

Abstract
Utp9p is a nucleolar protein that is part of a subcomplex containing several U3 snoRNA-associated proteins including Utp8p, which is a protein that shuttles aminoacyl-tRNAs from the nucleolus to the nuclear tRNA export receptors Los1p and Msn5p in Saccharomyces cerevisiae. Here we show that Utp9p is also an intranuclear component of the Msn5p-mediated nuclear tRNA export pathway. Depletion of Utp9p caused nuclear accumulation of mature tRNAs derived from intron-containing precursors, but not tRNAs made from intronless pre-tRNAs. Utp9p binds tRNA directly and saturably, and copurifies with Utp8p, Gsp1p, and Msn5p, but not with Los1p or aminoacyl-tRNA synthetases. Utp9p interacts directly with Utp8p, Gsp1p, and Msn5p in vitro. Furthermore, Gsp1p forms a complex with Msn5p and Utp9p in a tRNA-dependent manner. However, Utp9p does not shuttle between the nucleus and the cytoplasm. Because tRNA splicing occurs in the cytoplasm and the spliced tRNAs are retrograded back to the nucleus, we propose that Utp9p facilitates nuclear reexport of retrograded tRNAs. Moreover, the data suggest that Utp9p together with Utp8p translocate aminoacyl-tRNAs from the nucleolus to Msn5p and assist with formation of the Msn5p-tRNA-Gsp1p-GTP export complex.
INTRODUCTION
Trafficking of tRNA between the nucleus and cytoplasm is a complex process that plays an essential role in protein synthesis, regulation of cell division (Kruse et al., 2000; White, 2004a,b; Ernens et al., 2006; Ghavidel et al., 2007), nutrient-related regulation of gene expression (Shaheen and Hopper, 2005, 2007; Hurto et al., 2007; Whitney et al., 2007), replication of the HIV genome (Zaitseva et al., 2006), and neuronal development (Schmidt et al., 2007). In eukaryotes, tRNAs are made as precursors that undergo a number of processing steps to become fully functional (Hopper and Phizicky, 2003). These steps involve removal of the 5′- and 3′-extensions, base modifications, addition of C, C, and A to their 3′ ends, and removal of an intron for some tRNA species. For intronless pre-tRNAs, the maturation process occurs entirely in the nucleus in yeast, plants, and mammals (Hopper and Phizicky, 2003; Paushkin et al., 2004; Englert et al., 2007). Maturation of intron-containing pre-tRNAs occurs primarily in the nucleus; however, in Saccharomyces cerevisiae unlike in vertebrates and plants, the intron-containing precursors are exported to the cytoplasm for removal of the introns and the spliced tRNAs are returned to the nucleus for reasons that are not understood (Huh et al., 2003; Yoshihisa et al., 2003, 2007; Shaheen and Hopper, 2005; Takano et al., 2005).
Although evidence reported suggests that nuclear import of spliced tRNAs occurs by a constitutive process (Takano et al., 2005), the mechanism involved is poorly understood. Genetic studies have shown that inactivation of Rna1p blocks retrograde movement of tRNA when cells are starved of amino acids (Shaheen and Hopper, 2005). This suggests that the Gsp1p-GTP/Gsp1p-GDP cycle plays a role in this process and that a β-karyopherin may facilitate nuclear import of tRNA. The β-karyopherin Mtr10p, which is involved in nuclear import of the RNA component of the S. cerevisiae telomerase, is also thought to play a role in retrograde movement of tRNA from the cytoplasm to the nucleus (Shaheen and Hopper, 2005). In contrast, other studies found that inactivation of Rna1p had no effect on nuclear import of tRNA in mutant cells lacking the function of the nuclear tRNA export receptors Los1p and Msn5p when grown in compete medium (Takano et al., 2005). Furthermore, sodium azide or 2-dexoyglucose treatment of msn5 los1 mutant cells was found to abolish nuclear import of tRNA (Takano et al., 2005). These data suggest that nuclear import of tRNA requires energy and that ATP may be the energy source. Thus, there may be two retrograde pathways operating in yeast: one that is constitutive and dependent on ATP, and the other requiring the Gsp1p-GTP/Gsp1p-GDP cycle and operating in response to nutrient-related stress; interestingly, deprivation of rat hepatoma H4IIE cells of amino acids has been shown to result in retrograde movement of cytoplasmic tRNAs to the nucleus (Shaheen et al., 2007).
The fully processed tRNAs from the two classes of precursor tRNAs are subjected to aminoacylation in the nucleus to ensure that they are functional before export to the cytoplasm (Lund and Dahlberg, 1998; Sarkar et al., 1999; Grosshans et al., 2000; Azad et al., 2001; Steiner-Mosonyi and Mangroo, 2004). In S. cerevisiae, the tRNA aminoacylation quality assurance step has been shown to occur in the nucleolus (Steiner-Mosonyi and Mangroo, 2004). However, it is not known whether nuclear tRNA aminoacylation occurs in the nucleolus of other eukaryotes including mammals. tRNAs deemed functional in S. cerevisiae are picked up in the nucleolus by Utp8p and delivered to Los1p and Msn5p for translocation across the nuclear pore complex (NPC; Hellmuth et al., 1998; Steiner-Mosonyi et al., 2003; Takano et al., 2005; Strub et al., 2007). An unidentified protein is also responsible for movement of tRNAs across the NPC in S. cerevisiae based on the finding that a los1 msn5 mutant strain was not affected in growth or viability (Takano et al., 2005). In mammalian cells, translocation of tRNAs across the NPC is facilitated by Xpo-t and Xpo-5, the orthologues of Los1p and Msn5p, respectively (Kutay et al., 1998; Arts et al., 1998; Bohnsack et al., 2002; Calado et al., 2002). tRNAs arriving at the cytoplasmic side of the NPC in S. cerevisiae are collected and delivered to the translation apparatus by Cex1p (McGuire and Mangroo, 2007). This step in mammalian cells most likely involves Scyl1 the orthologue of Cex1p.
Although the overall mechanism of nuclear tRNA export is, in general, poorly understood, the details of specific steps are slowly emerging from studies carried out in both mammalian cells and S. cerevisiae to identify and characterize participants of the process. Recent biochemical studies in S. cerevisiae have provided evidence suggesting that Utp8p uses a channelling mechanism to collect aminoacyl-tRNAs from the aminoacyl-tRNA synthetases in the nucleolus and to deliver them to the nuclear tRNA export receptors located in the nucleoplasm and NPC (Strub et al., 2007). However, the molecular mechanism by which Utp8p trafficks tRNAs from the nucleolus to the export receptors is currently not known. Utp8p is a component of the U3 snoRNA-associated protein (Utp) complex involved in 18S rRNA biogenesis (Dragon et al., 2002). Furthermore, Utp8p, Utp17p, Utp15p, Utp10p, Utp9p, Utp5p, and Utp4p of the Utp complex have been found to form a subcomplex involved in regulating transcription of the rRNA gene (Gallagher et al., 2004; Krogan et al., 2004). It is therefore possible that other Utp proteins of the subcomplex could also be playing a role in the trafficking of tRNAs from the nucleolus to the export receptors.
In this report we provide biochemical evidence that Msn5p is an authentic nuclear export receptor for tRNA in S. cerevisiae. Furthermore, we show that Utp9p is a nucleolar tRNA-binding protein that plays an intranuclear role that is essential for nuclear export of mature tRNAs derived from intron-containing pre-tRNAs, but not export of tRNAs derived from intronless pre-tRNAs. Moreover, nutrient stress conditions that block nuclear reexport of mature tRNAs derived from intron-containing pre-tRNAs had no effect on nuclear export of tRNAs made from intronless precursors. These results combined with the finding that Utp9p interacts with Msn5p but not with Los1p suggest that Utp9p participates in the reexport of retrograded tRNAs derived from intron-containing pre-tRNAs by the Msn5p-mediated nuclear tRNA export pathway. Furthermore, the data imply that Utp9p together with Utp8p translocate aminoacylated tRNAs from the nucleolus to Msn5p and assist with the formation of the Msn5p-tRNA-Gsp1p-GTP export complex.
MATERIALS AND METHODS
Strains and Plasmids
The S. cerevisiae strains used in this study are listed in Table 1. Plasmids pET19b and pET23d were purchased from Novagen (San Diego, CA). The pGEX-GST-TEV plasmid was obtained from Dr. D. Heinrichs (University of Western Ontario, Canada). pGEX-4T-GSP1 and pGEX-GSP1p (Q71L) were provided by Dr. U. Stochaj (McGill University, Montreal, Canada). pET28GST-LIC was obtained from the Structural Genomics Consortium (Toronto, Canada). pET9D-5S, pRS315-RPL25-GFP, and pRS316-RPS2-GFP were obtained from Dr. E. Hurt (University of Heidelberg, Germany). The BG1805-GAL1-UTP13 plasmid was obtained from Open Biosystems (Huntsville, AL). pCEN-URA-GAL1-UTP10 was prepared by introducing the UTP10 open reading frame (ORF), obtained by PCR amplification of S. cerevisiae genomic DNA, into the XhoI site of pRS416-CEN-URA-GAL1. The plasmid pET19-UTP9 was constructed by inserting the UTP9 ORF into the NdeI and BamHI sites of pET19b. The UTP9 ORF obtained by PCR using S. cerevisiae genomic DNA was cloned into the BseRI site of pET28GST-LIC by ligation-independent cloning using the In-Fusion PCR Cloning kit from Clontech Laboratories (Palo Alto, CA). The plasmids pET23d-MSN5, pGEX-LOS1, pGEX-TYS1, pET19-UTP8, pGEX-UTP8, and pET19b-GSP1 were described previously (Strub et al., 2007). pET19b-GSP1 Q71L was prepared by obtaining the GSP1 ORF containing the Q71L mutation by PCR using pGEX-GSP1 (Q71L) as the template and cloning into the NdeI and BamHI sites of pET19b.
Table 1.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BY4741 | MATa, his3, leu2, met15, ura3 | Open Biosystems |
| UTP8-TAP | MATa, his3, leu2, met15, ura3, UTP8::TAP-HIS3 | Open Biosystems |
| UTP9-TAP | MATa, his3, leu2, met15, ura3, UTP9::TAP-HIS3 | Open Biosystems |
| UTP21-TAP | MATa, his3, leu2, met15, ura3, UTP21::TAP-HIS3 | Open Biosystems |
| LOS1-TAP | MATa, his3, leu2, met15, ura3, LOS1::TAP-HIS3 | Open Biosystems |
| MSN5-TAP | MATa, his3, leu2, met15, ura3, MSN5::TAP-HIS3 | Open Biosystems |
| CCA1-TAP | MATa, his3, leu2, met15, ura3, CCA1::TAP-HIS3 | Open Biosystems |
| MTR10-TAP | MATa, his3, leu2, met15, ura3, MTR10::TAP-HIS3 | Open Biosystems |
| msn5 | MATa, his3, leu2, met15, ura3, msn5::KanMX6 | Open Biosystems |
| utp9-ts | MATa, ura3, leu2, his3, can1::LEU2-MFA1pr::His3 utp9-ts::URA3 | Ben-Aroya et al. (2008) |
| utp8-ts | MATa, ura3, leu2, his3, lys2, met15, can1::LEU2-MFA1pr::His3 utp8-ts::URA3 | Ben-Aroya et al. (2008) |
| W303 | MATa, ade2-1, ura3-1, his3-11, trp1-1, leu2-3, 112, can100 | ATCC |
| kar1-1 | MATα | Shaheen and Hopper (2005) |
| YKL200 | W303, MATa, ade2-1, ura3-1, his3-1, trp1-1, leu2-3, can1-100,UBR1::GAL-HA-UBR1 (HIS3) | Euroscarf |
| utp9-td | W303, MATa, ade2-1, ura3-1, his3-11, trp1-1, leu2-3, can1-100,UBR1::GAL-HA-UBR1 (HIS3), YHR196w::CUP1-UBI4-myc-YHR196w (kanMX) | Euroscarf |
| utp8-td | W303, MATa, ade2-1, ura3-1, his3-11, trp1-1, leu2-3, can1-100,UBR1::GAL-HA-UBR1 (HIS3), YGR128c::CUP1-UBI4-myc-YGR128c (kanMX) | This study |
| xpo1-1ts | W303, MATa, xpo1::LEU2, pRS313-xpo1-1ts | Stade et al. (1997) |
| rio2-1 | MATa his3 leu2 trp1 ura3 rio2::kanMX4, pRS315-rio2-1 (LEU) | Schafer et al. (2003) |
| nmd3-2 | MATa trp1 his3 leu2 ura3 nmd3::kanMX4, pRS313-nmd3-2 (HIS) | Gadal et al. (2001) |
| BY4743 | MATa/MATα; his3D1/his3D1, leu2D0/leu2D0, met15D0/MET15,LYS2/lys2D0, ura3D0/ura3D0 | Open Biosystems |
| utp8: PGAL1-UTP8 | Derivative of BY4743, utp8::kanMX6, pRS416-GAL1-UTP8, MATa | Steiner-Mosonyi et al. (2003) |
| utp10: PGAL1-UTP10 | Derivative of BY4743, utp10::kanMX6, pRS416-GAL1-UTP10, MATα | This study |
| utp13: PGAL1-UTP13 | Derivative of BY4743, utp13::kanMX6, BG1805-GAL1-UTP13, MATa | This study |
Generation of utp8-td
The utp8-td strain was generated using a one-step PCR approach as described (Kanemaki et al., 2003). Briefly, a PCR product was generated using 70-mer primers that contain 50 bases at the 5′-end corresponding to sequences at the end of the UTP8 ORF and promoter region, and 20 bases at the 3′-end corresponding to sequences specific to the degron cassette using pKL187 plasmid (Euroscarf, Frankfurt, Germany) as the template. The PCR product was transformed into the diploid YMK165 strain (Euroscarf) containing one copy of UBR1::HIS3 under the control of the GAL1 promoter. The transformed diploid strain was sporulated in the presence of 0.1 mM CuSO4 and screened for haploids containing the HIS3 and KanMX6 markers. Integration of the degron cassette into the N-terminus of UTP8 was confirmed by PCR using primers specific to the degron cassette and the UTP8 ORF. Expression of the fusion protein was confirmed by Western blotting analysis using anti-Myc tag (Cell Signaling Technology, Beverly, MA) and anti-Utp8p antibodies.
Overexpression and Purification of His-tagged Proteins
Escherichia coli BL21 (DE3) Codon Plus RIL (Novagen) with pET19b-UTP9, pET19b-UTP8, pET19b-GSP1, or pET23d-MSN5 was grown in 1 l of 2YT broth containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol at 37°C to an A600 of 0.4. The cultures were shifted to 15°C and expression of the His-tagged proteins was induced with 200–500 μM isopropyl β-d-thiogalactoside (IPTG) for 16 h. The cells were collected by centrifugation and resuspended in 30 ml of binding buffer: 20 mM NaH2PO4, pH 7.5, buffer containing 500 mM NaCl, 5 mM imidazole, 10 μg/ml RNase A and a mixture of protease inhibitors (Complete EDTA-free; Roche Applied Science, Indianapolis, IN). The cells were lysed at 70,000 kPa using a French press, and the lysate was clarified by centrifugation at 27,000 × g for 30 min. The clarified lysate was applied onto a 1 ml HisTrap HP column (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom). The column was washed with 20 ml of 20 mM NaH2PO4, pH 7.5, buffer containing 500 mM NaCl and 50 mM imidazole and then with 20 mM NaH2PO4, pH 7.5, buffer containing 500 mM KCl to remove tRNA. The His-tagged proteins were eluted using a gradient of increasing concentration of imidazole in 20 mM NaH2PO4, pH 7.5, buffer containing 500 mM NaCl. Fractions containing purified protein were pooled and dialyzed against IPP150 buffer (25 mM Tris-HCl, pH 7.5, containing 150 mM NaCl, 20% glycerol [wt/vol], and 0.1% Nonidet P-40 [vol/vol]). The protein was concentrated and stored at −80°C. Utp9p-His was further purified by ion exchange chromatography using a HiTrap Q HP Sepharose column (GE Healthcare). HisTrap HP eluate containing Utp9p-His was dialyzed against 20 mM Tris-HCl, pH 7.5, buffer containing 100 mM NaCl and applied onto the HiTrap Q HP column. The column was eluted with a gradient of increasing concentration of NaCl up to 500 mM NaCl. Fractions containing purified protein were pooled and dialyzed against IPP150 and stored at −80°C after concentration. An aliquot of the concentrated proteins were subjected to phenol-chloroform extraction and RNA precipitation, and analyzed on a 2% agarose gel to check for the presence of tRNA. tRNA could not be detected with any of the purified proteins.
Overexpression and Purification of GST-tagged Proteins
E. coli BL21 (DE3) Codon Plus RIL (Novagen) containing pGEX-LOS1, pGEX-TYS1, or pGEX-UTP8 was grown in 1 l of 2YT medium containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol at 37°C to an A600 of 0.4. E. coli BL21 (DE3) Codon Plus RIL containing pET28GST-LIC-UTP9 was grown in the presence of 100 μg/ml kanamycin and 34 μg/ml chloramphenicol. The cultures were shifted to 15°C, and protein expression was induced using 0.2–1 mM IPTG for 16 h. The cells were collected by centrifugation and resuspended in 30 ml of binding buffer (10 mM Na2HPO4 and 1.8 mM KH2PO4, pH 7.3, containing 140 mM NaCl, 2.7 mM KCl, 1 mM dithiothreitol [DTT]), a mixture of protease inhibitors (Complete EDTA-free; Roche Applied Science), and 10 μg/ml RNase A. The cells were lysed by using a French pressure cell at 70,000 kPa, and the lysate was clarified by centrifugation at 27,000 × g for 30 min. The clarified lysate was applied onto a GSTrap column (GE Healthcare). The column was washed with binding buffer and then with 20 mM NaH2PO4, pH 7.5, buffer containing 500 mM KCl to remove tRNA. The glutathione S-transferase (GST)-tagged protein was eluted using 50 mM Tris-HCl, pH 8.0, buffer containing 10 mM reduced glutathione and 1 mM DTT. The eluates were dialyzed against IPP150 buffer and stored at −80°C. GST-Utp8p was further purified by ion exchange chromatography using a HiTrap Q HP Sepharose column (GE Healthcare). GSTrap eluate containing GST-Utp8p was dialyzed against 20 mM Tris-HCl, pH 7.5, buffer containing 100 mM NaCl and applied onto the HiTrap Q HP column. The column was eluted with a gradient of increasing concentration of NaCl up to 500 mM NaCl. Fractions containing purified protein were pooled and dialyzed against IPP150 and stored at −80°C after concentration. An aliquot of the concentrated proteins were subjected to phenol-chloroform extraction and RNA precipitation, and analyzed on a 2% agarose gel to check for the presence of tRNA. tRNA could not be detected with any of the purified proteins.
Fluorescence In Situ Hybridization Analysis
The utp9-td, utp8-td, and YKL200 strains were grown at 23°C in YPD medium supplemented with 0.1 mM CuSO4 to an A600 of 0.6–0.7. The cells were washed with water and shifted to YPG medium supplemented with 0.1 mM CuSO4 and grown at 23°C for 6 h. The cells were washed and shifted to YPG medium without CuSO4 and grown at 37°C for 1–6 h. The temperature-sensitive strains (utp8-ts and utp9-ts) were grown in YPD medium at 23°C to an A600 of 0.6; the cells were diluted to an A600 of 0.1 in YPD medium and grown at 37°C for 1–9 h. Fifty milliliters of culture was fixed and permeabilized as described (Grosshans et al., 2000; Steiner-Mosonyi and Mangroo, 2004) and incubated at 37°C for 1 h in hybridization buffer (4× SSC, 50% formamide, 10% dextran sulfate, 125 μg/ml E. coli 5S rRNA, 500 μg/ml salmon sperm DNA, 0.5 U/μl RNasin, 10 mM DTT, and 1× Denhardt's). Hybridization was carried out at 37°C for 12 h in hybridization buffer containing 0.5 pmol/μl 5′-end Cy3-labeled oligonucleotide or 5′-end Alexa488-labeled oligo-dT. The cells were washed once for 5 min at room temperature with 2× SSC, two times with 1× SSC, and once with 0.5× SSC. DAPI (1 μg/ml) was used to visualize nuclear DNA. The slides were viewed under a 100× objective lens of a Nikon Eclipse 6600 microscope (Melville, NY). The images were recorded using a Coolsnapfx monochrome CCD digital camera (Roper Scientific, Tucson, AZ) and processed using Metamorph (Universal Imaging, West Chester, PA).
Oligonucleotides Used in This Study
The cellular location of mature tRNATyr, tRNAGly, tRNAHis, and tRNALeu was detected using 5′-GCGAGTCGAACGCCCGATCTCAAGATTTACAGTCTTGCGCCTTAAACCAACTTGGCTACC-3′, 5′-GGCCCAACGATGGCAACG-3′, 5′-TCCTAGAATCGAACCAGGGTTTCATCGGCCACAACGATGTGTACTAACCACTATACTAAG-3′, and 5′-GCATCTTACGATACCTGAGCTTGAATCAGGCGC-3′, respectively. tRNATrp was detected using 5′-AACCTGCAACCCTTCGA-3′. This oligonucleotide hybridizes to all the different processing forms of tRNATrp. The 18S and 25S rRNAs were detected using 5′-CATGGCTTAATCTTTGAGAC-3′ and 5′-CTCCGCTTATTGATATGC-3′, respectively. mRNA was detected with a 30-mer poly-dT oligonucleotide.
Tandem Affinity Purification
Tandem affinity purification (TAP)-tagged strains were grown in 2 l of YPD medium to an A600 of 2.0 at 30°C. The cells were harvested by centrifugation, resuspended in 50 ml of NP-40 buffer (15 mM Na2HPO4, pH 7.2, buffer containing 10 mM NaH2PO4, 2% NP-40 [vol/vol], 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 0.1 mM Na3VO4, and protease inhibitors) and lysed using glass beads at 4°C. The lysate was clarified by ultracentrifugation at 142,000 × g for 75 min at 4°C and subjected to sequential affinity purification using IgG-Sepharose (GE Healthcare) and calmodulin-Sepharose as described (Puig et al., 2001). The proteins in the final eluate were precipitated using 25% trichloroacetic acid, solubilized in LDS sample buffer (Invitrogen, Carlsbad, CA) and separated on 4–12% Novex Bis-Tris gels (Invitrogen). The separated proteins were transferred electrophoretically to Immobilon PVDF membrane (Millipore, Bedford, MA) and probed with rabbit anti-Utp9p, anti-Utp8p, anti-human TysRS, anti-Cex1p, anti-Los1p, anti-Gsp1p, or mouse anti-human eEF-1A (Upstate Cell Signaling, Waltham, MA).
Protein Extraction and Western Blot Analysis to Monitor Protein Degradation
utp9-td, utp8-td, and the parental YKL200 strain were grown as above and shifted to YPG medium without CuSO4 and grown at 37°C for 1–3 h. Three milliliters of culture was pelleted, and the cell pellet was resuspended in breaking buffer (50 mM sodium phosphate, pH 7.4, 1 mM EDTA, 5% glycerol, and 1 mM PMSF). The cells were lysed using glass beads and clarified by centrifugation. An aliquot of the cell lysate was subjected to PAGE using a 4–12% Novex Bis-Tris gel, and the separated proteins were transferred to Immobilon-P PVDF membranes. Degradation of the target proteins was monitored by probing the membranes with anti-Utp9p, anti-Utp8p, or anti-Myc tag (Cell Signaling Technology). Expression of Ubr1p was monitored by probing the membranes with anti-hemagglutinin (HA) antibodies (Sigma-Aldrich, St. Louis, MO). The membranes were probed with anti-actin antibodies to confirm uniform loading of the samples.
Northern Hybridization Analysis
The utp9-td and YKL200 strains were grown at 23°C in YPD medium supplemented with 0.1 mM CuSO4 to an A600 of 0.6–0.7. The cells were washed with water and shifted to YPG supplemented with 0.1 mM CuSO4 and grown at 23°C for 6 h. The cells were washed and shifted to YPG medium without CuSO4 and grown at 37°C for 1–6 h. The cells were collected and total RNA was isolated using glass beads as described (Cleary and Mangroo, 2000). For analysis of tRNA processing and maturation, 2.5 μg of total RNA was separated on a 10% polyacrylamide gel containing 8 M urea using 1× TBE at room temperature. For analysis of the processing of rRNA, 2.5 μg of total RNA was separated on a 1.2% formaldehyde-MOPS agarose gels using 1× MOPS at room temperature. The separated RNAs were transferred electrophoretically onto Nytran membrane (Whatman, Clifton, NJ). The membranes were incubated at 37°C for 4 h in prehybridization solution consisting of 4× SSPE (1× SSPE = 0.18 M NaCl, 10 mM NaH2PO4, and 1 mM Na2EDTA), 250 μg/ml sheared and denatured salmon sperm DNA, 0.1% SDS, and 10× Denhardt's solution (1× Denhardt's = 0.02% bovine serum albumin, 0.02% polyvinylpyrrolidone 40, and 0.02% Ficoll). Hybridization was performed overnight at 37°C in prehybridization solution containing 5′-end 32P-labeled oligonucleotide (1–2 × 106 cpm/ml). The membranes were washed twice for 30 min at room temperature and once for 30 min at 38°C with 1× SSPE and 0.1% SDS and subjected to autoradiography.
Analysis of the Aminoacylation Status of tRNAs
The utp9-td, utp8-td, and YKL200 strains were grown at 23°C in YPD supplemented with 0.1 mM CuSO4 to an A600 of 0.6–0.7. The cells were washed with water and shifted to YPG medium supplemented with 0.1 mM CuSO4 and grown at 23°C for 6 h. The cells were washed and shifted to YPG medium without CuSO4 and grown at 37°C for 3 h. The cells were centrifuged and nuclear and postnuclear fractions were isolated as described (Steiner-Mosonyi and Mangroo, 2004). Total RNA from the nuclear and postnuclear fractions was separated by electrophoresis on a 6.5% polyacrylamide gel containing 8 M urea at 4°C using 100 mM sodium acetate buffer, pH 5.0, and transferred onto Nytran Plus membranes. Northern analysis was performed as described above. Deacylated tRNA marker was prepared by incubating nuclear and cytoplasmic RNA in 100 mM Tris-HCl, pH 9.5, at 37°C for 1 h.
Loading Gsp1p with [γ-32P]GTP
[γ-32P] GTP (65 μM, 6000Ci/mmol) and 140 μM GTP were incubated with 30 μM Gsp1p in PBSM (10 mM Na2HPO4, 1.75 mM KH2PO4, 137 mM NaCl, 5 mM KCl, and 0.5 mM MgCl2, pH 7.4, containing 10% glycerol) containing 5 mM EDTA at room temperature. After 30 min, MgCl2 was added to a concentration of 10 mM and placed on ice for 10 min (Lounsbury and Macara, 1997; Kutay et al., 1998). The mixture was then passed through a G50 spin column preequilibrated with PBSM to remove unbound nucleotide. The amount of Gsp1p-GTP [γ-32P] was determined by filtering an aliquot of the flow through fraction from the G50 resin through nitrocellulose membrane and determining the amount of radioactivity trapped on the membrane.
Heterokaryon Shuttling Assay
W303a cells transformed with pRS416-CEN-URA-GAL1-UTP9-GFP or pRS416-CEN-URA-GAL1-XPO1-GFP were grown in CS medium containing 2% galactose, 2% raffinose and lacking uracil to midlogarithmic phase at 30°C. Expression of the green fluorescent protein (GFP) proteins was repressed by shifting the cells to CS medium containing 2% dextrose (CSD) and lacking uracil for 1 h at 30°C. The donor cells were mixed with recipient kar1-1α cells grown in CSD medium and pelletted by centrifugation (Dilworth et al., 2001; Steiner-Mosonyi et al., 2003). The cell pellet was resuspended in CSD medium and incubated at room temperature for 30 min. An aliquot of the cells was placed on glass slides coated with CSD medium containing 2% agarose and sealed with a coverslip. The slides were incubated at room temperature and visualized at various times by direct fluorescence microscopy.
In Vitro Protein-binding Analyses
The GST fusion proteins Los1p, Tys1p, Utp9p, Utp8p, or GST were bound to glutathione (GT)-Sepharose in IPP150 buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.1% NP-40) containing a cocktail of protease inhibitors and 1 mM DTT at 4°C for 1 h. All washing steps were carried with IPP150 buffer, and when required, total yeast mature tRNA was provided at a 10-fold molar excess of the Kd of the protein. Binding was performed with a twofold molar excess of the interacting protein at 4°C. The GST-tagged proteins were eluted from the resin using TEV, thrombin or 10 mM reduced glutathione. The final eluates and unbound proteins in the washes were subjected to electrophoresis on 4–12% Novex Bis-Tris gels and transferred to Immobilon-P PVDF membranes. The proteins were detected by Western blot analysis followed by SYPRO Ruby staining of the blots.
Binding of Nucleotides to Gsp1p (Q71L) for In Vitro Protein-binding Analyses
Gsp1p (Q71L) was purified in a single step using Talon metal affinity chromatography. The purified protein was dialyzed against 25 mM Tris-HCl, pH 7.4, buffer containing 150 mM NaCl, and incubated with a 10-fold molar excess of GTP in 25 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 5 mM EDTA, and 1 mM DTT at room temperature for 30 min. Magnesium chloride was added to a final concentration of 10 mM, and the mix was incubated on ice for at least 15 min (Bischoff et al., 1994, 1995). Unbound nucleotide was removed by passing the mix through G50 spin columns.
RNA-binding Analysis
Interaction of Utp9p with tRNA was determined by substrate-induced intrinsic fluorescence quenching of tryptophan residues (Steiner-Mosonyi et al., 2003; McGuire and Mangroo, 2007). The analysis was performed in 20 mM HEPES, pH 7.4, buffer containing 100 mM NaCl, 0.25 μM Utp9p, and increasing amounts of mature yeast tRNA, 75-mer single-stranded DNA oligonucleotide, or E. coli 5S rRNA (1, 2, 4, 6.25, and 8 μM). The 5S rRNA was prepared by in vitro transcription of linearized pET9D-5S template using Ribomax kit from Promega (Madison, WI). Control reactions lacking the protein and containing tRNA or oligonucleotide alone were prepared as above. The reaction mixtures were incubated for 1 h at 4°C, and tryptophan fluorescence was measured using a Photon Technology International spectrofluorimeter (Lawrenceville, NJ) with excitation and emission slits set to 4 nm and excitation and emission wavelengths of 295 and 334 nm, respectively. The fluorescence intensity of each sample was subtracted from that of the appropriate control reaction and expressed as a percent reduction of the fluorescence intensity obtained with Utp9p alone. Msn5p interaction with tRNA was performed as described for Utp9p.
Fluorescence In Situ Hybridization Analysis in Conditional Mutants of utp10 and utp13
The utp10 pCEN-URA-GAL1-UTP10 and utp13 GB1805-CEN-URA-GAL1-UTP13 strains were prepared by sporulation and tetrad dissections of heterozygous strains with pCEN-URA-GAL1-UTP10 and BG1805-CEN-URA-GAL1-UTP13, respectively. The utp8 pCEN-URA-GAL1-UTP8 strain was described previously (Steiner-Mosonyi et al., 2003). The strains were grown in CS medium containing 2% galactose and 200 μg/ml G418, and lacking uracil to an OD600 of 0.6 at 30°C. The cells were washed and diluted in CS medium containing 2% dextrose and lacking uracil to an OD600 of 0.1 and grown at 30°C for 6 h. The cells were fixed at the indicated time points, and fluorescence in situ hybridization (FISH) analysis was performed to monitor the distribution of mature tRNATyr as described above.
Nuclear-Cytoplasmic Distribution of Ribosomal Subunits
The nmd3-2 Rpl25-GFP and rio2-1 Rps2-GFP strains were grown in CS medium containing 2% dextrose and lacking leucine or uracil, respectively at 23°C to an OD600 of 0.6. The cells were diluted to an OD600 of 0.3 in prewarmed medium and shifted to 37°C for 4 h. The cells were treated with DAPI (2 μg/ml) for 30 min and washed with 1× PBS. YKL200- and utp9-td–expressing Rpl25-GFP or Rps2-GFP were grown in CS medium containing 2% dextrose and lacking leucine or uracil, respectively, at 23°C to an OD600 of 0.6. Expression of Ubr1p and depletion of Utp9p were induced as described earlier. The cells were treated with DAPI (2 μg/ml) stain for 30 min and washed with 1× PBS. Localization of Rpl25 and Rsp2 was monitored by fluorescence microscopy.
RESULTS
Loss of Utp9p Function Affects Nuclear Export of Mature tRNAs Derived from Intron-containing Pre-tRNAs But Not Export of tRNAs Obtained from Intronless Precursors
As part of our ongoing efforts to identify proteins that participate in nuclear tRNA export in S. cerevisiae, we focused on investigating whether Utp proteins of the Utp8p-containing subcomplex could be playing a role in the nuclear tRNA export process. The Utp protein chosen for investigation is Utp9p. It is an essential protein located in the nucleolus (Giaever et al., 2002; Huh et al., 2003), and like Utp8p, an orthologue of Utp9p is not found in higher eukaryotes by PSI-BLAST searches.
To test the possibility that Utp9p is involved in the nuclear tRNA export process, we investigated whether depletion of Utp9p affects nuclear-cytoplasmic tRNA trafficking. Because Utp9p is essential, this analysis was conducted using a conditional mutant strain of Utp9p (utp9-td). In this strain, the chromosomal gene of Utp9p is under the control of the copper-inducible CUP1 promoter and is tagged at the 5′-end with a heat inducible degron cassette (Kanemaki et al., 2003). The degron cassette consists of a mutant form of the mouse dihydrofolate reductase, which is stable at 23°C but denatures at 37°C, exposing lysine residues that are recognized by the ubiquitin-mediated proteosome degradation pathway. To control the degradation, the chromosomal UBR1 gene encoding the yeast ubiquitin ligase is under the control of the GAL1 promoter. Thus, depletion of Utp9p is achieved by repression of the CUP1 promoter, induction of the expression of Ubr1p, and incubation of the cells at 37°C. This strategy is clearly capable of depleting Utp9p, because utp9-td did not grow at 37°C on YP medium containing galactose (YPG) and without copper (Supplemental Figure S1). Similarly, utp8-td, which was prepared for use as a control, did not grow under these conditions (Supplemental Figure S1).
The time required to deplete Utp9p or Utp8p was determined by Western blot analyses. utp9-td (Figure 1A, left panel) and utp8-td (Figure 1A, right panel) were grown at 23°C in YP medium containing glucose (YPD) and copper (lane 1) and transferred to YPG medium with copper (lane 2) for 6 h at 23°C. The cells were then switched to YPG medium lacking copper and grown at 37°C for 1 (lane 3) or 3 h (lane 4). The level of actin was used to monitor the amount of cell extracts analyzed. The results indicate that Utp9p was maximally depleted within 1 h of incubation of utp9-td at 37°C in YPG medium lacking copper. In contrast, it took ∼3 h to significantly decrease the level of Utp8p. Western blot analysis also indicate that depletion of the proteins coincide with the induced expression of Ubr1p (Figure 1A, left).
Figure 1.

Depletion of Utp9p resulted in nuclear accumulation of tRNATyr. The wild-type YKL200 strain, utp9-td and utp8-td were grown at 23°C in YPD medium supplemented with 0.1 mM CuSO4. Expression of Ubr1p was then induced by growing the cells at 23°C for 6 h in YPG medium supplemented with 0.1 mM CuSO4. Degradation of Utp9p or Utp8p was induced by growing the cells in YPG medium without CuSO4 at 37°C for 1 and 3 h. Western blot analyses were performed to monitor the level of Utp9p (A, left), Ubr1p (A, left), actin (A) and Utp8p (A, right) in cell extracts prepared from utp9-td and utp8-td grown under the different conditions. The cellular location of tRNATyr in YKL200 (B), utp9-td (C), and utp8-td (D) was monitored by FISH and the DNA was visualized by DAPI staining.
The effect of depletion of Utp9p on nuclear-cytoplasmic tRNA trafficking was assessed by using FISH to monitor the cellular location of tRNATyr, a tRNA derived from intron-containing pre-tRNA. tRNATyr was uniformly distributed in the wild-type isogenic YKL200 strain (Figure 1B), utp9-td (Figure 1C), and utp8-td (Figure 1D) grown in YPD medium with copper or YPG medium containing copper at 23°C. Furthermore, the nuclear-cytoplasmic distribution of tRNATyr in YKL200 incubated at 37°C in YPG medium without copper for 1 or 3 h was not affected. In contrast, nuclear accumulation of tRNATyr was detected when Utp9p or Utp8p was depleted for 1 or 3 h.
To investigate whether nuclear accumulation of tRNATyr was due specifically to the loss of Utp9p function or to degradation of a protein involved in nuclear tRNA export and associates with the degron-tagged Utp9p, nuclear-cytoplasmic trafficking of tRNATyr was assessed in a temperature-sensitive mutant of Utp9p (Ben-Aroya et al., 2008; Figure 2). FISH analyses indicate that the tRNA was distributed uniformly in the wild-type BY4741 strain (Figure 2A), utp9-ts (Figure 2B), utp8-ts (Figure 2C), and XPO1 (Figure 2D) grown at 23°C. Nuclear accumulation of tRNATyr was observed in utp9-ts and utp8-ts, but not in BY4741 incubated at 39°C for 3 h or xpo1-1ts grown at 37°C for 3 h. Furthermore, tRNATyr was detected in the nucleus of utp9-ts and utp8-ts, but not in BY4741 grown at 39°C for 6 or 9 h (Supplemental Figure S2). The data suggest that the nuclear accumulation of tRNATyr observed in utp9-td was due specifically to the loss of Utp9p function.
Figure 2.

Loss of Utp9p function also caused nuclear accumulation of tRNATyr in a temperature-sensitive utp9 mutant strain. The wild-type BY4741 strain (A), utp9-ts (B), and utp8-ts (C) were grown in YPD medium at 23°C and shifted to 39°C for 3 h. The W303 XPO1 and xpo1-1ts strains (D) were grown at 23°C and shifted to 37°C for 3 h. The cellular location of tRNATyr was monitored by FISH and the DNA was visualized by DAPI staining.
To ascertain whether the Utp8p associated subcomplex is involved in nuclear tRNA export, the effect of depletion of Utp10p, which is a member of the subcomplex, on nuclear export of tRNATyr was monitored. Furthermore, to exclude the possibility that depletion of any Utp protein will result in a defect in nuclear tRNA export, nuclear-cytoplasmic tRNA trafficking was investigated in a conditional mutant of Utp13p, a Utp protein that is not part of the Utp8p containing subcomplex. The wild-type BY4741 pCEN-URA-GAL1, utp8 pCEN-URA-GAL1-UTP8, utp10 pCEN-URA-GAL1-UTP10, or utp13 BG1805-CEN-URA-GAL1-UTP13 strain was grown in CS medium containing galactose and then in CS medium containing dextrose for 6 h to repress expression of the Utp proteins (Figure 3). FISH analyses show that shifting the wild-type BY4741 strain (Figure 3A) from galactose to glucose did not affect nuclear export of tRNATyr. Similarly, the nuclear-cytoplasmic distribution of tRNATyr was not affected when Utp10p (Figure 3C) or Utp13p (Figure 3D) was depleted for 6 h. Furthermore, a defect in nuclear tRNA export was not observed when Utp10p or Utp13p was depleted over a 9-h period (data not shown). However, loss of the function of Utp8p resulted in nuclear retention of tRNATyr (Figure 3B). These data suggest that only certain members of the Utp8p containing subcomplex are components of the S. cerevisiae nuclear tRNA export machinery.
Figure 3.

Depletion of Utp10p, a member of the Utp8p-containing subcomplex, or Utp13p, a Utp protein involved in 18S rRNA biogenesis does not affect nuclear tRNA export. The wild-type BY4741 strain harboring pCEN-URA-GAL1 (A), utp8 pCEN-URA-GAL1-UTP8 (B), utp10 pCEN-URA-GAL1-UTP10 (C), and utp13 BG1805-CEN-URA-GAL1-UTP13 (D) were grown in CS medium containing 2% galactose and lacking uracil to A600 of 0.6 at 30°C. The cells were washed and grown in CS medium containing 2% dextrose and lacking uracil at 30°C for 6 h. FISH was then used to monitor the nuclear-cytoplasmic distribution of tRNATyr. DNA was visualized by DAPI staining.
Depletion of Utp9p has been shown to cause a marked decrease in transcription of the rRNA gene (Gallagher et al., 2004). Northern blot analyses show that the levels of the 18S and 25S rRNAs decreased in total RNA isolated from utp9-td depleted of Utp9p over a 6-h period at 37°C (Supplemental Figure S3), which is consistent with previous reports (Gallagher et al., 2004). Furthermore, the loss of Utp9p function appears to affect nuclear tRNA export and rRNA biogenesis concurrently. Thus, it is probable that the observed nuclear accumulation of tRNATyr when Utp9p was depleted could be due to a general effect on nuclear export processes caused by a defect in ribosome biogenesis. To exclude this possibility, the nuclear-cytoplasmic distribution of mRNA in utp9-td was compared with that in the temperature-sensitive strain of Xpo1p, which is a nuclear export receptor that influences nuclear mRNA export in S. cerevisiae (Stade et al., 1997). FISH analyses show that mRNA was uniformly distributed in YKL200 (Figure 4A), utp9-td (Figure 4B) grown at 23°C in YPG (Figure 4), or YPD (Supplemental Figure S4) containing copper, and in xpo1-1ts (Figure 4C) incubated at 23°C. The nuclear-cytoplasmic distribution of mRNA was not affected in YKL200 incubated at 37°C for 1 h (Supplemental Figure S4) or 3 h (Figure 4). As expected, inactivation of Xpo1p in xpo1-1ts at 37°C for 3 h resulted in nuclear accumulation of mRNA (Stade et al., 1997). However, depletion of Utp9p in utp9-td at 37°C for 1 (Supplemental Figure S4) or 3 h (Figure 4) had no effect on nuclear mRNA export.
Figure 4.

Depletion of Utp9p did not affect mRNA export. The wild-type YKL200 strain (A) and utp9-td (B) were grown at 23°C in YPD medium supplemented with 0.1 mM CuSO4. Expression of Ubr1p was then induced by growing the cells in YPG medium supplemented with 0.1 mM CuSO4 at 23°C for 6 h. Degradation of the Utp9p was induced by growing the cells in YPG medium without CuSO4 at 37°C for 3 h. (C) The W303 XPO1 and xpo1-1ts strains were grown in YPD medium at 23°C and shifted to 37°C for 3 h. Nuclear-cytoplasmic distribution of mRNA was monitored by FISH using Alexa-488–labeled oligo-dT, and the DNA was visualized by DAPI staining.
To verify further that depletion of Utp9p does not have a general effect on nuclear export processes, nuclear export of the large and small ribosomal subunits was monitored in utp9-td (Figure 5). Rps2-GFP and Rpl25-GFP were used as markers to monitor nuclear export of the small and large ribosomal subunits, respectively. Fluorescence microscopy shows that Rps2-GFP was distributed uniformly in YKL200 (Figure 5A, middle column), utp9-td (right column), and rio2-1 (left column), a temperature-sensitive mutant that affects nuclear export of the small ribosomal subunit (Schafer et al., 2003), grown at 23°C. The nuclear-cytoplasmic distribution of Rsp2-GFP was not affected in YKL200 grown at 37°C or in utp9-td depleted of Utp9p for 3 h. As expected, loss of the function of Rio2p resulted in nuclear accumulation of Rsp2-GFP.
Figure 5.

Depletion of Utp9p does not affect nuclear-cytoplasmic distribution of the large and small ribosomal subunits. Fluorescence microscopy was used to monitor the distribution of the small ribosomal subunit protein Rps2-GFP (A) in rio2-1, YKL200 and utp9-td and the large ribosomal subunit protein Rpl25-GFP (B) in nmd3-2, YKL200, and utp9-td at the permissive (23°C) and nonpermissive (37°C) temperatures. DAPI staining was performed to visualize the nucleus.
Rpl25-GFP was also found primarily in the cytoplasm in YKL200 (Figure 5B, middle column), utp9-td (right column), and nmd3-2 (left column), a temperature-sensitive strain that affects nuclear export of the large ribosomal subunit (Gadal et al., 2001), grown at 23°C. However, a punctuate signal was observed in YKL200 and utp9-td but not in nmd3-2 grown at 23°C. The distribution pattern of Rpl25-GFP was not affected in YKL200 grown at 37°C or in utp9-td depleted of Utp9p for 3 h. In contrast, nuclear retention of Rpl25-GFP was detected in nmd3-2 grown at the nonpermissive temperature for 4 h. These data suggest that depletion of Utp9p does not affect nuclear export of the large and small ribosomal subunits. This is consistent with previous studies showing that depletion of Utp8p also did not affect nuclear export of the two ribosomal subunits (Steiner-Mosonyi et al., 2003). Moreover, the data taken together suggest that the loss of Utp9p function affects nuclear-cytoplasmic trafficking of tRNA specifically, and that this effect may be unrelated to the defect observed in rRNA biogenesis.
To ascertain whether depletion of Utp9p also affects nuclear export of mature tRNAs derived from intronless pre-tRNAs, the cellular location of tRNAGly in utp9-td and utp9-ts was investigated (Figure 6). Both tRNATyr, which is made from intron-containing pre-tRNA (left panels), and tRNAGly (right panels) are distributed throughout the cells expressing Utp9p at 23°C (Figure 6 and Supplemental Figure S5). As observed before, tRNATyr was retained in the nucleus of utp9-td and utp9-ts depleted of Utp9p (Figure 6, A and C, right and S5) for various times. In contrast, nuclear retention of tRNAGly was not observed when Utp9p was depleted in utp9-td for 1 (Supplemental Figure S5) or 3 h (Figure 6, B and D, right) at 37°C and in utp9-ts for 3 (Figure 6), 6 or 9 h (Supplemental Figure S5) at 39°C. The data show that Utp9p is only required for nuclear export of mature tRNAs derived from intron-containing pre-tRNAs. Furthermore, nuclear accumulation of tRNATyr resulting from depletion of Utp9p was not caused by a defect in nuclear tRNA aminoacylation, which has been shown to affect the efficiency of nuclear tRNA export (Azad et al., 2001; Supplemental Figure S6) or tRNA splicing (Supplemental Figure S7). It is therefore possible that Utp9p participates in the reexport of mature tRNAs derived from tRNAs that were returned to the nucleus after undergoing splicing in the cytoplasm.
Figure 6.

Loss of Utp9p function resulted in nuclear accumulation of mature tRNATyr derived from intron-containing pre-tRNA but not mature tRNAGly obtained from intronless precursor. utp9-td (top panels) was grown at 23°C in YPD medium supplemented with 0.1 mM CuSO4. Expression of Ubr1p was then induced by growing the cells in YPG medium supplemented with 0.1 mM CuSO4 at 23°C for 6 h. Degradation of the Utp9p was induced by growing the cells in YPG medium without CuSO4 at 37°C for 3 h. utp9-ts (bottom panels) was grown in YPD medium at 23°C and shifted to 39°C for 3 h. The cellular location of tRNATyr (left panels) and tRNAGly (right panels) was detected by FISH and the DNA was visualized by DAPI staining.
To test further that Utp9p is required for nuclear reexport of mature tRNAs derived from intron-containing pre-tRNAs, nuclear export of mature tRNALeu made from intron-containing pre-tRNA and mature tRNAHis, which is derived from intronless precursor was investigated in utp9-td (Figure 7). FISH analyses indicate that nuclear export of mature tRNALeu (Figure 7A) or tRNAHis (Figure 7B) was not affected in cells expressing Utp9p at 23°C. However, depletion of Utp9p in utp9-td at 37°C for 2 h results in nuclear retention of tRNALeu (Figure 7A, right) but not tRNAHis (Figure 7B, right). This data further establish that Utp9p participates in a pathway that is specific for nuclear export of mature tRNAs derived from intron-containing tRNAs.
Figure 7.

Depletion of Utp9p results in nuclear accumulation of mature tRNALeu derived from intron-containing pre-tRNA but not mature tRNAHis obtained from intronless precursor. YKL200 (left panels) and utp9-td (right panels) were grown at 23°C in YPD medium supplemented with 0.1 mM CuSO4. Expression of Ubr1p was then induced by growing the cells in YPG medium supplemented with 0.1 mM CuSO4 at 23°C for 6 h. Degradation of the Utp9p was induced by growing the cells in YPG medium without CuSO4 at 37°C for 2 h. The cellular location of tRNALeu (top panels) and tRNAHis (bottom panels) was detected by FISH and the DNA was visualized by DAPI staining.
Nitrogen or amino acid starvation of S. cerevisiae has been shown to cause nuclear accumulation of mature tRNAs derived from intron-containing pre-tRNAs (Shaheen and Hopper, 2005; Whitney et al., 2007). These observations led to the suggestion that nutrient deprivation blocks nuclear reexport of retrograded tRNAs. To test whether nutrient deprivation also blocks nuclear export of mature tRNAs that are made entirely in the nucleus by processing of intronless precursors, the cellular location of tRNAGly was monitored by FISH in cells starved of amino acids (top) or nitrogen (bottom). Starvation of UTP9 of amino acids (Figure 8A) or nitrogen (bottom panels) for as little as 30 min results in nuclear accumulation of tRNATyr (Figure 8, A and C) but not in accumulation of tRNAGly (Figure 8, B and D). Moreover, starvation of the cells for another 30 min did not affect nuclear-cytoplasmic trafficking of tRNAGly. Nuclear accumulation of tRNALeu, which is made from intron-containing pre-tRNA, but not tRNAHis derived from intronless precursor was also observed when the cells are deprived of amino acids (Supplemental Figure S8). The data show that amino acid or nitrogen deprivation only affects nuclear export of retrograded tRNAs, supporting the notion that Utp9p may be involved in reexport of retrograded tRNAs from the nucleus to the cytoplasm.
Figure 8.

Nutrient stress blocks nuclear export of retrograded mature tRNA obtained from intron-containing pre-tRNAs, but not export of mature tRNA made from intronless precursor. S. cerevisiae cells were starved of amino acids (top panels) or nitrogen (bottom panels) in synthetic medium containing glucose for the times indicated, and the cellular distribution of mature tRNATyr (left panels) and tRNAGly (right panels) was monitored by FISH. The DNA was visualized by DAPI staining.
Utp9p Binds tRNA Directly and Saturably In Vitro But Does Not Function as a Nuclear tRNA Export Receptor
To understand how Utp9p influences nuclear-cytoplasmic trafficking of mature tRNAs made from intron-containing pre-tRNAs, the ability of Utp9p to interact with tRNA was determined using substrate-induced intrinsic fluorescence quenching of tryptophan residues (Figure 9). The analysis shows that recombinant Utp9p binds mature yeast tRNA directly and saturably (Figure 9A). The calculated affinity of Utp9p for tRNA is 2 μM. Utp9p also interacts with a 76-base pair DNA oligonucleotide (Figure 9B) and 5S rRNA (Figure 9C), but this binding was not saturable using the same concentrations used for tRNA binding. The data suggest that Utp9p has a tRNA-binding site, but it will bind nucleic acid nonspecifically in vitro. This behavior is commonly observed for bona fide prokaryotic and eukaryotic tRNA-binding proteins, including Utp8p, Cex1p, and Arc1p (Gite and RajBhandary, 1997; Wang and Schimmel, 1999; Steiner-Mosonyi et al., 2003; McGuire and Mangroo, 2007; McGuire et al., 2009). Thus, the data suggest that tRNA is a substrate for Utp9p in vivo.
Figure 9.

Utp9p binds tRNA directly and saturably. Substrate-induced intrinsic fluorescence quenching of Trp residues was used to determine whether Utp9p interacts with tRNA (A), DNA (B), or 5S rRNA (C).
Loss of the function of Los1p and Msn5p has been shown to affect the efficiency of nuclear tRNA export in S. cerevisiae, but not growth of the cells (Takano et al., 2005). This led to the suggestion that an unidentified receptor is also involved in nuclear tRNA export. To ascertain whether Utp9p functions as a nuclear tRNA export receptor, a heterokaryon shuttling assay was used to investigate whether Utp9p shuttles between the nucleus and cytoplasm (Dilworth et al., 2001; Steiner-Mosonyi et al., 2003). Xpo1p, a nuclear receptor that is required for nuclear export of proteins with a leucine-rich nuclear export signal, was used as a control for a protein known to shuttle between the nucleus and cytoplasm (Stade et al., 1997). The shuttling assay involves monitoring the movement of a protein from a donor nucleus to a recipient nucleus in heterokaryons. To avoid nuclear import of newly synthesized Utp9p and Xpo1p into the recipient nucleus, the donor strain harboring a low copy number plasmid with UTP9-GFP or XPO1-GFP under the control of the GAL1 promoter was first grown in synthetic medium containing galactose to induce expression of the fusion proteins, and then briefly in medium containing glucose to repress the GAL1 promoter. The donor strain was then mated with a kar1-1 mutant strain, which is defective in nuclear fusion, on medium containing glucose. Movement of Utp9-GFP and Xpo1-GFP between nuclei was monitored by direct fluorescence microscopy (Figure 10). Utp9-GFP was found in a single nucleus in all heterokaryons analyzed over a 5-h period after mating was initiated (Figure 10A). In contrast, Xpo1-GFP was detected in both nuclei of heterokaryons over the same period (Figure 10B). The data suggest that Utp9p most likely does not function as an export receptor or an adaptor protein for a nuclear tRNA export receptor.
Figure 10.
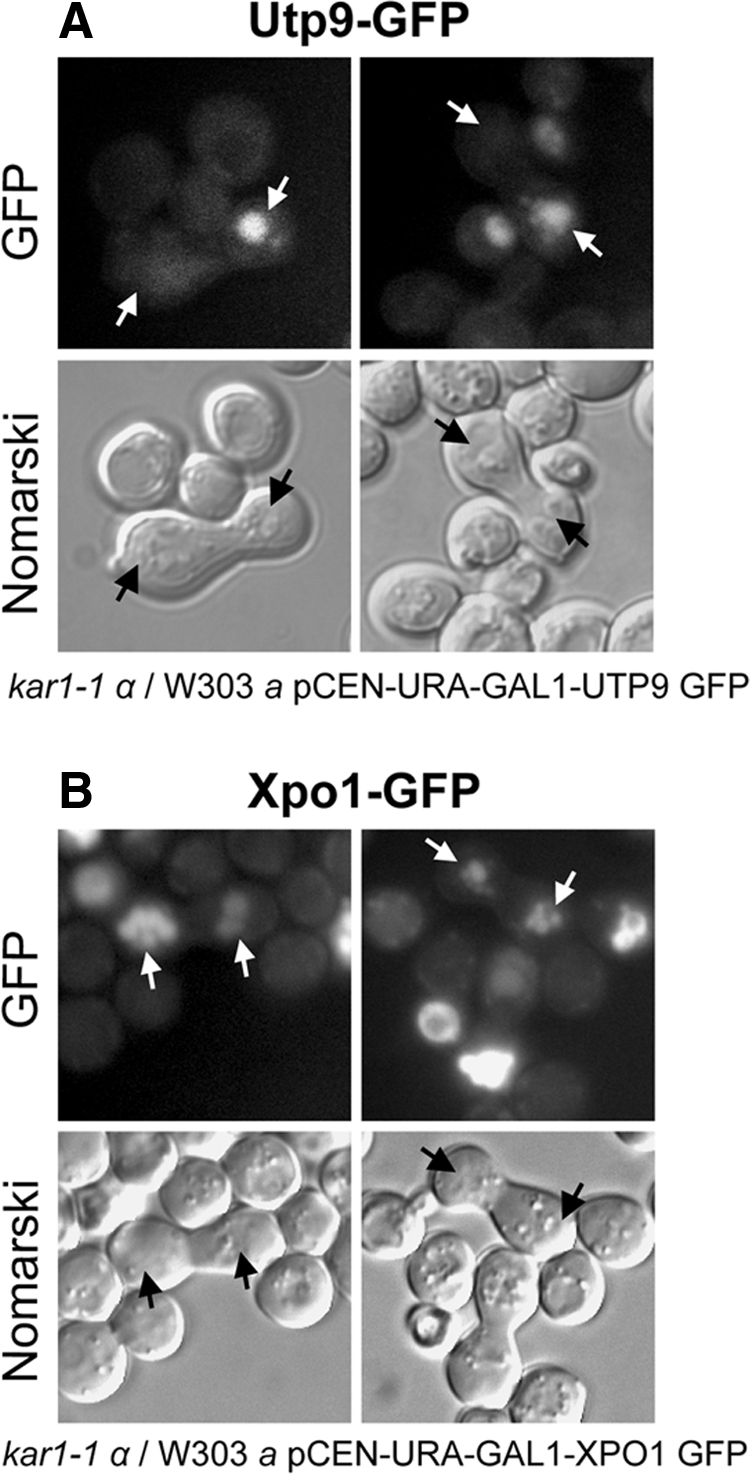
Utp9p does not function as a nuclear tRNA export receptor. A heterokaryon shuttling assay was used to test whether Utp9p has the ability to shuttle between the nucleus and cytoplasm. Heterokaryons were identified by bright field microscopy and then analyzed for the location of Utp9-GFP (A) and Xpo1-GFP (B).
Utp9p Interacts with Utp8p But Not with Aminoacyl-tRNA Synthetases
TAP followed by mass spectrometric analyses have shown that Utp8p forms a complex with Utp17p, Utp15p, Utp10p, Utp9p, Utp5p, and Utp4p in vivo (Krogan et al., 2004). We have also established by TAP and Western blot analyses using anti-Utp8p that Utp8p copurified with Utp17p, Utp15p, Utp10p, Utp9p, Utp5p, and Utp4p (data not shown). Furthermore, we have verified that Utp9p copurifies with Utp8-TAP by Western blot analysis (see TAP eluate in Figure 11A). The proteins that copurify with Utp8-TAP were also detected by SYPRO Ruby staining of the blot (Figure 11B). To ascertain whether Utp9p interacts directly with Utp8p, in vitro protein-binding studies were conducted (Figure 11C). GST-Utp8p bound to GT-Sepharose with (lane 3) or without (lane 4) tRNA and bound GST (lane 5) were incubated with a twofold molar excess of Utp9p. The resins were washed and GST-Utp8p was eluted with glutathione. Purified Utp9p (lane 1) and GST-Utp8p (lane 2), the bound proteins and unbound Utp9p in the wash eluates were subjected to Western blot analyses to detect Utp9p (top two rows) and Utp8p (middle row). Densitometric analyses of the blots indicate that the same amount of Utp9p interacts with Utp8p loaded with tRNA or free of tRNA (cf. lanes 3 and 4). Furthermore, no interaction was detected between Utp9p and GST (lane 5). SYPRO Ruby staining of the blot also shows that Utp9p interacts with Utp8p in a tRNA-independent manner (bottom row), and that this interaction is fairly weak. The data suggest that Utp9p may act together with Utp8p to facilitate a step after the tRNAs have undergone aminoacylation in the nucleolus.
Figure 11.

Utp9p interacts directly with Utp8p but not with aminoacyl-tRNA synthetases in vivo. Cell extract prepared from UTP8-TAP (top) or UTP9-TAP (bottom) was subjected to TAP using IgG-Sepharose and calmodulin-Sepharose. Utp9p or Tys1p in cell lysate (A, lane 1), the flow through from the IgG-Sepharose chromatography step (A, lane 2) and the eluate of proteins bound to calmodulin-Sepharose (TAP Eluate; A, lane 3) were detected by Western blot analysis. The blots were then stained with SYPRO Ruby to detect proteins that copurified with Utp8p and Utp9p, respectively (B). To test whether Utp9p interacts directly with Utp8p, GST-Utp8p (100 μg, 1 nmol) bound to GT-Sepharose in the presence (C, lane 3) or absence (C, lane 4) of 6 μM tRNA was incubated with a twofold molar excess of Utp9p (130 μg, 2 nmol). The same amount of Utp9p was incubated with bound GST (C, lane 5). The resins were washed and eluted with 10 mM glutathione. Purified Utp9p (lane 1) and GST-Utp8p (lane 2), the bound proteins, and unbound Utp9p in wash eluates were subjected to Western blot analyses to detect Utp9p (top panels) and GST-Utp8p (middle panel), followed by SYPRO Ruby staining of the blot (bottom panel).
Utp8p was shown to interact with Tys1p by TAP and protein-binding studies in vitro (Strub et al., 2007). Furthermore, this interaction was found to occur in the nucleolus using a split GFP system (Strub et al., 2007). Therefore, TAP was used to ascertain whether Utp9p interacts with aminoacyl-tRNA synthetases in vivo by monitoring the tyrosyl-tRNA synthetase Tys1p (Figure 11A). Utp9-TAP was purified from cell extract prepared from UTP9-TAP by sequential chromatography on IgG-Sepharose and calmodulin-Sepharose. Western blot analysis detected Tys1p in the cell lysate (lane 1) and flow-through from the IgG-Sepharose resin (lane 2), but none was found in the final eluate from the calmodulin-Sepharose resin (TAP eluate; lane 3). The blot was then stained with SYPRO Ruby to detect the proteins that copurify with Utp9p (Figure 11B). In vitro protein-binding analyses also show that Utp9p did not interact with Tys1p in the absence or presence of tRNA (Supplemental Figure S9). The data suggest that Utp9p may not interact with Utp8p to collect aminoacyl-tRNAs from the aminoacyl-tRNA synthetases. Based on the function of Utp8p, it is possible that Utp9p interacts with Utp8p to translocate aminoacylated tRNAs out of the nucleolus and deliver them to the nuclear tRNA export receptors.
Utp9p May Deliver Retrograded tRNAs to Msn5p for Translocation across the NPC
To understand the role of Utp9p in tRNA export, TAP was conducted to determine whether Utp9p copurifies with the known and putative nuclear tRNA export receptors Los1p and Msn5p, respectively, and Cca1p, which is also thought to facilitate translocation of mature tRNA across the NPC. Lysates were prepared from cells expressing Msn5-TAP and Mtr10-TAP, which is a nuclear import receptor shown to be involved in nuclear tRNA import (Shaheen and Hopper, 2005), Cca1-TAP or Los1-TAP, and subjected to TAP using IgG-Sepharose and calmodulin-Sepharose. Aliquots of the cell lysate (lane 1), proteins that did not bind to IgG-Sepharose (lane 2, flow-through), and proteins bound to calmodulin-Sepharose (lane 3) were subjected to Western blot analysis (Figure 12A), followed by SYPRO Ruby staining of the blots to detect the proteins directly (Figure 12C). Utp9p was detected in the cell lysate (lane 1) and the flow-through from the IgG-Sepharose resin (lane 2). In addition, Utp9p was found to copurify with Msn5p (lane 3) but not with Mtr10p, Cca1p, or Los1p.
Figure 12.

Utp9p copurifies with Msn5p but not with Los1p, Mtr10p, Cca1p, eEF-1A, or Cex1p. TAP was performed using cell extract from MSN5-TAP, LOS1-TAP, MTR10-TAP, CCA1-TAP, and UTP9-TAP. Total cell lysate (lane 1), the flow through from the IgG-Sepharose chromatography step (lane 2), and the eluate of proteins bound to calmodulin-Sepharose (lane 3) were subjected to Western blot analyses to detect Utp9p (A) and Los1p, eEF-1A, and Cex1p (B). The blots were then stained with SYPRO Ruby to detect copurifying proteins (C).
TAP using cell extract prepared from cells expressing Utp9-TAP was also conducted to test whether Cex1p, a cytoplasmic component of the S. cerevisiae nuclear tRNA export process, or eukaryotic elongation factor eEF-1A, which delivers aminoacyl-tRNAs to the ribosomes, copurifies with Utp9p (Figure 12B). Western blot analyses detected Los1p, eEF-1A, and Cex1p in the cell lysate (lane 1) and IgG-Sepharose flow-through (lane 2). However, neither eEF-1A nor Cex1p copurified with Utp9-TAP (lane 3). As observed before, Los1p did not copurify with Utp9-TAP (lane 3). SYPRO Ruby staining of the blot (Figure 12C) shows that a number of proteins copurified with Utp9p. The data taken together indicate that Utp9p copurifies specifically with Msn5p.
To determine whether the interaction between Utp9p and Msn5p is direct, in vitro protein-binding studies were conducted (Figure 13A). GST-Utp9p bound to GT-Sepharose in the presence (lane 3) or absence (lane 4) of tRNA and bound GST (lane 5) were incubated with a twofold molar excess of Msn5p. The resins were washed, and Utp9p was released from GST using thrombin. Purified GST-Utp9p (lane 1) and Msn5p (lane 2), the bound proteins and unbound Msn5p in the wash eluates were subjected to Western blot analysis to monitor Msn5p (top panels) and Utp9p (middle panel). Utp9p was found to interact with Msn5p in the presence (lane 3) and absence (lane 4) of tRNA. However, Msn5p appears to interact better with Utp9p containing tRNA, because the amount of Msn5p bound to Utp9p loaded with tRNA is ∼1.5-fold higher compared with that bound to Utp9p free of tRNA (cf. lanes 3 and 4). In contrast, Msn5p did not interact with GST (lane 5). SYPRO Ruby staining of the blot indicates that only a small amount of Msn5p interacted with Utp9p, suggesting that the binding was weak irrespective of the presence or absence of tRNA (bottom panel).
Figure 13.

Utp9p interacts directly and specifically with Msn5p. GST-Utp9p (100 μg, 1.1 nmol) was bound to GT-Sepharose in the presence (lane 3) or absence (lane 4) of 40 μM tRNA, and incubated with a twofold molar excess of Msn5p (319 μg, 2.2 nmol) (A). The same amount of Msn5p was incubated with bound GST (lane 5). GST-Los1p (100 μg, 800 pmol) was bound to GT-Sepharose in the presence (lane 3) or absence (lane 4) of 6 μM tRNA and incubated with a twofold molar excess of Utp9p (104 μg, 1.6 nmol) (B). The same amount of Utp9p was incubated with bound GST (lane 5). The resins were washed, and Utp9p and Los1p were released from bound GST using thrombin and TEV, respectively. Purified GST-Utp9p (A, lane 1), Msn5p (A, lane 2), Utp9p (B, lane 1), and GST-Los1p (B, lane 2), the bound proteins and unbound proteins in the wash eluates were subjected to Western blot analyses to detect Msn5p (A, top panels), GST-Utp9p (A, middle panel), Utp9p (B, top panels), and Los1p (B, middle panel). The blots were then stained with SYPRO Ruby to detect the proteins directly (A and B, bottom panels).
To verify that the interaction between Msn5p and Utp9p is specific, Utp9p was tested for an association with Los1p in vitro (Figure 13B). GST-Los1p bound to GT-Sepharose in the presence (lane 3) or absence (lane 4) of tRNA and bound GST were incubated with a twofold molar excess of Utp9p. The resins were washed and Los1p was released from GST using TEV protease. Purified Utp9p (lane 1) and GST-Los1p (lane 2), the bound proteins and unbound Utp9p in the wash eluates were subjected to SDS-PAGE. Western blot analysis (top panel and middle panels), and SPYRO Ruby staining of the blot (bottom panel) were used to detect Utp9p (top panels) and Los1p (middle panel). Both analyses show that Utp9p did not interact with Los1p irrespective of the presence or absence of tRNA.
In addition to being the orthologue of the mammalian Xpo-5, genetic studies led to the suggestion that Msn5p functions as a nuclear tRNA export receptor (Takano et al., 2005), and this notion is consistent with in vitro and in vivo studies showing that Msn5p interacts with Utp8p and Cex1p (McGuire and Mangroo, 2007; Strub et al., 2007). However, biochemical evidence verifying that Msn5p is a nuclear tRNA export receptor has not been reported. The use of substrate-induced intrinsic fluorescence quenching of tryptophan residues to test whether Msn5p is capable of interacting with tRNA shows that it binds tRNA nonsaturably with a calculated Kd of 753 μM (Figure 14A). The data suggest that Msn5p is interacting nonspecifically with the tRNA, because this method has been used to show that Utp8p, Cex1p, and Utp9p bind tRNA saturably and with very good affinity (Figure 9; Steiner-Mosonyi et al., 2003; McGuire and Mangroo, 2007).
Figure 14.

Loading of Msn5p with tRNA is dependent on Gsp1p. (A) Determination of the affinity of Msn5p for tRNA. Substrate-induced intrinsic fluorescence quenching of Trp residues was used to determine the tRNA binding affinity of Msn5p. (B) Interaction of Msn5p with Gsp1p in vitro is not dependent on tRNA. GST-Msn5p (100 μg, 0.7 nmol) was bound to GT-Sepharose in the presence (lane 3) or absence (lane 4) of 40 μM tRNA and incubated with a twofold molar excess of Gsp1p (Q71L)-GTP (35 μg, 1.4 nmol). The same amount of Gsp1p was incubated with bound GST (lane 5). The resins were washed and GST-Msn5p was released using 10 mM reduced glutathione. Purified Gsp1p (Q71L) (lane 1) and GST-Msn5p (lane 2), the bound proteins, and unbound Gsp1p in the wash eluates were subjected to Western blot analyses to detect Gsp1p (Q71L) (top panels) and GST-Msn5p (middle panel). SYPRO Ruby staining was performed on the membrane to detect the proteins directly (bottom panel). (C) Msn5p binds tRNA in a Gsp1p-dependent manner in a Gsp1p GTPase protection assay. The assays were performed in 350 μl of PBSM buffer. Gsp1 [γ-32P]-GTP (5 pmol) was incubated with 0.5 μM Msn5p (◇), 0.5 μM Msn5p and 2.5 μM tRNA(□), or 0.5 μM Msn5p and 2.5 μM aminoacylated tRNA (○) for 10 min, and Rna1p was added to a final concentration of 0.2 nM. Gsp1p [γ-32P]-GTP (5 pmol) was incubated in the absence (▴) or presence (■) of 0.2 nM Rna1p. At the times specified, 50-μl aliquots were diluted into 200 μl of 20 mM phosphoric acid containing 5% (wt/vol) activated charcoal. The reactions were then centrifuged at maximum speed for 10 min in 1.5-ml tubes, and 100-μl aliquots were removed for scintillation counting to determine the amount of GTP hydrolyzed.
Like other β-karyopherin export receptors including the nuclear tRNA export receptor Los1p, loading of Msn5p with protein cargo has been shown to be dependent on the Ran GTPase Gsp1p in the GTP bound form (Hellmuth et al., 1998; Quan et al., 2006). Consequently, protein-binding studies were conducted in vitro to ascertain whether the interaction between Msn5p and tRNA is dependent on Gsp1p-GTP (Figure 14B). For this analysis the Gsp1p (Q71L) mutant was used, because it is unable to hydrolyze GTP (Maurer et al., 2001; Quan et al., 2006). GST-Msn5p bound to GT-Sepharose was incubated in the presence (lane 3) or absence (lane 4) of tRNA and in the presence of a twofold molar excess of Gsp1p (Q71L) loaded with GTP. The same amount of Gsp1p (Q71L) was incubated with bound GST (lane 5). The resins were washed and GST-Msn5p was released using glutathione. Purified Gsp1p (Q71L; lane 1) and GST-Msn5p (lane 2), the bound proteins, and unbound Gsp1p in the wash eluates were subjected to Western blot analyses to detect Gsp1p (top panels) and GST-Msn5p (middle panel). Gsp1p (Q71L) was found to interact with Msn5p in the presence (lane 3) or absence (lane 4) of tRNA, but not with bound GST (lane 5). A smaller amount of Gsp1p was bound to Msn5p in the absence of tRNA compared with that bound to Msn5p in the presence of tRNA. However, this reduction in Gsp1p binding in the absence of tRNA is due to less GST-Msn5p bound to the resin. Quantification by densitometric analyses of the blots indicate that the same amount of Gsp1p was bound to Msn5p in the absence or presence of tRNA (cf. lanes 3 and 4). Thus, the interaction between Msn5p and Gsp1p occurs independently of tRNA. SYPRO Ruby staining of the blot (bottom panel) detected GST-Msn5p but not Gsp1p, indicating that the interaction between the two proteins is very weak.
A number of studies have shown that Ran/Gsp1p-GTP bound to a β-karyopherin is not accessible to RanGAP/Rna1p activation of its GTPase activity in vitro (Kutay et al., 1997, 1998; Lounsbury and Macara, 1997; Hellmuth et al., 1998). This enzymatic activity-based assay has been used to show the specific and cooperative formation of the Los1p, tRNA, Gsp1p-GTP export complex (Hellmuth et al., 1998). Therefore, this assay was used to ascertain whether tRNA is an export cargo for Msn5p (Figure 14C). Incubation of Gsp1p-GTP with Rna1p results in rapid GTP hydrolysis by Gsp1p. Similarly, the presence of Msn5p in the absence of tRNA did not significantly block Rna1p-induced Gsp1p GTP hydrolysis. However, the presence of Msn5p and aminoacylated or nonaminoacylated tRNA significantly reduced the rate of Rna1p-induced GTP hydrolysis by Gsp1p, indicating that Msn5p binds both forms of tRNA to about the same extent in a Gsp1p-GTP-dependent manner. These rates of GTP hydrolysis were slightly higher than that observed for Gsp1p alone. The data show that, like Los1p, binding of tRNA to Msn5p is dependent on Gsp1p-GTP, validating that Msn5p is an authentic nuclear tRNA export receptor. Thus, the interaction observed between Utp9p and Msn5p in vitro and in vivo suggests that Utp9p may deliver retrograded tRNAs to Msn5p for export to the cytoplasm.
Utp8p has been shown previously to interact with Gsp1p in vivo and in vitro and to form a complex with Los1p and Gsp1p-GTP in a tRNA-dependent manner in vitro (Strub et al., 2007). We therefore surmise that if Utp9p is delivering tRNA to Msn5p, it may interact with Gsp1p. To test this possibility, TAP was performed using extract prepared from UTP9-TAP or UTP21-TAP followed by Western blot analysis to detect Gsp1p (Figure 15A). Gsp1p was detected in the cell lysate (lane 1) and flow-through from IgG-Sepharose (lane 2). In addition, Gsp1p was found to copurify with Utp9p but not with Utp21p (lane 3). SYPRO Ruby staining of the blots shows the proteins that copurify with Utp9p and Utp21p (panel B). The data suggest that the association between Utp9p and Gsp1p in vivo is specific.
Figure 15.

Utp9p interacts directly with Gsp1p in a tRNA-dependent manner. TAP was performed using cell extract from UTP9-TAP and UTP21-TAP. Total cell lysate (lane 1), the flow through from the IgG-Sepharose chromatography step (lane 2), and the eluate of proteins bound to calmodulin-Sepharose (lane 3) were subjected to Western blot analyses to detect Gsp1p (A). The blots were then stained with SYPRO Ruby to detect proteins that copurified with Utp9p and Utp21p (B). To ascertain whether Utp9p interacts directly with Gsp1p, protein-binding analyses were conducted in vitro (C). GST-Utp9p (100 μg, 1.1 nmol) bound to GT-Sepharose in the presence (lane 3) or absence (lane 4) of 40 μM tRNA was incubated with a twofold molar excess (55 μg, 2.2 nmol) of Gsp1p (Q71L) in the GTP bound form. The same amount of Gsp1p (Q71L)-GTP was incubated with bound GST (lane 5). The resins were washed and incubated with 10 mM reduced glutathione to release GST-Utp9p. Purified Gsp1p (Q71L) (lane 1) and GST-Utp9p (lane 2), the bound proteins, and Gsp1p in the wash eluates were subjected to Western blot analyses to detect Gsp1p (top panels) and GST-Utp9p (middle panel). The blot was then stained with SYPRO Ruby to detect the protein directly (bottom panel).
Protein-binding analyses were conducted in vitro to ascertain whether Utp9p interacts directly with Gsp1p (Figure 15C). GST-Utp9p bound to GT-Sepharose was incubated with (lane 3) or without (lane 4) tRNA and with a twofold molar excess of Gsp1p (Q71L) loaded with GTP. Bound GST was incubated with the same amount of Gsp1p (Q71L; lane 5). The resins were washed and incubated with glutathione to release Utp9p. Purified Gsp1p (Q71L; lane 1) and GST-Utp9p (lane 2), the bound proteins and unbound Gsp1p (Q71L) in the wash eluates were subjected to Western blot analysis to detect Gsp1p (top panels) and Utp9p (middle panel). Gsp1p (Q71L) interacts with Utp9p in the presence (lane 3) or absence (lane 4) of tRNA, and a small amount of Gsp1p (Q71L) was found to interact with GST (lane 5). However, densitometric analyses of the blots indicate that the amount of Gsp1p (Q71L) bound to Utp9p in the presence of tRNA was twofold higher compared with that bound to Utp9p in the absence of tRNA (cf. lanes 3 and 4). The data show that Utp9p interacts directly with Gsp1p, and that this interaction is dependent on tRNA. SYPRO Ruby staining of the blot detected GST-Utp9p but not Gsp1p, indicating that the interaction between the two proteins is very weak (bottom panel).
Utp9p Plays a Key Role in the Formation of the Msn5p-tRNA-Gsp1p-GTP Export Complex
The findings that Utp9p interacts directly with tRNA, Msn5p and Gsp1p in vitro and that Utp8p interacts with Msn5p and Utp9p in vivo and in vitro suggest that in vivo Utp9p and Utp8p may participate in the formation of the Msn5p-tRNA-Gsp1p-GTP export complex. To test this possibility, sequential protein-binding analyses were conducted to ascertain whether Utp8p (Figure 16A) or Utp9p (Figure 16B) forms a complex with Msn5p and Gsp1p-GTP in a tRNA-dependent manner. To determine whether Utp8p forms a complex with Msn5p and Gsp1p, GST-Utp8p bound to GT-Sepharose in the presence (lanes 1 and 3) or absence (lanes 2 and 4) of tRNA was incubated with a twofold molar excess of Msn5p (lanes 1 and 2) or Gsp1p (Q71L) loaded with GTP (lanes 3 and 4). The resins were washed and incubated with a twofold molar excess of Gsp1p (Q71L)-GTP (lanes 1 and 2) or Msn5p (lanes 3 and 4). Bound GST was incubated with the same amount of Msn5p (lane 5) or Gsp1p (Q71L; lane 6). The resins were washed and Utp8p was released using TEV. The bound proteins and unbound Gsp1p (Q71L) and Msn5p in the wash eluates were subjected to Western blot analyses to detect Gsp1p (top panels), Msn5p (middle panels) and Utp8p (bottom panel). Msn5p and Gsp1p (Q71L) bound to Utp8p to the same extent in the presence (lanes 1 and 3) or absence (lanes 2 and 4) of tRNA irrespective of the order of addition of Msn5p and Gsp1p. A small amount of Gsp1p (Q71L) bound to GST (lane 6), but no binding was observed between Msn5p and GST (lane 5). The data show that Utp8p, Msn5p, and Gsp1p form a complex in a tRNA-independent manner.
Figure 16.

Utp9p plays a more important role than Utp8p in the assembly of the Msn5p-tRNA-Gsp1p-GTP export complex. (A) Utp8p forms a complex with Gsp1p and Msn5p in a tRNA-independent manner. GST-Utp8p (100 μg, 1 nmol) bound to GT-Sepharose was incubated with (lanes 1 and 3) or without (lanes 2 and 4) 6 μM tRNA. The resins were washed and incubated with a twofold molar excess of Msn5p (284 μg, 2 nmol; lanes 1 and 2) or Gsp1p (Q71L)-GTP (50 μg, 2 nmol; lanes 3 and 4). The resins were washed and incubated with a twofold molar excess of Gsp1p (Q71L)-GTP (50 μg, 2 nmol; lanes 1 and 2) or Msn5p (284 μg, 2 nmol; lanes 3 and 4). Bound GST was incubated with the same amount of Msn5p (lane 5) or Gsp1p (Q71L) (lane 6). Utp8p was released from the washed resins using TEV, and the bound proteins and unbound proteins in the washes were subjected to Western blot analysis to detect Gsp1p (top panels), Msn5p (middle panels), and Utp8p (bottom panel). (B) Formation of a complex between Utp9p, Msn5p, and Gsp1p is dependent on tRNA and the order of addition of the interacting proteins. GST-Utp9p (100 μg, 1.11 nmol) bound to GT-Sepharose was incubated with (lanes 1 and 3) or without (lanes 2 and 4) 40 μM tRNA. The resins were washed and incubated with a twofold molar excess of Msn5p (312 μg, 2.2 nmol; lanes 1 and 2) or Gsp1p (Q71L)-GTP (55 μg, 2.2 nmol; lanes 3 and 4). The resins were washed and incubated with a twofold molar excess of Gsp1p (Q71L)-GTP (55 μg, 2.2 nmol; lanes 1 and 2) or Msn5p (312 μg, 2.2 nmol; lanes 3 and 4). The same amount of Msn5p (lane 5) or Gsp1p (Q71L) (lane 6) was incubated with bound GST. Utp9p was released from the washed resins using thrombin. The bound proteins and unbound proteins in the wash eluates were analyzed for Gsp1p (Q71L) (top panels), Msn5p (middle panels), and Utp9p (bottom panel) by Western blot analyses.
Sequential binding analyses were conducted to determine whether Utp9p forms a complex with Msn5p and Gsp1p (Figure 16B). GST-Utp9p bound to GT-Sepharose in the presence (lanes 1 and 3) or absence (lanes 2 and 4) of tRNA was incubated with a twofold molar excess of Msn5p (lanes 1 and 2) or Gsp1p (Q71L)-GTP (lanes 3 and 4). The resins were washed and incubated with a twofold molar excess of Gsp1p (Q71L)-GTP (lanes 1 and 2) or Msn5p (lanes 3 and 4). Bound GST was incubated with same amount of Msn5p (lane 5) or Gsp1p (Q71L; lane 6). The resins were washed and Utp9p was released using thrombin. The bound proteins and unbound Msn5p and Gsp1p (Q71L) in the wash eluates were subjected to Western blot analyses to detect Gsp1p (top panels), Msn5p (middle panels) and Utp9p (bottom panel). The same amount of Msn5p bound to Utp9p in the presence or absence of tRNA and irrespective of the order of addition of Msn5p and Gsp1p. In contrast, a larger amount of Gsp1p (Q71L)-GTP bound to Utp9p in the presence than in the absence of tRNA when Msn5p was added first (cf. lanes 1 and 2). The amount of Gsp1p bound to Utp9p in the presence of tRNA when it was added after Msn5p (lane 1) was fourfold higher than that observed in the absence of tRNA (lane 2). In contrast, Gsp1p interacted with Utp9p to the same extent in the presence or absence of tRNA when it was added before Msn5p (cf. lanes 3 and 4). Both Msn5p (lane 5) and Gsp1p (Q71L; lane 6) did not interact with GST alone. The data suggest that Utp9p is playing a more important role than Utp8p in the formation of the tRNA export complex.
DISCUSSION
A small percentage of eukaryotic tRNA genes contain an intron, and recent studies have demonstrated that removal of the intron from the pre-tRNA transcript occurs in the cytoplasm of S. cerevisiae (Yoshihisa et al., 2003, 2007). Furthermore, it has been shown that the spliced tRNAs then undergo retrograde transport from the cytoplasm to the nucleus (Shaheen and Hopper, 2005; Takano et al., 2005), followed by reexport to the cytoplasm after quality assurance by aminoacylation in the nucleolus. However, retrograded tRNAs have been shown to accumulate in the nucleus of S. cerevisiae deprived of glucose, nitrogen, amino acids, or phosphate (Shaheen and Hopper, 2005; Shaheen et al., 2007; Hurto et al., 2007; Whitney et al., 2007). This appears to occur because of a block at a step specifically involved in nuclear reexport of retrograded tRNAs, as nuclear accumulation of tRNATyr (Figure 8) and tRNALeu (Supplemental Figure S8) made from intron-containing pre-tRNAs, but not tRNAGly (Figure 8) and tRNAHis (Supplemental Figure S8), which are tRNAs derived from intronless pre-tRNAs, was observed in S. cerevisiae starved of amino acids or nitrogen. It is possible that inhibition of nuclear reexport of retrograded tRNAs occurs after the nuclear tRNA aminoacylation quality assurance step, as retrograded tRNAs are found in the aminoacylated form (Whitney et al., 2007). Importantly, these data imply that separate pathways are used for nuclear reexport of retrograded tRNAs and nonretrograded tRNAs, which are tRNAs derived from intronless pre-tRNAs and are matured in the nucleus.
Although the mechanism responsible for nuclear reexport of retrograded tRNAs is not understood, several lines of evidence obtained by this study suggest that Utp9p is an intranuclear component of this pathway that acts at a step after the tRNAs have undergone maturation and aminoacylation in the nucleus. 1) Depletion of Utp9p affects nuclear export of mature tRNAs derived from intron-containing pre-tRNAs but not tRNAs made from intronless precursors (Figures 6 and 7). 2) Amino acid or nitrogen starvation results in nuclear accumulation of tRNAs made from intron-containing pre-tRNAs but not tRNAs made from intronless precursors (Figures 8 and S8). 3) Loss of the function of Utp9p did not affect splicing or nuclear aminoacylation of a tRNA derived from intron-containing precursor (Figures S6 and S7). 4) Nutrient deprivation of Utp9p-depleted cells did not abolish nuclear accumulation of tRNATyr (data not shown). 5) Utp9p does not function as a nuclear tRNA export receptor or an adaptor protein for a nuclear tRNA export receptor, because it does not shuttle between the nucleus and cytoplasm (Figure 10A). In addition, the finding that Utp8p interacts with Utp9p in vivo and in vitro suggests that Utp8p is an intranuclear component that is required for nuclear reexport of retrograded tRNAs (Figure 11); however, it also participates in the pathway that is involved in nuclear export of mature tRNAs derived from intronless precursors, as depletion of Utp8p blocks export of mature tRNAs obtained from both classes of pre-tRNAs (Steiner-Mosonyi et al., 2003). Furthermore, Msn5p appears to be one of the export receptors that facilitate nuclear export of retrograded tRNAs based on in vivo and in vitro data showing that it interacts with Utp9p (Figures 12, 13, and 16). The cytoplasmic Cex1p may also participate in this pathway to collect aminoacylated retrograded tRNAs from the export receptor, because it was found to copurify with Msn5p by TAP (McGuire and Mangroo, 2007). However, it is unlikely that Utp9p, Utp8p, Msn5p, and Cex1p are the only proteins involved in the nuclear tRNA reexport pathway, because the functions of Msn5p and Cex1p are not essential.
Utp8p has been shown to collect aminoacyl-tRNAs from the aminoacyl-tRNA synthetases and translocate them from the nucleolus to the nuclear tRNA export receptors located in the nucleoplasm and NPC by using a channelling mechanism (Strub et al., 2007). Like Utp8p (Steiner-Mosonyi et al., 2003), Utp9p binds tRNA directly and saturably in vitro (Figure 9A). Furthermore, Utp9p copurifies with Utp8p by TAP and interacts directly with Utp8p in vitro (Figure 11, A and B). However, unlike Utp8p (Strub et al., 2007), Utp9p does not appear to interact with aminoacyl-tRNA synthetases in vivo, as it does not interact with the tyrosyl-tRNA synthetase in vivo and in vitro (Figure 11A and Supplemental Figure S9). An interpretation that is consistent with the data is that Utp9p does not facilitate transfer of aminoacylated retrograded tRNAs from the aminoacyl-tRNA synthetases to Utp8p. Instead, it most likely acts in concert with Utp8p to translocate the tRNAs from the nucleolus to a nuclear tRNA export receptor of a pathway specific for nuclear reexport of retrograded tRNAs. However, it is not known how Utp9p discriminates between retrograded tRNAs derived from intron-containing precursors and nonretrograded tRNAs made from intronless precursors. The finding that the interaction between Utp8p and Utp9p in vitro is not dependent on tRNA suggests that in vivo an unidentified protein may recruit Utp9p to Utp8p carrying aminoacylated retrograded tRNA or that a protein transfers aminoacylated retrograded tRNAs from the synthetases to Utp9p, followed by recruitment of Utp8p for translocation of the Utp9p-tRNA complex to the nuclear export receptor of the nuclear tRNA reexport pathway.
Los1p has been unambiguously demonstrated to be a nuclear tRNA export receptor (Hellmuth et al., 1998). In contrast, Msn5p was presumed to be involved in translocation of tRNAs across the NPC based on the finding that an msn5 los1 strain, which is not affected in growth, exhibits a reduction in the efficiency of nuclear export of tRNA (Takano et al., 2005). Because Msn5p also functions as a nuclear import receptor for proteins (Quan et al., 2006), it is possible that the reduced efficiency of nuclear tRNA export observed in the msn5 los1 strain could be due to a defect in nuclear import of a protein that plays a redundant role in the nuclear tRNA export process. We established biochemically that tRNA is a poor substrate for Msn5p in the absence of Gsp1p-GTP (Figure 14, A and C), but like other nuclear export receptors including Los1p, Msn5p binding to tRNA requires Gsp1p-GTP (Figure 14C), suggesting that Msn5p also functions as a nuclear tRNA export receptor. This notion is consistent with in vitro and in vivo studies showing that Msn5p interacts with Utp8p and Cex1p (McGuire and Mangroo, 2007; Strub et al., 2007). Msn5p may export both aminoacylated and nonaminoacylated tRNA, because it can bind both forms of tRNA in a Gsp1p-GTP-dependent manner (Figure 14C). Moreover, the finding that Utp9p interacts with Msn5p but not with Los1p in vivo and in vitro (Figures 12 and 13) suggests that Msn5p facilitates reexport of retrograded tRNAs from the nucleus to the cytoplasm. However, it is unlikely that Msn5p is the only receptor responsible for nuclear reexport of retrograded tRNAs, as the loss of Msn5p function does not affect the efficiency of nuclear export of tRNAs derived from intron-containing pre-tRNAs or cell growth (data not shown). It is possible that an unidentified export receptor also participates in nuclear export of retrograded tRNAs. Utp9p may also function with this nuclear export receptor, because loss of Utp9p function has a more prominent impact on nuclear reexport of retrograded tRNA than the loss of Msn5p function alone. The finding that Utp9p is not required for Los1p-mediated nuclear tRNA export does not exclude the possibility that Los1p can facilitate nuclear reexport of retrograded tRNAs.
Previous studies have shown that Utp8p loaded with tRNA, Los1p, and Gsp1p form a complex. However, this complex is not formed when Utp8p lacks tRNA, and the tRNA-dependent formation of the complex only occurs when Los1p is added to Utp8p containing tRNA followed by addition of Gsp1p-GTP (Strub et al., 2007). These results led to the suggestion that Utp8p facilitates the formation of the Los1p-tRNA-Gsp1p-GTP export complex. We found that Utp9p copurifies with Msn5p and Gsp1p by TAP (Figures 12A and 15A). Utp9p also interacts directly with Msn5p and Gsp1p in vitro (Figures 13A and 15C). We therefore surmise that Utp8p and Utp9p may be required for formation of the Msn5p-tRNA-Gsp1p-GTP export complex in vivo. Measurement of the protection of the Rna1p-induced GTPase activity of Gsp1p demonstrates that in vitro formation of the Msn5p-tRNA-Gsp1p-GTP complex can occur without the assistance of additional factors (Figure 14C). Furthermore, by using this enzymatic assay we could not detect any effect of Utp8p and Utp9p on the efficiency of complex formation (data not shown). However, in vitro protein-binding analyses indicate that Utp8p, Msn5p, and Gsp1p form a complex that was not dependent on tRNA or the order of addition of Msn5p and Gsp1p to Utp8p (Figure 16A). In contrast, Gsp1p was found to interact with Msn5p and Utp9p in a tRNA-dependent manner when Msn5p was added first to the Utp9p-tRNA complex, but not when it was added to the Utp9p-tRNA complex before the addition of Msn5p (Figure 16B). Based on the finding from in vitro protein-binding analyses that the interaction between Msn5p and Gsp1p was not dependent on tRNA, and the binding between Utp9p and Gsp1p was tRNA-dependent (Figure 15) suggest that the tRNA-dependency of binding of Gsp1p to the Msn5p-Utp9p-tRNA complex is most likely related to Utp9p. Taken together, the data suggest that both Utp8p and Utp9p are involved in formation of the export complex in vivo. However, Utp9p appears to play a more important role than Utp8p in the assembly of the export complex. Utp9p bound to tRNA may, in part, facilitate binding of Gsp1p-GTP to Msn5p. A model that is consistent with the data suggests that both Utp8p and Utp9p bound to tRNA interact with Msn5p to indicate the presence of tRNA; Utp9p interacts with Gsp1p-GTP to recruit it to Msn5p, allowing Msn5p to interact with the tRNA. Gsp1p then interacts with Utp8p to facilitate release of the tRNA, and dissociation of the Utp8p-Utp9p complex. However, further studies are required to fully understand the mechanism of Utp8p- and Utp9p-mediated loading of Msn5p with tRNA, and the significance of the interaction of Gsp1p with Utp8p and Utp9p.
We have shown that Utp9p and Msn5p are components of a pathway that facilitates reexport of retrograded tRNAs from the nucleus to the cytoplasm in S. cerevisiae. Furthermore, the results suggest that Utp9p acts in concert with Utp8p to translocate aminoacylated retrograded tRNAs from the nucleolus to the tRNA export receptor Msn5p and assist with the formation of the Msn5p-tRNA-Gsp1p-GTP export complex. Why nuclear export of retrograded tRNAs should involve a pathway that is different from that required for nuclear export of nonretrograded tRNAs is not understood. Furthermore, the significance of the involvement of Utp9p in both transcription of rRNA gene and nuclear reexport of retrograded tRNA is not understood. A possible explanation is that Utp9p also plays a regulatory role that coordinates nuclear reexport of retrograded tRNA and ribosome biogenesis in the nucleus. This communication between the two processes may be important for controlling the protein translation rate, which is dictated by a number of external stimuli such as nutrient availability. This hypothesis is consistent with the finding that both nuclear reexport of retrograded tRNAs (Shaheen and Hopper, 2005; Shaheen et al., 2007; Whitney et al., 2007) and ribosome biogenesis are affected by nutrient-related stress (Jorgensen and Tyers, 2004; Li et al., 2006; Zaman et al., 2008).
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. U. Stochaj and E. Hurt for their generous gifts of plasmids, strains, and antibodies. We also thank B. Strub for technical assistance. This work was supported by an operating grant from the Canadian Institutes of Health Research (CIHR). J.B.P. and A.T.M. are recipients of Natural Sciences and Engineering Research Council of Canada (NSERC) Doctoral Scholarships.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-06-0490) on October 7, 2009.
REFERENCES
- Arts G. J., Fornerod M., Mattaj I. W. Identification of a nuclear export receptor for tRNA. Curr. Biol. 1998;8:305–314. doi: 10.1016/s0960-9822(98)70130-7. [DOI] [PubMed] [Google Scholar]
- Azad A. K., Stanford D. R., Sarkar S., Hopper A. K. Role of nuclear pools of aminoacyl-tRNA synthetases in tRNA nuclear export. Mol. Biol. Cell. 2001;12:1381–1392. doi: 10.1091/mbc.12.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Aroya S., Coombes C., Kwok T., O'Donnell K. A., Boeke J. D., Hieter P. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol. Cell. 2008;30:248–258. doi: 10.1016/j.molcel.2008.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff F. R., Klebe C., Kretschmer J., Wittinghofer A., Ponstingl H. Rangap1 induced Gtpase activity of nuclear Ras-related Ran. Proc. Natl. Acad. Sci. USA. 1994;91:2587–2591. doi: 10.1073/pnas.91.7.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff F. R., Krebber H., Smirnova E., Dong W. H., Ponstingl H. Coactivation of Rangtpase and Inhibition of Gtp dissociation by Ran Gtp-binding protein Ranbp1. EMBO J. 1995;14:705–715. doi: 10.1002/j.1460-2075.1995.tb07049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack M. T., Regener K., Schwappach B., Saffrich R., Paraskeva E., Hartmann E., Gorlich D. Exp5 exports eEF1A via tRNA from nuclei and synergizes with other transport pathways to confine translation to the cytoplasm. EMBO J. 2002;21:6205–6215. doi: 10.1093/emboj/cdf613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado A., Treichel N., Muller E. C., Otto A., Kutay U. Exportin-5-mediated nuclear export of eukaryotic elongation factor 1A and tRNA. EMBO J. 2002;21:6216–6224. doi: 10.1093/emboj/cdf620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary J. D., Mangroo D. Nucleotides of the tRNA D-stem that play an important role in nuclear-tRNA export in Saccharomyces cerevisiae. Biochem. J. 2000;347(Pt 1):115–122. [PMC free article] [PubMed] [Google Scholar]
- Dilworth D. J., Suprapto A., Padovan J. C., Chait B. T., Wozniak R. W., Rout M. P., Aitchison J. D. Nup2p dynamically associates with the distal regions of the yeast nuclear pore complex. J. Cell Biol. 2001;153:1465–1478. doi: 10.1083/jcb.153.7.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragon F., et al. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature. 2002;417:967–970. doi: 10.1038/nature00769. [DOI] [PubMed] [Google Scholar]
- Englert M., Latz A., Becker D., Gimple O., Beier H., Akama K. Plant pre-tRNA splicing enzymes are targeted to multiple cellular compartments. Biochimie. 2007;89:1351–1365. doi: 10.1016/j.biochi.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Ernens I., Goodfellow S. J., Innes F., Kenneth N. S., Derblay L. E., White R. J., Scott P. H. Hypoxic stress suppresses RNA polymerase III recruitment and tRNA gene transcription in cardiomyocytes. Nucleic Acids Res. 2006;34:286–294. doi: 10.1093/nar/gkj402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadal O., Strauss D., Kessl J., Trumpower B., Tollervey D., Hurt E. Nuclear export of 60s ribosomal subunits depends on Xpo1p and requires a nuclear export sequence-containing factor, Nmd3p, that associates with the large subunit protein Rpl10p. Mol. Cell. Biol. 2001;21:3405–3415. [Google Scholar]
- Gallagher J.E.G., Dunbar D. A., Granneman S., Mitchell B. M., Osheim Y., Beyer A. L., Baserga S. J. RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev. 2004;18:2506–2517. doi: 10.1101/gad.1226604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavidel A., Kislinger T., Pogoutse O., Sopko R., Jurisica I., Emili A. Impaired tRNA nuclear export links DNA damage and cell-cycle checkpoint. Cell. 2007;131:915–926. doi: 10.1016/j.cell.2007.09.042. [DOI] [PubMed] [Google Scholar]
- Giaever G., et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Gite S., RajBhandary U. L. Lysine 207 as the site of cross-linking between the 3′-end of Escherichia coli initiator tRNA and methionyl-tRNA formyltransferase. J. Biol. Chem. 1997;272:5305–5312. doi: 10.1074/jbc.272.8.5305. [DOI] [PubMed] [Google Scholar]
- Grosshans H., Hurt E., Simos G. An aminoacylation-dependent nuclear tRNA export pathway in yeast. Genes Dev. 2000;14:830–840. [PMC free article] [PubMed] [Google Scholar]
- Hellmuth K., Lau D. M., Bischoff F. R., Kunzler M., Hurt E., Simos G. Yeast Los1p has properties of an exportin-like nucleocytoplasmic transport factor for tRNA. Mol. Cell. Biol. 1998;18:6374–6386. doi: 10.1128/mcb.18.11.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper A. K., Phizicky E. M. tRNA transfers to the limelight. Genes Dev. 2003;17:162–180. doi: 10.1101/gad.1049103. [DOI] [PubMed] [Google Scholar]
- Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., O'Shea E. K. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Hurto R. L., Tong A.H.Y., Boone C., Hopper A. K. Inorganic phosphate deprivation causes tRNA nuclear accumulation via retrograde transport in Saccharomyces cerevisiae. Genetics. 2007;176:841–852. doi: 10.1534/genetics.106.069732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P., Tyers M. How cells coordinate growth and division. Curr. Biol. 2004;14:R1014–R1027. doi: 10.1016/j.cub.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Kanemaki M., Sanchez-Diaz A., Gambus A., Labib K. Functional proteomic identification of DNA replication proteins by induced proteolysis in vivo. Nature. 2003;423:720–724. doi: 10.1038/nature01692. [DOI] [PubMed] [Google Scholar]
- Krogan N. J., et al. High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell. 2004;13:225–239. doi: 10.1016/s1097-2765(04)00003-6. [DOI] [PubMed] [Google Scholar]
- Kruse C., Willkomm D. K., Grunweller A., Vollbrandt T., Sommer S., Busch S., Pfeiffer T., Brinkmann J., Hartmann R. K., Muller P. K. Export and transport of tRNA are coupled to a multi-protein complex. Biochemical J. 2000;346:107–115. [PMC free article] [PubMed] [Google Scholar]
- Kutay U., Bischoff F. R., Kostka S., Kraft R., Gorlich D. Export of importin alpha from the nucleus is mediated by a specific nuclear transport factor. Cell. 1997;90:1061–1071. doi: 10.1016/s0092-8674(00)80372-4. [DOI] [PubMed] [Google Scholar]
- Kutay U., Lipowsky G., Izaurralde E., Bischoff F. R., Schwarzmaier P., Hartmann E., Gorlich D. Identification of a tRNA-specific nuclear export receptor. Mol. Cell. 1998;1:359–369. doi: 10.1016/s1097-2765(00)80036-2. [DOI] [PubMed] [Google Scholar]
- Li H., Tsang C. K., Watkins M., Bertram P. G., Zheng X.F.S. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature. 2006;442:1058–1061. doi: 10.1038/nature05020. [DOI] [PubMed] [Google Scholar]
- Lounsbury K. M., Macara I. G. Ran-binding protein 1 (RanBP1) forms a ternary complex with Ran and karyopherin beta and reduces Ran GTPase-activating protein (RanGAP) inhibition by karyopherin beta. J. Biol. Chem. 1997;272:551–555. doi: 10.1074/jbc.272.1.551. [DOI] [PubMed] [Google Scholar]
- Lund E., Dahlberg J. E. Proofreading and aminoacylation of tRNAs before export from the nucleus. Science. 1998;282:2082–2085. doi: 10.1126/science.282.5396.2082. [DOI] [PubMed] [Google Scholar]
- Maurer P., Redd M., Solsbacher J., Bischoff F. R., Greiner M., Podtelejnikov A. V., Mann M., Stade K., Weis K., Schlenstedt G. The nuclear export receptor Xpo1p forms distinct complexes with NES transport substrates and the yeast ran binding protein 1 (Yrb1p) Mol. Biol. Cell. 2001;12:539–549. doi: 10.1091/mbc.12.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire A. T., Keates R.A.B., Cook S., Mangroo D. Structural modeling identified the tRNA-binding domain of Utp8p, an essential nucleolar component of the nuclear tRNA export machinery of Saccharomyces cerevisiae. Biochem. Cell Biol. Biochim. Biol. Cell. 2009;87:431–443. doi: 10.1139/o08-145. [DOI] [PubMed] [Google Scholar]
- McGuire A. T., Mangroo D. Cex1p is a novel cytoplasmic component of the Saccharomyces cerevisiae nuclear tRNA export machinery. EMBO J. 2007;26:288–300. doi: 10.1038/sj.emboj.7601493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin S. V., Patel M., Furia B. S., Peltz S. W., Trotta C. R. Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3′ end formation. Cell. 2004;117:311–321. doi: 10.1016/s0092-8674(04)00342-3. [DOI] [PubMed] [Google Scholar]
- Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Quan X. X., Tsoulos P., Kuritzky A., Zhang R., Stochaj U. The carrier Msn5p/Kap142p promotes nuclear export of the hsp70 Ssa4p and relocates in response to stress. Mol. Microbiol. 2006;62:592–609. doi: 10.1111/j.1365-2958.2006.05395.x. [DOI] [PubMed] [Google Scholar]
- Sarkar S., Azad A. K., Hopper A. K. Nuclear tRNA aminoacylation and its role in nuclear export of endogenous tRNAs in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 1999;96:14366–14371. doi: 10.1073/pnas.96.25.14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer T., Strauss D., Petfalski E., Tollervey D., Hurt E. The path from nucleolar 90S to cytoplasmic 40S pre-ribosomes. EMBO J. 2003;22:1370–1380. doi: 10.1093/emboj/cdg121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt W. M., et al. Mutation in the Scyl1 gene encoding amino-terminal kinase-like protein causes a recessive form of spinocerebellar neurodegeneration. EMBO Rep. 2007;8:691–697. doi: 10.1038/sj.embor.7401001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen H. H., Hopper A. K. Retrograde movement of tRNAs from the cytoplasm to the nucleus in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 2005;102:11290–11295. doi: 10.1073/pnas.0503836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen H. H., Horetsky R. L., Kimball S. R., Murthi A., Jefferson L. S., Hopper A. K. Retrograde nuclear accumulation of cytoplasmic tRNA in rat hepatoma cells in response to amino acid deprivation. Proc. Natl. Acad. Sci. USA. 2007;104:8845–8850. doi: 10.1073/pnas.0700765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stade K., Ford C. S., Guthrie C., Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90:1041–1050. doi: 10.1016/s0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- Steiner-Mosonyi M., Leslie D. M., Dehghani H., Aitchison J. D., Mangroo D. Utp8p is an essential intranuclear component of the nuclear tRNA export machinery of Saccharomyces cerevisiae. J. Biol. Chem. 2003;278:32236–32245. doi: 10.1074/jbc.M302779200. [DOI] [PubMed] [Google Scholar]
- Steiner-Mosonyi M., Mangroo D. The nuclear tRNA aminoacylation-dependent pathway may be the principal route used to export tRNA from the nucleus in Saccharomyces cerevisiae. Biochem. J. 2004;378:809–816. doi: 10.1042/BJ20031306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strub B. R., Eswara M.B.K., Pierce J. B., Mangroo D. Utp8p is a nucleolar tRNA-binding protein that forms a complex with components of the nuclear tRNA export machinery in Saccharomyces cerevisiae. Mol. Biol. Cell. 2007;18:3845–3859. doi: 10.1091/mbc.E06-11-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano A., Endo T., Yoshihisa T. tRNA actively shuttles between the nucleus and cytosol in yeast. Science. 2005;309:140–142. doi: 10.1126/science.1113346. [DOI] [PubMed] [Google Scholar]
- Wang C. C., Schimmel P. Species barrier to RNA recognition overcome with nonspecific RNA binding domains. J. Biol. Chem. 1999;274:16508–16512. doi: 10.1074/jbc.274.23.16508. [DOI] [PubMed] [Google Scholar]
- White R. J. RNA polymerase III transcription—a battleground for tumour suppressors and oncogenes. Eur. J. Cancer. 2004a;40:21–27. doi: 10.1016/j.ejca.2003.09.027. [DOI] [PubMed] [Google Scholar]
- White R. J. RNA polymerase III transcription and cancer. Oncogene. 2004b;23:3208–3216. doi: 10.1038/sj.onc.1207547. [DOI] [PubMed] [Google Scholar]
- Whitney M. L., Hurto R. L., Shaheen H. H., Hopper A. K. Rapid and reversible nuclear accumulation of cytoplasmic tRNA in response to nutrient availability. Mol. Biol. Cell. 2007;18:2678–2686. doi: 10.1091/mbc.E07-01-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihisa T., Ohshima C., Yunoki-Esaki K., Endo T. Cytoplasmic splicing of tRNA in Saccharomyces cerevisiae. Genes Cell. 2007;12:285–297. doi: 10.1111/j.1365-2443.2007.01056.x. [DOI] [PubMed] [Google Scholar]
- Yoshihisa T., Yunoki-Esaki K., Ohshima C., Tanaka N., Endo T. Possibility of cytoplasmic pre-tRNA splicing: the yeast tRNA splicing endonuclease mainly localizes on the mitochondria. Mol. Biol. Cell. 2003;14:3266–3279. doi: 10.1091/mbc.E02-11-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva L., Myers R., Fassati A. tRNAs promote nuclear import of HIV-1 intracellular reverse transcription complexes. Plos Biol. 2006;4:1689–1706. doi: 10.1371/journal.pbio.0040332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman S., Lippman S. I., Zhao X., Broach J. R. How Saccharomyces Responds to Nutrients. Annu. Rev. Genet. 2008;42:27–81. doi: 10.1146/annurev.genet.41.110306.130206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.