and highly specific inhibitor of V-ATPases (Bowman and Bowman, 2002). Acidified regions of contact between agLDL and macrophages were neutralized by addition of bafilomycin A1 to the cells (Figure 2, E and F). Immunolocalization studies did not indicate that the V-ATPase is enriched in the SCC compared with the rest of the plasma membrane (data not shown). These data demonstrate that V-ATPase in the macrophage plasma membrane is responsible for acidification of SCCs. This acidification enables hydrolysis by lysosomal enzymes, including LAL.
To show that these acidified regions were still topologically outside the cell, we labeled cells with Alexa555-CtB immediately after measuring the pH (Figure 2, G and H). Regions of contact between dual-labeled agLDL and macrophages were imaged in live cells. After data acquisition, Alexa555-CtB was added to cells on the microscope stage, and each field immediately reimaged. Acidified sites of contact with the agLDL were outlined by CtB, indicating that this compartment is connected to the cell surface and topologically outside of the cell. The acidification likely occurs in small invaginations off the large open SCC shown in Figure 2H. This mechanism of acidification is consistent with the detailed SCC morphology revealed by electron microscopy (EM; Figure 3, A–C). To further confirm that the acidified regions were extracellular, we examined the interaction of macrophages with micropatterned arrays of agLDL. Micropatterned regions of biotin formed on glass coverslips (Wu et al., 2004) were incubated with streptavidin-derivatized CypHer 5E and Alexa488 dual-labeled LDL. After incubation, the coverslips were treated with sphingomyelinase to induce aggregation (Devlin et al., 2008) and washed extensively. J774 macrophages were incubated with the micropatterned arrays of agLDL for 30 min. When macrophages interacted with the agLDL tightly bound to the coverslip, regions of low pH were seen at contact sites (Figure 2I), further demonstrating that the acidic domains are extracellular. Figure 2J displays histograms of the pH values of macrophage-associated patterns (red trace) and patterns not in contact with cells (blue trace) in Figure 2I. The pH of aggregates not touching cells is neutral, whereas the cell-associated aggregates display a range of acidic pH values.
Figure 3.

Portions of SCCs are almost sealed. J774 cells were incubated with colloidal gold-labeled agLDL for 1 h and EM images acquired to investigate the morphology of the SCC. (A) A single computed tomogram slice. (B and C) Enlarged views of nearly sealed portions of the SCC, highlighted by boxes in A. Regions of close apposition of the macrophage membrane are indicated by arrows. Scale bar, 500 nm. (D–L) J774 macrophages were incubated with Alexa546-agLDL for 2 h and treated with Alexa488-CtB on ice for 3 min to label the plasma membrane. Cells were fixed and Alexa488-CtB staining in SCCs visualized. (D) Alexa488-CtB, (E) Alexa546-agLDL, and (F) merged image. (G–L) Enlarged views, highlighted by a box in F, depicting variable degrees of CtB staining within a single SCC (arrow). These data suggest that portions of SCCs have different permeabilities to the extracellular space. Images in D–I are single confocal slices, with axial resolution 0.8 μm. (J–L) Axial slices through the 6-μm-thick SCC at the position of the dashed line in I. Scale bar, 10 μm.
SCCs Are Dynamic and Change in Permeability
We used EM to investigate the morphology of the SCC. J774 cells were incubated with colloidal gold-labeled agLDL for 1 h. Cells were prepared for EM, 250-nm sections cut, images were acquired at different tilt angles, and a tomographic reconstruction was performed (Winkler and Taylor, 2006). Figure 3A shows an SCC from a single computed EM slice. Extensive plasma membrane ruffling is observed in the region of contact between the macrophage and agLDL, reminiscent of the actin rich protrusions and ruffled border seen during osteoclast bone resorption (Baron et al., 1985). The SCC is clearly contiguous with the extracellular space but a close apposition of the macrophage membrane (arrows) was observed. Figure 3, B and C, shows enlarged views of nearly sealed portions of the SCC, highlighted by boxes in Figure 3A. Although this is only a single two-dimensional section, almost sealed regions like this may explain how the SCC is able to maintain a proton gradient while remaining outside the boundaries of the plasma membrane.
Our preliminary EM data suggests that some portions of the SCC are less permeable than others. Consistent with this, SCCs showed varying degrees of accessibility to CtB. J774 macrophages were incubated with Alexa546-agLDL (Figure 3, E, H, and K) for 2 h and then treated with Alexa488-CtB on ice to label the plasma membrane. Cells were fixed, and Alexa488-CtB staining in SCCs was visualized (Figure 3, D, G, and J). Because the cells were not permeabilized, the macromolecular CtB can only label glycolipids on the plasma membrane. Figure 3G shows an enlarged view of the region indicated by a box in Figure 3F, which depicts variable degrees of CtB staining within a single SCC (arrow). Figure 3J shows an axial slice through the SCC at the position of the dashed line in Figure 3I. These data suggest that portions of SCCs have different permeability to the extracellular space. It follows that regions of the SCC that are less accessible to CtB may correspond to those portions that are most effectively acidified.
Measurements of pH in small regions showed time-dependent fluctuations indicating that portions of the SCC that are acidified are only transiently sealed. Figure 4 shows selected images from a time-lapse data set (Supplemental Movie I). Figure 4, B and C (top to bottom) shows enlarged views, highlighted by boxes in Figure 4A. This data set was acquired for 22 min, with 10 s between frames. Over the period of observation, significant portions of the aggregate were digested. Some portions were internalized and could be seen moving as independent vesicles (examples are indicated by arrows in panel B). The fluorescence of these internalized lipoproteins is lost rapidly from the cells, indicating that these lipoproteins are catabolized completely after internalization.
Figure 4.

Time-lapse images of acidic domains formed during interaction of J774 cells with agLDL. Cells were incubated for 30 min with agLDL dual-labeled with CypHer 5E and Alexa488. (A) Selected ratiometric images from a time-lapse data set. The data set was acquired for 22 min with 10 s between frames. Scale bar, 25 μm. (B and C) Enlarged images from the time-lapse data set highlighted by boxes in column A. Some portions of the aggregate were internalized and could be seen moving as independent vesicles (examples are indicated by arrows in column 4B). Scale bars, 10 μm. (D) Acidity profile (red trace), averaged over the region indicated by a circle in column B (t = 0 min), as a function of time. The pH measured in a region of agLDL far away from cells (blue trace), indicated in column C (t = 0 min), is included for comparison. (E) Acidity profile (red trace), averaged over the region indicated by a circle in column C (t = 0 min), as a function of time. Fluctuations in pH are on the order of minutes and are typical of most data sets.
The fluctuations in pH in the regions indicated by circles in Figure 4, B and C (t = 0 min) are shown in Figure 4, D and E, respectively. The pH measured in a region of agLDL far away from cells, indicated in Figure 3C (t = 0 min), was stable near a value of 7.4 (blue trace). In cell-associated regions, fluctuations in pH occurred on the time scale of minutes (red traces). The traces shown do not extend for the full time course of the experiment because the signal was lost as the agLDL was digested. It should be noted that the locally averaged pH values shown in Figure 4, D and E, were not as acidic as some individual pixels in Figure 4, A–C, suggesting that the most highly acidified regions may be small and transient. Such temporal fluctuations are consistent with the dynamic nature of SCCs.
Figure 5 shows selected images from another time-lapse data set (Supplemental Movie II). This data set was acquired for 5 min, with 1 s between frames. An acidic domain, highlighted by a circle in the ratiometric image shown at t = 0 min, persists for the period of observation. The red trace displays fluctuations in pH in the acidic region. The pH measured in a region of agLDL far away from the cell, the blue trace, is shown for comparison. Again, fluctuations in pH were on the time scale of minutes, which was typical of most data sets.
Figure 5.

Time-lapse images of acidic domains formed during interaction of J774 cells with agLDL. Cells were incubated for 30 min with agLDL dual-labeled with CypHer 5E and Alexa488. (A) Selected ratiometric images from a time-lapse data set. Images were acquired for 5 min with 1 s between frames. Scale bar, 15 μm. An enlarged panel highlighting the SCC is shown in the lower right-hand corner of each image. (B) Acidity profile (red trace), averaged over the region indicated by a circle in the ratiometric image shown at t = 0 min, as a function of time. The pH measured in a region of agLDL far away from the cell (blue trace), also indicated by a circle, is included for comparison.
FC Increases in SCCs during Macrophage Uptake of agLDL
To show that agLDL undergoes catabolism in SCCs, agLDL reconstituted with cholesteryl-[4-14C]-oleate was added to a monolayer of macrophages. After a 30-min incubation with radiolabeled agLDL, the supernatant was gently aspirated to remove any unbound aggregates. Degradation of cell-engaged aggregates proceeded for an additional 60 min. Cells were then washed vigorously to remove extracellular aggregates from SCCs, and the fraction of hydrolyzed cholesterol in the released agLDL was measured. FC was found in the agLDL released from the cells (Figure 6), showing that lipoprotein CE hydrolysis occurs extracellularly. Although a relatively small percentage of FC was detected, it represents only that portion of the hydrolyzed CE that was not transferred to the cell during the 90-min incubation. Consistent with this, biochemical data indicates a large amount of FC is internalized during the 90-min incubation (>60% of the cell associated radiolabeled cholesterol is in the form of FC). The production of FC in the SCC was blocked by the V-ATPase inhibitor, bafilomycin A1 (Figure 6).
Figure 6.
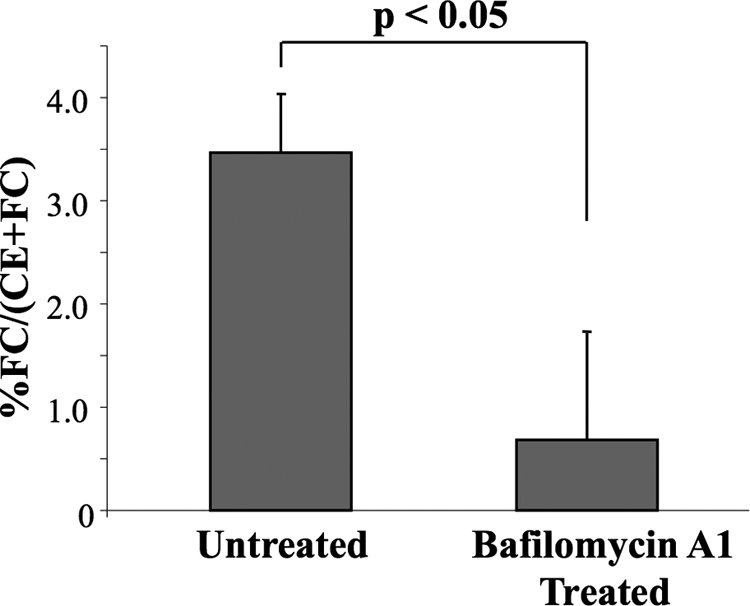
The presence of free cholesterol in cell-engaged but noninternalized aggregates. AgLDL reconstituted with cholesteryl-[4-14C]-oleate was added to a monolayer of macrophages. After a 90-min exposure, the cells were washed vigorously to remove any cell-engaged but extracellular aggregates. Wash/supernatant was assayed for both radiolabeled FC and CE. FC was generated extracellularly during macrophage interaction with agLDL. The extracellular generation of FC was blocked upon addition of bafilomycin A1, a V-ATPase inhibitor. Error bars, ±SEM. Significance was determined with an unpaired t test. A lactose dehydrogenase assay was used to ensure that cell detachment during washing was not responsible for the increase in FC seen in the supernatant/wash. No detectable lactose dehydrogenase was found in the material released by the vigorous washing, indicating that cells were not detached by this procedure (data not shown).
DISCUSSION
A key pathogenic event in the development of atherosclerosis is the retention of lipoprotein particles in the subintima (Tabas et al., 2007). These lipoprotein particles undergo oxidative modification, association with extracellular matrix proteoglycans, and aggregation. Our studies elucidate the mechanism of a novel pathway for catabolism of agLDL by macrophages. We observed the delivery of lysosomal contents to SCCs and their acidification, thereby enabling activation of LAL and other hydrolases. Our data show that portions of SCCs have different permeability to the extracellular space and that those portions that are actively acidified are dynamic and only transiently sealed. Further, an increase in FC, which was inhibited by the addition of bafilomycin A1, was observed in aggregates contained in SCCs. Together, these results demonstrate that during macrophage engagement of agLDL, at least a portion of CE hydrolysis occurs in an extracellular lysosome-like compartment, which can be described as a lysosmal synapse. This mechanism of degradation is supported by the extensive degradation of extracellular agLDL that can be seen in time-lapse imaging (Figure 4). This interaction with agLDL, which differs substantially from receptor-mediated endocytosis, is designed to mimic the initial events that occur as macrophages encounter lipoproteins in vivo. On the basis of our findings, we propose that rapid CE hydrolysis and the transfer of FC directly from LDL aggregates to the macrophage may occur when macrophages encounter subendothelial lipoproteins in developing atherosclerotic lesions.
Prolonged incubation of macrophages with agLDL leads to massive CE accumulation (Khoo et al., 1988; Suits et al., 1989). Studies from our laboratory and others have shown that sterols that are delivered to the plasma membrane can be redistributed to intracellular organelles within minutes (Hao et al., 2002; Wustner et al., 2005). Thus, the sterol released extracellularly may become incorporated into the plasma membrane and then delivered rapidly to the endoplasmic reticulum where it is reesterified by ACAT, thereby contributing to foam cell formation.
Changes in plasma membrane cholesterol content dramatically affect the biology of cells, including alteration of several signal transduction processes (Maxfield and Tabas, 2005). Such changes could play an important role in determining the properties of SCCs. It is known that actin cytoskeleton-mediated processes are required for agLDL catabolism (Grosheva et al., 2009) and that uptake of agLDL is very sensitive to treatment of cells with actin-depolymerizing agents or inhibitors of certain Rho GTPases (Sakr et al., 2001). Elevation of plasma membrane cholesterol in macrophages (by incubation of cells with cholesterol chelated to a carrier) results in dramatic changes in the actin cytoskeleton, such as membrane ruffling and cell spreading (Qin et al., 2006; Nagao et al., 2007). Cell spreading can be seen in macrophages contacting agLDL (Figure 4B). This suggests that changes induced by agLDL cause alterations in cellular plasma membrane cholesterol levels and that such local increases in cholesterol may mimic the effects of global plasma membrane cholesterol loading.
We have emphasized the role of CE hydrolysis and cholesterol delivery to macrophages in altering cellular properties such as actin assembly. Although increased levels of cholesterol can promote actin assembly independent of the ligation of specific receptors (Yeung and Grinstein, 2007), receptor interactions could also play an important role in the formation and properties of SCCs. However, previous studies have shown that the LDL receptor is not required for catabolism of agLDL, and the lack of competition by a vast excess of oxidized LDL suggests that SR-A and SR-B are also not critical (Buton et al., 1999). One receptor that does play a role in the uptake of agLDL is the LDL receptor–related protein (LRP). A significant inhibition of agLDL degradation was observed with the use of LRP inhibitors, indicating that LRP partially mediates the interaction of macrophages with agLDL (Sakr et al., 2001). Recent work has also indicated a role for syndecan-4 in the uptake of lipolytically modified LDL (Boyanovsky et al., 2009). Other cell surface molecules may also play important roles in interaction with agLDL.
We propose a model for a pathogenic circle of events, depicted in Figure 7. When subintimal macrophages come into contact with LDL aggregates, an extracellular, acidic, hydrolytic compartment (a lysosomal synapse) is formed. The low pH of the compartment is maintained by the V-ATPase in the macrophage plasma membrane. FC may be transferred to the macrophage plasma membrane either directly from the aggregate or after hydrolysis of LDL CEs by LAL that has been delivered to the extracellular space by lysosomal exocytosis. FC in the plasma membrane activates signaling events, such as modulation of Rho GTPase activities, which lead to polymerization of actin. This drives membrane extension and thus a broader area of cell surface contact with the aggregate, which may promote more FC delivery to the membrane, additional actin polymerization, and the continuation of the cycle. This lysosomal synapse allows rapid catabolism of even large aggregates or aggregates that are tightly linked to the extracellular matrix that could not be internalized even by phagocytic mechanisms. From our time-lapse imaging, it appears that after partial catabolism outside the cell, remnants can be internalized by the macrophages and digested in lysosomes. This model of agLDL catabolism may explain why cholesterol-enriched extracellular lipid particles have been found in human atheromas (Kruth, 1985; Chao et al., 1988).
Figure 7.

Schematic diagram of agLDL macrophage interactions. (a and b) Monomeric LDL in the blood stream is deposited in the subintimal space, where it becomes oxidatively modified, retained and aggregated. (c) When subintimal macrophages (shown in purple) interact with agLDL, an extracellular, acidic, hydrolytic compartment is formed. The low pH of the compartment is maintained by V-ATPases in the macrophage plasma membrane. (d) FC may be transferred to the macrophage plasma membrane either directly from the agLDL or after hydrolysis of LDL CE by LAL that has been delivered to the SCC from lysosomes. (e) FC in the plasma membrane may initiate or potentiate signaling events, such as modulation of Rho GTPase activities, which lead to polymerization of actin. (f) Actin polymerization drives membrane extension and thus more extensive membrane contact with agLDL, which promotes more FC delivery to the membrane, additional actin polymerization, and the continuation of the cycle. Portions of agLDL are internalized into endosomes, where further degradation occurs.
Eventually, foam cells in lesions undergo apoptosis releasing their cholesterol load as well as several chemoattractants (Tabas, 2004). This cholesterol load would again need to be removed by macrophages if a lesion were to regress. However, it has been found that clearance of remnants of foam cells eventually ceases, leading to mature atherosclerotic plaques with unresolved necrotic cores (Tabas, 2007). Better understanding of the mechanisms by which macrophages interact with agLDL and other cholesterol-rich components of plaques may lead to improved strategies to prevent lesion formation or promote lesion regression (Llodra et al., 2004; Williams et al., 2008).
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Ira Tabas (Columbia University, New York, NY) for ACAT inhibitor, Dr. Chunbo Qin (Weill Medical College of Cornell University) for the hMDMs, and Lee Cohen-Gould (Weill Medical College of Cornell University), and Dr. William Rice (New York Structural Biology Center) for help with EM experiments. This work was supported by National Institutes of Health (NIH) Grants R37-DK27083, R01 AI018306, P01-HL072942, RO1 HL 093324, and R01 HL057560, by the National Science Foundation, Science and Technology Centers, Nanobiotechnology Center Grant ECS9876771, and by an NIH National research Service Awards postdoctoral training fellowship (A.S.H.).
Abbreviations used:
- ACAT
acyl-coenzyme A cholesterol acyltransferase
- agLDL
aggregated LDL
- Alexa
AlexaFluor
- CE
cholesteryl ester
- CtB
cholera toxin subunit B
- CypHer 5E
CypHer 5E Mono N-hydroxysuccinimide ester
- FC
free cholesterol
- hMDM
human monocyte-derived macrophages
- SCC
surface-connected compartment
- V-ATPase
vacuolar (H+)-ATPase.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-07-0559) on October 7, 2009.
REFERENCES
- Azhar S., Reaven E. Scavenger receptor class BI and selective cholesteryl ester uptake: partners in the regulation of steroidogenesis. Mol. Cell. Endocrinol. 2002;195:1–26. doi: 10.1016/s0303-7207(02)00222-8. [DOI] [PubMed] [Google Scholar]
- Baron R., Neff L., Louvard D., Courtoy P. J. Cell-mediated extracellular acidification and bone resorption: evidence for a low pH in resorbing lacunae and localization of a 100-kD lysosomal membrane protein at the osteoclast ruffled border. J. Cell Biol. 1985;101:2210–2222. doi: 10.1083/jcb.101.6.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beletskii A., Cooper M., Sriraman P., Chiriac C., Zhao L., Abbot S., Yu L. High-throughput phagocytosis assay utilizing a pH-sensitive fluorescent dye. Biotechniques. 2005;39:894–897. doi: 10.2144/000112001. [DOI] [PubMed] [Google Scholar]
- Boren J., Gustafsson M., Skalen K., Flood C., Innerarity T. L. Role of extracellular retention of low density lipoproteins in atherosclerosis. Curr. Opin. Lipidol. 2000;11:451–456. doi: 10.1097/00041433-200010000-00002. [DOI] [PubMed] [Google Scholar]
- Bowman B. J., Bowman E. J. Mutations in subunit C of the vacuolar ATPase confer resistance to bafilomycin and identify a conserved antibiotic binding site. J. Biol. Chem. 2002;277:3965–3972. doi: 10.1074/jbc.M109756200. [DOI] [PubMed] [Google Scholar]
- Boyanovsky B. B., Shridas P., Simons M., van der Westhuyzen D. R., Webb N. R. Syndecan-4 mediates macrophage uptake of group V secretory phospholipase A2-modified LDL. J. Lipid. Res. 2009;50:641–650. doi: 10.1194/jlr.M800450-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. S., Goldstein J. L., Krieger M., Ho Y. K., Anderson R. G. Reversible accumulation of cholesteryl esters in macrophages incubated with acetylated lipoproteins. J. Cell Biol. 1979;82:597–613. doi: 10.1083/jcb.82.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buton X., Mamdouh Z., Ghosh R., Du H., Kuriakose G., Beatini N., Grabowski G. A., Maxfield F. R., Tabas I. Unique cellular events occurring during the initial interaction of macrophages with matrix-retained or methylated aggregated low density lipoprotein (LDL). Prolonged cell-surface contact during which ldl-cholesteryl ester hydrolysis exceeds ldl protein degradation. J. Biol. Chem. 1999;274:32112–32121. doi: 10.1074/jbc.274.45.32112. [DOI] [PubMed] [Google Scholar]
- Chao F. F., Amende L. M., Blanchette-Mackie E. J., Skarlatos S. I., Gamble W., Resau J. H., Mergner W. T., Kruth H. S. Unesterified cholesterol-rich lipid particles in atherosclerotic lesions of human and rabbit aortas. Am. J. Pathol. 1988;131:73–83. [PMC free article] [PubMed] [Google Scholar]
- de Winther M. P., Gijbels M. J., van Dijk K. W., van Gorp P. J., Suzuki H., Kodama T., Frants R. R., Havekes L. M., Hofker M. H. Scavenger receptor deficiency leads to more complex atherosclerotic lesions in APOE3Leiden transgenic mice. Atherosclerosis. 1999;144:315–321. doi: 10.1016/s0021-9150(98)00332-3. [DOI] [PubMed] [Google Scholar]
- Devlin C. M., Leventhal A. R., Kuriakose G., Schuchman E. H., Williams K. J., Tabas I. Acid sphingomyelinase promotes lipoprotein retention within early atheromata and accelerates lesion progression. Arterioscler. Thromb. Vasc. Biol. 2008;28:1723–1730. doi: 10.1161/ATVBAHA.108.173344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein J. L., Brown M. S., Anderson R. G., Russell D. W., Schneider W. J. Receptor-mediated endocytosis: concepts emerging from the LDL receptor system. Annu. Rev. Cell Biol. 1985;1:1–39. doi: 10.1146/annurev.cb.01.110185.000245. [DOI] [PubMed] [Google Scholar]
- Goldstein J. L., Ho Y. K., Basu S. K., Brown M. S. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA. 1979;76:333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinstein S., Nanda A., Lukacs G., Rotstein O. V-ATPases in phagocytic cells. J. Exp. Biol. 1992;172:179–192. doi: 10.1242/jeb.172.1.179. [DOI] [PubMed] [Google Scholar]
- Grosheva I., Haka A.S., Qin C., Pierini L.M., Maxfield F.R. Aggregated LDL in contact with macrophages induces local increases in free cholesterol levels that regulate local actin polymerization. Arterioscler. Thromb. Vasc. Biol. 2009;29:1615–1621. doi: 10.1161/ATVBAHA.109.191882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakala J. K., Oksjoki R., Laine P., Du H., Grabowski G. A., Kovanen P. T., Pentikainen M. O. Lysosomal enzymes are released from cultured human macrophages, hydrolyze LDL in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2003;23:1430–1436. doi: 10.1161/01.ATV.0000077207.49221.06. [DOI] [PubMed] [Google Scholar]
- Handley D. A., Arbeeny C. M., Witte L. D., Chien S. Colloidal gold–low density lipoprotein conjugates as membrane receptor probes. Proc. Natl. Acad. Sci. USA. 1981;78:368–371. doi: 10.1073/pnas.78.1.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M., Lin S. X., Karylowski O. J., Wustner D., McGraw T. E., Maxfield F. R. Vesicular and non-vesicular sterol transport in living cells. The endocytic recycling compartment is a major sterol storage organelle. J. Biol. Chem. 2002;277:609–617. doi: 10.1074/jbc.M108861200. [DOI] [PubMed] [Google Scholar]
- Havel R. J., Eder H. A., Bragdon J. H. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurdic P., Saltel F., Chabadel A., Destaing O. Podosome and sealing zone: specificity of the osteoclast model. Eur. J. Cell Biol. 2006;85:195–202. doi: 10.1016/j.ejcb.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Khoo J. C., Miller E., McLoughlin P., Steinberg D. Enhanced macrophage uptake of low density lipoprotein after self-aggregation. Arteriosclerosis. 1988;8:348–358. doi: 10.1161/01.atv.8.4.348. [DOI] [PubMed] [Google Scholar]
- Krieger M. Reconstitution of the hydrophobic core of low-density lipoprotein. Methods Enzymol. 1986;128:608–613. doi: 10.1016/0076-6879(86)28094-5. [DOI] [PubMed] [Google Scholar]
- Kruth H. S. Subendothelial accumulation of unesterified cholesterol. An early event in atherosclerotic lesion development. Atherosclerosis. 1985;57:337–341. doi: 10.1016/0021-9150(85)90045-0. [DOI] [PubMed] [Google Scholar]
- Kruth H. S. Sequestration of aggregated low-density lipoproteins by macrophages. Curr. Opin. Lipidol. 2002;13:483–488. doi: 10.1097/00041433-200210000-00003. [DOI] [PubMed] [Google Scholar]
- Llodra J., Angeli V., Liu J., Trogan E., Fisher E. A., Randolph G. J. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc. Natl. Acad. Sci. USA. 2004;101:11779–11784. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A., Cruz D., Asamoah N., Buxbaum A., Sohar I., Lobel P., Maxfield F. R. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol. Biol. Cell. 2007;18:1490–1496. doi: 10.1091/mbc.E06-10-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning-Tobin J. J., Moore K. J., Seimon T. A., Bell S. A., Sharuk M., Alvarez-Leite J. I., de Winther M. P., Tabas I., Freeman M. W. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler. Thromb. Vasc. Biol. 2009;29:19–26. doi: 10.1161/ATVBAHA.108.176644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield F. R., Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- Mukherjee S., Ghosh R.N., Maxfield F.R. Endocytosis Physiol. Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- Muller W. A., Weigl S. A. Monocyte-selective transendothelial migration: dissection of the binding and transmigration phases by an in vitro assay. J. Exp. Med. 1992;176:819–828. doi: 10.1084/jem.176.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers J. N., Tabas I., Jones N. L., Maxfield F. R. Beta-very low density lipoprotein is sequestered in surface-connected tubules in mouse peritoneal macrophages. J. Cell Biol. 1993;123:1389–1402. doi: 10.1083/jcb.123.6.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao T., Qin C., Grosheva I., Maxfield F. R., Pierini L. M. Elevated cholesterol levels in the plasma membranes of macrophages inhibit migration by disrupting RhoA regulation. Arterioscler. Thromb. Vasc. Biol. 2007;27:1596–1602. doi: 10.1161/ATVBAHA.107.145086. [DOI] [PubMed] [Google Scholar]
- Punturieri A., Filippov S., Allen E., Caras I., Murray R., Reddy V., Weiss S. J. Regulation of elastinolytic cysteine proteinase activity in normal and cathepsin K-deficient human macrophages. J. Exp. Med. 2000;192:789–799. doi: 10.1084/jem.192.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C., Nagao T., Grosheva I., Maxfield F. R., Pierini L. M. Elevated plasma membrane cholesterol content alters macrophage signaling and function. Arterioscler. Thromb. Vasc. Biol. 2006;26:372–378. doi: 10.1161/01.ATV.0000197848.67999.e1. [DOI] [PubMed] [Google Scholar]
- Sakr S. W., Eddy R. J., Barth H., Wang F., Greenberg S., Maxfield F. R., Tabas I. The uptake and degradation of matrix-bound lipoproteins by macrophages require an intact actin cytoskeleton, Rho family GTPases, and myosin ATPase activity. J. Biol. Chem. 2001;276:37649–37658. doi: 10.1074/jbc.M105129200. [DOI] [PubMed] [Google Scholar]
- Schissel S. L., Keesler G. A., Schuchman E. H., Williams K. J., Tabas I. The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J. Biol. Chem. 1998;273:18250–18259. doi: 10.1074/jbc.273.29.18250. [DOI] [PubMed] [Google Scholar]
- Sheriff S., Du H., Grabowski G. A. Characterization of lysosomal acid lipase by site-directed mutagenesis and heterologous expression. J. Biol. Chem. 1995;270:27766–27772. doi: 10.1074/jbc.270.46.27766. [DOI] [PubMed] [Google Scholar]
- Smith E. B., Massie I. B., Alexander K. M. The release of an immobilized lipoprotein fraction from atherosclerotic lesions by incubation with plasmin. Atherosclerosis. 1976;25:71–84. doi: 10.1016/0021-9150(76)90049-6. [DOI] [PubMed] [Google Scholar]
- Stenbeck G. Formation and function of the ruffled border in osteoclasts. Semin. Cell Dev. Biol. 2002;13:285–292. doi: 10.1016/s1084952102000587. [DOI] [PubMed] [Google Scholar]
- Suits A. G., Chait A., Aviram M., Heinecke J. W. Phagocytosis of aggregated lipoprotein by macrophages: low density lipoprotein receptor-dependent foam-cell formation. Proc. Natl. Acad. Sci. USA. 1989;86:2713–2717. doi: 10.1073/pnas.86.8.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- Tabas I. Nonoxidative modifications of lipoproteins in atherogenesis. Annu. Rev. Nutr. 1999;19:123–139. doi: 10.1146/annurev.nutr.19.1.123. [DOI] [PubMed] [Google Scholar]
- Tabas I. Apoptosis and plaque destabilization in atherosclerosis: the role of macrophage apoptosis induced by cholesterol. Cell Death Differ. 2004;11(Suppl 1):S12–S16. doi: 10.1038/sj.cdd.4401444. [DOI] [PubMed] [Google Scholar]
- Tabas I. Apoptosis and efferocytosis in mouse models of atherosclerosis. Curr. Drug Targets. 2007;8:1288–1296. doi: 10.2174/138945007783220623. [DOI] [PubMed] [Google Scholar]
- Tabas I., Williams K. J., Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- Williams K. J., Feig J. E., Fisher E. A. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat. Clin. Pract. Cardiovasc. Med. 2008;5:91–102. doi: 10.1038/ncpcardio1086. [DOI] [PubMed] [Google Scholar]
- Winkler H., Taylor K. A. Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt series in electron tomography. Ultramicroscopy. 2006;106:240–254. doi: 10.1016/j.ultramic.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Wu M., Holowka D., Craighead H. G., Baird B. Visualization of plasma membrane compartmentalization with patterned lipid bilayers. Proc. Natl. Acad. Sci. USA. 2004;101:13798–13803. doi: 10.1073/pnas.0403835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wustner D., Mondal M., Tabas I., Maxfield F. R. Direct observation of rapid internalization and intracellular transport of sterol by macrophage foam cells. Traffic. 2005;6:396–412. doi: 10.1111/j.1600-0854.2005.00285.x. [DOI] [PubMed] [Google Scholar]
- Yeung T., Grinstein S. Lipid signaling and the modulation of surface charge during phagocytosis. Immunol. Rev. 2007;219:17–36. doi: 10.1111/j.1600-065X.2007.00546.x. [DOI] [PubMed] [Google Scholar]
- Zhang W. Y., Gaynor P. M., Kruth H. S. Aggregated low density lipoprotein induces and enters surface-connected compartments of human monocyte-macrophages. Uptake occurs independently of the low density lipoprotein receptor. J. Biol. Chem. 1997;272:31700–31706. doi: 10.1074/jbc.272.50.31700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.