

Abstract
Photodynamic therapy (PDT) is an approved therapeutic procedure that exerts cytotoxic activity towards tumor cells by inducing production of reactive oxygen species such as singlet oxygen. PDT leads to oxidative damage of cellular macromolecules, including numerous proteins that undergo multiple modifications such as fragmentation, cross-linking and carbonylation that result in protein unfolding and aggregation. Since the major mechanism for elimination of carbonylated proteins is their degradation by proteasomes, we hypothesized that a combination of PDT with proteasome inhibitors might lead to accumulation of carbonylated proteins in endoplasmatic reticulum (ER), aggravated ER stress and potentiated cytotoxicity towards tumor cells. Indeed, we observed that Photofrin-mediated PDT leads to robust carbonylation of cellular proteins and induction of unfolded protein response (UPR). Pre-treatment of tumor cells with three different proteasome inhibitors, including bortezomib, MG132 and PSI gave increased accumulation of carbonylated and ubiquitinated proteins in PDT-treated cells. Proteasome inhibitors effectively sensitized tumor cells of murine (EMT6 and C-26) as well as human (HeLa) origin to PDT-mediated cytotoxicity. Significant retardation of tumor growth with 60-100% complete responses was observed in vivo in two different murine tumor models (EMT6 and C-26) when PDT was combined with either bortezomib or PSI. Altogether these observations indicate that combination of PDT with proteasome inhibitors leads to potentiated antitumor effects. The results of these studies are of immediate clinical application as bortezomib is a clinically approved drug that undergoes extensive clinical evaluations for the treatment of solid tumors.
Keywords: photodynamic therapy, Photofrin, proteasome, bortezomib, cancer
Introduction
Reactive oxygen species (ROS), and in particular singlet oxygen (1O2) are responsible for the cytotoxic effects induced in photosensitized cells during photodynamic therapy (PDT) (1, 2). 1O2 readily reacts with proteins, lipids (mainly unsaturated fatty acids, but also cholesterol), and DNA (3-7). Proteins are among the most important targets of 1O2 as they constitute about 70% of the dry weight of cells (5). Radical-induced protein modifications include: fragmentation, dimerization or multimerization, misfolding and structural alterations resulting in functional inactivation or changes in mechanical properties, aggregation, changes in binding of co-factors and metal ions, formation of further reactive species or accelerated degradation (8).
Multiple mechanisms have evolved in aerobic organisms to interact with various oxidants and to form less reactive products. These include enzymes that directly react with ROS such as superoxide dismutases, catalase, glutathione peroxidase and a number of secondary scavengers that participate in the reduction of oxidized biomolecules. However, during robust oxidative stress conditions, such as those occurring during PDT, these cytoprotective mechanisms are insufficient and a significant damage to cellular constituents may ensue. Moreover, only cysteine and methionine can be easily reduced to their initial forms, whereas other oxidized amino acids cannot be repaired (9). Therefore, other mechanisms have developed to enable restoration of normal protein function within the cell. Oxidized proteins, which lose their physiological function due to conformational changes, can be re-folded by molecular chaperones such as heat shock proteins (HSPs). Notably, PDT was found to induce expression of a variety of HSPs including HS1, HSP27, HSP60, HSP70, HSP90, GRP78 and GRP94, and for some of these proteins a protective role in PDT-treated cells has been shown (10-14). However, rapid degradation is often the only way to remove oxidatively damaged proteins from the cellular milieu (15, 16). One of the typical biomarkers for oxidative protein damage is carbonylation. Carbonyl derivatives are formed by a direct metal-catalyzed oxidative modification of several amino acids (15), as well as covalent modification by reactive intermediates (17). Carbonylation exposes hydrophobic residues that are normally hidden in the interior of soluble proteins. Generation of hydrophobic patches results in partial protein unfolding, which favours their ubiquitination followed by recognition and degradation by proteasomes (18). Ubiquitin-proteasome system (UPS) forms the most important pathway for the degradation of oxidatively modified proteins (16, 18, 19). The proteasome is a large multisubunit protease complex endowed with three main proteolytic activities: trypsin-like, chymotrypsin-like and caspase-like (also known as peptidyl-glutamyl-hydrolysing activity) (20, 21).
Accumulation of incorrectly folded and/or misfolded proteins within endoplasmic reticulum (ER) is referred to as ER stress (22). ER stress triggers several independent but partially overlapping rescue responses that lead to restoration of protein homeostasis. These rescue responses are collectively referred to as unfolded protein response (UPR). UPR is triggered by at least three signalling pathways: activation of PERK (protein kinase RNA-like ER kinase), ATF6 (activating transcription factor 6) and IRE1 (inositol requiring enzyme 1) (23). Activation of IRE1 leads to unusual cytosolic splicing of mRNA for X-box-binding protein 1 (XBP-1), which upon translation produces sXBP-1, a transcription factor that is translocated to the nucleus for activation of gene transcription. The genes induced during UPR encode chaperones (including numerous heat shock proteins), enzymes that participate in redox reactions and management of oxidative stress and many others. Excessive ER stress or a failure of UPR to counteract ER stress both can trigger cell death. ER-associated degradation (ERAD) is a pathway which is constitutively active in cells to clear misfolded proteins from the ER by directing them to cytoplasmic proteasomes (24). Robust carbonylation of proteins results in the formation of large protein aggregates or “aggresomes” which entrap elements of the UPS decreasing cellular capacity of proteasomal proteolysis (25). Also, treatment with proteasome inhibitors is associated with increased ER stress and UPR induction in different tumor models (26, 27).
It was previously demonstrated that PDT with Purpurin-18 induces protein carbonylation in tumor cells (28). However, the fate of carbonylated proteins in tumor cells has not been investigated. Therefore, we decided to study the proteasome-mediated degradation of PDT-induced protein carbonyls in order to better understand the mechanisms of PDT-mediated cytotoxicity, and to find out whether inhibition of this degradation would affect antitumor effects of PDT.
Materials and Methods
Tumor cells
Human cervical cancer (HeLa) and murine breast carcinoma (EMT6) cell lines were purchased from ATCC (Manassas, VA, USA). Murine colon adenocarcinoma (C-26) was obtained from Dr. Danuta Dus (Institute of Immunology and Experimental Medicine, Wroclaw, Poland). HeLa cells stably expressing HA-tagged δCD3, HA-tagged αTCR, α1-antitrypsin Hong Kong mutant (α1AT), and UbG76VGFP have been established and characterized previously (29). Expression plasmids encoding reporter substrates were obtained from Dr. Allan Weissman (δCD3), Dr. Kazuhiro Nagata (α1AT), Dr. Ron Kopito (αTCR), and Dr. Maria Masucci (UbG76VGFP) and their sequences were verified before transfection. Stably transfected HeLa clones that were most efficient in expressing each reporter substrate were chosen for experiments. Cells were cultured in RPMI 1640 medium (C-26) or Dulbecco's modified Eagle's medium (HeLa, EMT6) supplemented with 10% heat-inactivated fetal bovine serum (all from Invitrogen, Carlsbad, CA, USA), antibiotic/antimycotic solution (Sigma, St. Louis, MO), and 400 μg/ml Geneticin (Sigma, transfected HeLa cells) to ensure stable expression of plasmid vectors.
Mice
BALB/c mice, 8–12 weeks of age, were used in the experiments. Breeding pairs were obtained from the Animal House of the Polish Academy of Sciences, Medical Research Center (Warsaw, Poland). All in vivo experiments were performed in accordance with the guidelines approved by the Ethical Committee of the Medical University of Warsaw.
Reagents
Photofrin (Axcan Pharma Inc., Houdan, France), Verteporfin (a generous gift of QLT PhotoTherapeutics, Inc., Vancouver, BC, Canada), ALA (Sigma) and hypericin [prepared, purified and stored as described (30)] were used as photosensitizers. Tunicamycin, thapsigargin, MG132 and PSI were purchased from Calbiochem/EMD (San Diego, CA), and were dissolved in cell culture quality DMSO (Sigma). Bortezomib (MilleniumPharmaceuticals, Cambridge, MA) was dissolved in 0.9% NaCl.
Cytotoxic assays
Cell cultures for in vitro experiments were illuminated with either He-Ne laser at 632.8-nm (Amber, Warsaw, Poland) or with a 50 W sodium lamp (Phillips) through a red filter as described (31, 32), or as described in (33) when hypericin was used as the photosensitizer. Briefly, tumor cells were dispensed into a 24-well flat-bottomed plate at a concentration of 5 × 103 cells/well and allowed to attach overnight. Then, cells were treated with investigated compounds or with a control medium. After a 24-h incubation with 10 μg/ml Photofrin or indicated photosensitizers, the medium in each well was replaced with PBS, and each well was exposed to laser light. The illumination area matched the size of the wells. After the illumination PBS was removed, cells were trypsinized and seeded into a 96-well microtiter plate. Alternatively, tumor cells were dispensed into 35-mm plates at a concentration of 2.5 × 105 cells/dish and allowed to attach overnight, followed by addition of Photofrin or indicated photosensitizers, and illumination with a sodium lamp. For the evaluation of cytotoxic effects crystal violet staining and MTT assays were used as described previously (32, 34).
Western blotting
For Western blotting analysis cells were cultured with 10 μg/ml Photofrin for 24 h before illumination. After washing with PBS, the cells were illuminated with a 50 W sodium lamp using red filter. At indicated times the cells were washed with PBS and lysed with RIPA buffer (50 mM Tris base, 150 mM NaCl, 1% NP40, 0.25% sodium deoxycholate, and 1 mM EDTA) supplemented with Complete® protease inhibitors cocktail (Roche Diagnostics, Mannheim, Germany). Protein concentration was measured using BCA protein assay (Pierce, Rockford, IL). Equal amounts of proteins were separated on 12% SDS-polyacrylamide gel, transferred onto Protran® nitrocellulose membranes (Schleicher and Schuell BioScience Inc., Keene, NH, USA), blocked with TBST [Tris buffered saline (pH 7.4) and 0.05% Tween 20] with 5% nonfat milk and 5% FBS. The following antibodies were used for the overnight incubation: anti-HA.11 (mouse monoclonal, Covance, Princeton, NJ), anti-GFP (mouse monoclonal, Covance), anti-ubiquitin (mouse monoclonal, Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-actin (rabbit polyclonal, Sigma), anti-KDEL/BiP (mouse monoclonal, Stressgen, Ann Arbor, MI), anti-α-calnexin (mouse monoclonal, Stressgen). After extensive washing with TBST the membranes were incubated for 45 min in corresponding HRP-coupled secondary antibodies (Jackson Immuno Research, West Grove, PA). The reaction was developed using SuperSignal WestPico Kit® (Pierce). After scanning, densitometric analysis of Western blots was performed using the Image Quant 5.2 software (Amersham Bioscience, Piscataway, NJ).
Protein carbonylation assay
Carbonyl content of proteins was determined by DNPH method (35) with some modifications. Cells were washed with PBS and suspended in a buffer consisting of 10 mM HEPES, 1.1 mM KH2PO4, 137 mM NaCl, 0.6 mM MgSO4, 4.4 mM KCl, 1.1 mM EDTA supplemented with Complete® protease inhibitors. Ten-microgram samples of proteins were precipitated with 10% TCA. The precipitates were treated with either 2N HCl alone (control) or 2N HCl containing 5 mg/ml 2,4-DNPH at RT for 30 min. The resulting hydrazones were precipitated in 10% TCA and then washed three times with ethanol-ethyl acetate (1:1). Final precipitates were dissolved in 6 M guanidine chloride. Equal amounts of whole-cell proteins were separated on 12% SDS-polyacrylamide gel, transferred onto nitrocellulose membranes blocked with TBST [Tris-buffered saline (pH 7.4) and 0.05% Tween 20] supplemented with 5% nonfat milk. Anti-DNPH antibodies (Sigma) in dilution of 1:20 000 were used for a 2 h incubation. After washing with TBST, the membranes were incubated with horseradish peroxidase-coupled secondary antibodies. The reaction was developed using SuperSignal WestPico Kit® (Pierce).
Immunoprecipitation
For immunoprecipitation, cells collected from 60-mm plates 24 hours after PDT were lysed in 1 ml of lysis buffer (50 mM HEPES–KOH, pH 7.4 at 4°C, 100 mM NaCl, 1.5 MgCl2 and 0.1% NP-40) using repeated freeze-thaw cycles. After centrifugation (15 min, 16 000 × g at 4°C), protein concentration was determined with BCA protein assay (Pierce) and all lysates were diluted to the same concentration. Next, 1 ml of lysate from each group was precleared with agarose beads, incubated with 5 μl of antibodies for 1 h at 4°C on a rotary wheel, and then 50 μl of protein G bead slurry (HiTrap™, Amersham Pharmacia Biotech AB, Uppsala, Sweden) was added for overnight incubation at 4°C on a rotary wheel. Afterwards, beads were washed 5 times in lysis buffer. Antigen-bound antibodies were released from protein G by 5 min wash in 0.1 M glycine (pH 2.6) and the suspension was used for detection of protein carbonylation.
Transmission electron microscopy
For transmission electron microscopy (TEM) cells collected from 100-mm plates were fixed in 3% glutaraldehyde, and postfixed in 1% OsO4 both in the 0.1 M cacodylate buffer pH 7.4, dehydrated in increasing concentrations (50-100%) of ethanol and in propylene oxide and embedded in Poly/Bed® 812 (Polysciences, Inc., Warrington, PA). Resin blocks were cut with a diamond knife on a RMC type MTXL ultramicrotome. Ultrathin sections were mounted on Formvar carbon-coated grids, stained with lead citrate and uranyl acetate, and observed in a Jeol JEM-100S transmission electron microscope (Jeol, Tokyo, Japan).
RT-PCR
RNA was isolated using Chomczynski's modified method (36) from HeLa or C-26 cells after treatment with PDT and/or proteasome inhibitors. RT-PCR was performed with AMV reverse transcriptase (Promega) according to manufacturers protocol. Next, PCR was performed using GoFlexiTaq DNA Polymerase (Promega, Madison, WI) using pairs of primers amplifying huXBP1: 5′-CCT TGT AGT TGA GAA CCA GG-3′ (forward), 5′-GGG GCT TGG TAT ATA TGT GG-3′ (reverse), and muXBP1: 5′-CCT TGT GGT TGA GAA CCA GG-3′ (forward), 5′-GAG GCT TGG TGT ATA CAT GG-3′ (reverse) as described before (37). For the XBP-1 transcript, the primers are complementary to the region that includes the 26-base pair deletion dependent on IRE-1 endonuclease activity (38). Primers used for amplification of human and murine actin were: 5′-TTC CTT CCT GGG CAT GGA GT-3′ (forward) and 5′-ATC CAC ATC TGC TGG AAG GT-3′ (reverse).
Immunofluorescence microscopy
For immunofluorescence microscopy the cells were dispensed in 8-well chamber slides (Nunc, Roskilde, Denmark) and cultured with 10 μg/ml of Photofrin and other reagents for 24 h before illumination. After washing with PBS, the cells were exposed to laser light. After consecutive 24 h of culture in the fresh medium the cells were washed with PBS. Slides were methanol-fixed for 30 min in -20°C, blocked with 5% normal donkey serum and incubated overnight at 4°C with primary antibodies [anti-α1AT (Sigma), anti-Sec61α, anti-ubiquitin FK2 (both from Stressgen), anti-HA.11, and anti-GFP (both from Covance)] in 5% normal donkey serum in PBS. Slides were washed three times in PBS and incubated with donkey anti-mouse Alexa555- or donkey anti-rabbit Alexa 488-conjugated antibody (Invitrogen-Molecular Probes, Carlsbad, CA; 1:200 for 2 h at room temperature). The slides were washed, mounted in DAPI-enriched Vectashield (Vector Laboratories, Burlingame, CA) and observed under fluorescence confocal microscope (Leica TCS SP2).
Proteasome activity
The 20 S proteasomes were isolated from T2 cells as described (39). Briefly, cells were lysed with 1 mM dithiothreitol, and the stroma-free supernatant was applied to DEAE-Sepharose (Toyopearls). Then, 20S proteasome was eluted with a NaCl gradient in TEAD (20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM azide, and 1 mM dithiothreitol) from 100 to 350 mM NaCl. The 20S proteasome was concentrated by ammonium sulfate precipitation (between 40 and 70% of saturation) and separated in a 10–40% sucrose gradient by centrifugation at 40,000 rpm for 16 h (SW40; L7; Beckman & Coulter, Fullerton, CA). Finally, 20S proteasome was purified on MonoQ column and eluted with a NaCl gradient at 280 mM NaCl. The fractions containing purified 20S proteasome were dialyzed against 50 mM NaCl in TEAD and stored on ice. The purity was determined by SDS-PAGE. One hundred nanograms of purified proteasomes were incubated with 10 μg/ml Photofrin for 30 min. Then, the mixtures were illuminated with 632.8 nm He-Ne ion laser (Amber, Warsaw, Poland) and proteasome activities were determined with 100 μM fluorogenic substrates: Z-Gly-Gly-Arg-βNA (Z = benzyloxycarbonyl and βNA = β-naphtylamide) for tryptic-like activity, Z-Leu-Leu-Glu-βNA for caspase-like activity, and Suc–Leu-Leu-Val-Tyr-AMC (Suc = succinyl, and AMC = 7-amido-4-methylcoumarin) for chymotryptic-like activity. All peptides were purchased from Bachem (Weil am Rhein, Germany). For the measurements of proteasome activity in total cellular lysates C-26 cells were seeded in 100-mm plates at a density of 2 × 106 cells/plate. After incubation with Photofrin for 24 h cells were washed with PBS and illuminated with the sodium light. Then, at indicated time points cells were washed with PBS and lysed [20 mM Tris-HCl (pH 6.8), 50 mM NaCl, 2 mM MgCl2, 0.1% NP40, and Complete® protease inhibitors coctail (Roche)]. Protein content was estimated by BCA (Pierce) and proteasome activities were determined as above.
Tumor treatment and monitoring
All animal experiments were approved by the institutional animal care ethical review board. For in vivo experiments exponentially growing EMT6 and C-26 cells were harvested, resuspended in PBS and injected into the footpad of the right hind limb of experimental mice. Tumor cell viability measured by trypan blue exclusion was above 98%. Photofrin (in 5% dextrose) was administered intraperitoneally at a dose of 10 mg/kg 24 h before illumination with 632.8 nm light (day 6 after inoculation of tumor cells). Control mice received 5% dextrose. The light source was a He-Ne ion laser. The light at fluence rate of 40 mW/cm2 was delivered on day 7 of the experiments using a fiber optic light delivery system as described previously (40, 41). Total light dose delivered to the tumors was 90 J/cm2. During illumination mice were anesthetized with ketamine (87 mg/kg) and xylazine (13 mg/kg) and were restrained in a specially designed holder at 37°C. PSI at a dose of 20 nmoles (dissolved in DMSO) was administered intratumorally (i.t.) for 7 consecutive days, with the first dose given on day 7 of the experiment. Control animals received DMSO. Bortezomib at a dose of 1 mg/kg was administered intraperitoneally (i.p.) in two different schedules: bortezomib was administered on days 5 and 7 after inoculation of tumor cells (before PDT) or on days 7, 9, 11, and 13 after inoculation of tumor cells (after PDT). Control mice received 0.9 % NaCl i.p. Tumor treatment was started when all mice developed tumors with a minimum size of 3×4 mm. Local tumor growth was determined with calipers as described previously (42, 43) by the formula: tumor volume (mm3) = (longer diameter)×(shorter diameter)2.
Statistical analyses
Data were calculated using Microsoft™ Excel 2007. Differences in in vitro cytotoxicity assays and tumor volume were analyzed for significance by Student's t test. Kaplan-Meier plots were generated using days of animal death (after inoculation of tumor cells) as a criterion, and survival time of animals was analyzed for significance by log-rank survival analysis. Significance was defined as a two-sided p < 0.05. The nature of the interaction observed between proteasome inhibitors and PDT was analyzed using the Calcusyn software (Biosoft, Cambridge, England) which uses the combination index (CI) method of Chou and Talalay (44), based on multiple drug effect equation. The advantage of this method is the automatic construction of a fraction affected-CI table, graph, and calculation of dose reduction indices by the software. CIs of <1 indicate greater than additive effects (synergism; the smaller the value, the greater the degree of synergy), CIs equal to 1 indicate additivity, and CIs>1 indicate antagonism.
Results
PDT induces carbonylation and accumulation of ubiquitinated proteins
ROS-induced protein lesions include formation of carbonyl groups, which can be detected using 2,4-DNPH, a reagent that specifically binds to protein carbonyl groups, and can be revealed immunochemically using specific antibodies (35). A time course experiment with approximately equitoxic light fluencies revealed that PDT led to a robust protein carbonylation detectable 8 h after illumination in HeLa and C-26 cells. The amount of carbonylated proteins decreased during the following 16 h (Fig. 1A and C). The ubiquitin-proteasome system (UPS) is responsible for the removal of carbonylated proteins. Therefore, we examined, whether increased protein carbonylation coincided with the accumulation of polyubiquitinated proteins. Within 8 h after illumination there was a marked accumulation of polyubiquitinated proteins in PDT-treated cells (Fig. 1B and D). However, despite a decrease in the amount of carbonylated proteins in cellular lysates, there was a further accumulation of polyubiquitinated proteins 24 h after illumination.
Figure 1. The influence of PDT on carbonylation and ubiqutination of cellular proteins.
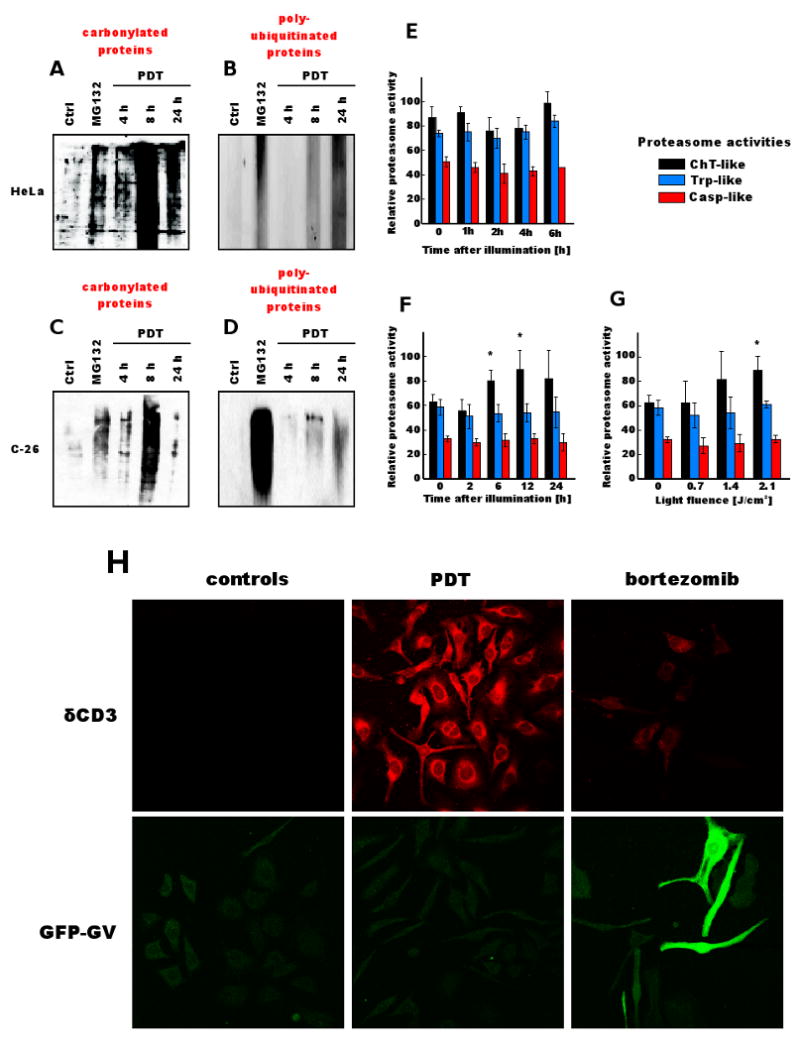
[A] HeLa cells or [B] C-26 cells were incubated with 10 μg/ml of Photofrin for 24 h and exposed to 2.4 J/cm2 of light. At indicated time points, tumor cells were collected and protein carbonylation was determined by DNPH method as described in Materials and Methods. MG132 at 250 nM concentration was used as a positive control.
[C] HeLa cells or [D] C-26 cells were incubated with 10 μg/ml of Photofrin for 24 h and exposed to 2.4 J/cm2 of light. At indicated time points total cell lysates were prepared from tumor cells, and Western blot analysis was performed using anti-ubiquitin antibodies. MG132 at 250 nM concentration was used as a positive control.
[E] Purified proteasome subunits were incubated for 30 min with 10 μg/ml of Photofrin and illuminated with laser light at a fluence of 2.4 J/cm2. Chymotrypsin-like (ChT), trypsin-like (Trp), and caspase-like (Casp) activities were measured with fluorogenic substrates. Relative proteasome activity represents the fluorescence intensity per μg of protein.
[F] C-26 cells were incubated with 10 μg/ml of Photofrin for 24 h and exposed to 1.4 J/cm2 of light. At indicated time points tumor cells were collected and proteasome activities were measured in whole tumor cell lysates using fluorogenic substrates.
[G] C-26 cells were incubated with 10 μg/ml of Photofrin for 24 h and exposed to indicated light fluencies. Proteasome activities were measured in whole tumor cell lysates collected 24 h after illumination using fluorogenic substrates.
[H] HeLa cells stably transfected with expression plasmids encoding reporter proteins (δCD3 and GFP-GV) were incubated with 4 ng/ml of bortezomib or with 10 μg/ml of Photofrin for 24 h and exposed to 2.4 J/cm2 of light. Indirect immunofluorescence microscopy was performed using a laser scanning confocal microscope 24 h after illumination with anti-HA-tag (to detect δCD3) and anti-GFP primary antibodies.
To study the influence of photosensitization on proteasome activity, purified 20S proteasome complexes were incubated with Photofrin and illuminated with laser light. PDT did not affect trypsin-, chymotrypsin-, nor caspase-like activities of purified proteasomes (Fig. 1E). However, despite the lack of direct effect of photosensitization on proteasome activity there was a time- (Fig. 1F), and dose-dependent (Fig. 1G) induction of chymotrypsin-like activity of proteasomes in whole cell lysates of PDT-treated C-26 cells. These observations indicate that PDT does not directly affect the activity of proteasomes (found in extramembraneuos compartments of cells, mainly in the nucleus and cytoplasm). The increased activity of proteasomes in PDT-treated cells might be an inducible and adaptive mechanism to remove oxidatively damaged proteins.
To investigate the sub-cellular localization of photodamaged and polyubiquitinated proteins we used two HeLa cells stably transfected with reporter constructs: an ER membrane-associated model protein (HA-tagged δCD3 chain) or a cytosolic UbG76VGFP protein (GFP-GV), which have been previously described (29). Most δCD3 resides on the cytosolic side of the ER membrane and is a substrate for ER-associated degradation (ERAD) pathway. GFP-GV is a cytosolic protein that has a non-cleavable ubiquitin fused in frame with GFP that targets the protein for degradation through the ubiquitin-fusion degradation (UFD) pathway. Photofrin-mediated PDT induced accumulation of ER-associated δCD3, but not cytosolic GFP-GV proteins (although accumulation of GFP-GV was marked in a positive-control group incubated with 4 ng/ml bortezomib) (Fig. 1H). Since Photofrin is a hydrophobic photosensitizer that accumulates in membrane compartments (45) it is likely that PDT is inducing a selective damage to membrane-associated proteins that leads to ER overloading and thereby impairment of ERAD.
PDT induces ER stress
Oxidative damage and impaired degradation of ER-associated proteins might lead to ER stress. Therefore, we examined the influence of PDT on the activation of signaling pathways associated with unfolded protein response (UPR). During UPR there is an initial global downregulation of protein translation resulting from transient phosphorylation of eIF2α. Nevertheless, UPR also leads to the transcriptional activation of several target genes (through activation of X-box binding protein 1 (XBP1) and other transcription factors) involved in an adaptation that increases the capacity of ER to handle unfolded proteins. Induced proteins include ER chaperones such as GRP78/BiP and calnexin (46). We observed only a modest increase in eIF2α phosphorylation after PDT in HeLa cells (data not shown), but there was a stronger influence of PDT on the expression of BiP and calnexin (Fig. 2A and B). RT-PCR revealed that PDT induced a time-dependent unconventional cytosolic splicing of XBP1, which was detectable within 4 h after illumination in both HeLa and C-26 cells (Fig. 2C).
Figure 2. PDT induces ER stress and unfolded protein response.

[A] HeLa cells were incubated with 10 μg/ml of Photofrin for 24 h and exposed to 1.2 J/cm2 of light. At indicated time points total cell lysates were prepared from tumor cells, and Western blot analysis was performed using anti-BiP, anti-calnexin or anti-β-actin antibodies. MG132 at 250 nM concentration was used as a positive control.
[B] Densitometric analysis of BiP and calnexin expression in HeLa cells from three independent experiments.
[C] HeLa and C-26 cells were incubated with 10 μg/ml of Photofrin for 24 h and exposed to 2.4 or 3.6 J/cm2 of light. At indicated time points mRNA was isolated and RT-PCR was perform to detect alternative XBP1 splicing. Tunicamycin at a 10 μM concentration was used as a positive control for 8 h incubation.
[D] EMT6 cells were incubated with 10 μg/ml of Photofrin for 24 h and exposed to 2.4 J/cm2 of light. For electron microscopy cells were collected and fixed 24 h after PDT as described under Materials and Methods section. Black arrows indicate distended ER, black arrowhead indicates autophagosomal structure, red arrows indicate swollen mitochondria, and red arrowheads show autophagosomes/lysosomes containing mitochondrial debris.
Electron microscopy studies of EMT6 revealed that PDT led to widening of the lumens of both smooth and rough ER and to occasional vacuolization of the cytoplasm, especially in perinuclear regions of the cells. These changes were accompanied by mitochondrial swelling and formation of infrequent lysosomal vacuoles that could represent autophagosomal vesicles (Fig. 2D and Supplementary Figure 1). Similar changes were noticed in PDT-treated HeLa cells (see below).
Pre-incubation of tumor cells with proteasome inhibitors augments the accumulation of carbonylated and polyubiquitinated proteins
Carbonylated proteins tend to form insoluble aggregates (47). Cells remove carbonylated proteins to avoid toxic effects of protein aggregation by directing them for proteasome-mediated degradation (15, 48). Therefore, we decided to examine whether inhibition of proteasomal degradation of proteins would affect the amount of carbonylated proteins in PDT-treated tumor cells. EMT6 cells were pre-incubated with bortezomib and Photofrin for 24 h and illuminated at a fluence of 2.4 J/cm2. A marked accumulation of carbonylated proteins was observed within 8 h after PDT, which significantly decreased during the next 16 h. In the presence of bortezomib the amount of carbonylated proteins was higher at 8 h after illumination and their amount further increased during the next 16 h. Similar effects were detected in HeLa cells (Fig. 3A). Immunofluorescence studies revealed that illumination of EMT6 cells pre-incubated with PSI increases the amount of polyubiquitinated proteins that accumulate in the cytoplasm (Fig. 3B). Similarly, using Western blotting we observed that two different proteasome inhibitors (PSI and bortezomib) significantly increased the amount of polyubiquitinated proteins that accumulate in PDT-treated tumor cells (Fig. 3C).
Figure 3. Proteasome inhibitors increase accumulation of carbonylated and polyubiquitinated proteins in PDT-treated cells.

[A] EMT6 and HeLa cells were incubated with 10 μg/ml of Photofrin and/or 4 ng/ml of bortezomib for 24 h and exposed to 1.2 J/cm2 of light. At indicated time points tumor cells were collected and protein carbonylation was determined by DNPH method as described in Materials and Methods.
[B] EMT6 cells were incubated with 10 μg/ml of Photofrin, and/or 20 nM PSI for 24 h and exposed to 1.2 J/cm2 of light. Indirect immunofluorescence microscopy was performed using a fluorescence microscope 24 h after illumination with anti-ubiquitin (red), anti-Sec61α (green) primary antibodies and DAPI staining (blue).
[C] EMT6 cells were incubated with 10 μg/ml of Photofrin, 20 nM PSI, and/or 4 ng/ml of bortezomib for 24 h and exposed to 1.2 J/cm2 of light. Total cell lysates were prepared from tumor cells 24 h after illumination, and Western blot analysis was performed using anti-ubiquitin antibodies.
Combination of PDT and bortezomib induces accumulation of ER-membrane associated proteins
To further study accumulation of polyubiquitinated proteins in tumor cells pre-incubated with proteasome inhibitor and exposed to PDT we performed immunofluorescence studies with HeLa cells stably transfected with ER-localized model proteins: δCD3, α1-antitrypsin Hong Kong mutant (α1AT), and αTCR. All these proteins are ERAD substrates and in normal conditions they undergo proteasomal degradation and are undetectable by immunofluorescence. Impairment of ERAD leads to accumulation of these proteins. α1AT is an entirely lumenal ERAD substrate (with no association to ER membrane), while α-TCR and δCD3 are single-span transmembrane proteins. Most δCD3 resides on the cytosolic side of the ER, while most αTCR is within the ER lumen, and both proteins are substrates of ERAD pathway (29). Both bortezomib and PDT used alone induced slight accumulation of reporter proteins in tumor cells, but only ER membrane-localized proteins (δCD3 and α-TCR) accumulated to a significantly higher extent in HeLa cells pre-incubated with proteasome inhibitor and exposed to low dose (1.2 J/cm2) PDT (Fig. 4A).
Figure 4. Bortezomib increases accumulation of ER-membrane associated proteins in PDT-treated cells.

[A] HeLa cells stably transfected with expression plasmids encoding reporter proteins (α1AT, δCD3 and αTCR) were incubated with 4 ng/ml of bortezomib and/or with 10 μg/ml of Photofrin for 24 h and exposed to 1.2 J/cm2 of light. Indirect immunofluorescence microscopy was performed using fluorescence microscope 24 h after illumination with anti-α1AT (green), anti-HA-tag (to detect δCD3 and αTCR) primary antibodies (red).
[B] HeLa cells were incubated with 10 μg/ml of Photofrin and/or 4 ng/ml of bortezomib for 24 h and exposed to 1.2 J/cm2 of light. After a 24-h incubation immunopreciptiation was performed with whole tumor cell lysates using anti-ubiquitin antibodies. Protein carbonylation was determined by DNPH method as described in Materials and Methods.
[C] HeLa cells stably transfected with expression plasmids encoding δCD3 were incubated with 4 ng/ml of bortezomib and/or with 10 μg/ml of Photofrin for 24 h and exposed to 1.2 J/cm2 of light. After a 24-h incubation immunopreciptiation was performed with whole tumor cell lysates using anti-HA antibodies. Protein carbonylation was determined by DNPH method as described in Materials and Methods. Equal amounts of immunoprecipitates were analysed using Western blotting for detection of δCD3 with anti-HA antibodies.
Therefore, we decided to check whether proteins that accumulated in cells treated with the combination of PDT and proteasome inhibitors were also carbonylated. Indeed, immunoprecipitation of cellular lysates with anti-ubiquitin antibodies followed by immunochemical detection of carbonyl groups revealed that ubiquitinated proteins that accumulate in the treated cells were also carbonylated (Fig. 4B). Similarly, immunoprecipitated δCD3 was heavily carbonylated (Fig. 4C). The antibody for α1AT is not suitable for immunoprecipitation. Therefore, we immunoprecipitated cytoplasmic GFP-GV protein. Its level of carbonylation did not change in the combination group (not shown).
Pre-incubation of tumor cells with proteasome inhibitors and ER stress-inducing agents potentiates cytotoxic effects of PDT in vitro
Considering that PDT induces robust oxidative protein damage followed by carbonylation, and that proteasomes participate in the degradation of carbonylated proteins, we decided to investigate the outcome of the combination treatment including PDT and proteasome inhibitors. To this end, tumor cells (EMT6, C-26, and HeLa) were pre-incubated with three different proteasome inhibitors (bortezomib, MG132 and PSI) as well as with Photofrin for 24 h. Then, tumor cells were illuminated with light at various fluencies that produced suboptimal rates of tumor cell death. Potentiated cytotoxic effects of the combination of PDT and proteasome inhibitors in all studied cell lines were observed (Fig. 5). Proteasome inhibitors concentrations shown are the lowest that produced potentiated cytotoxicity. At higher concentrations of bortezomib, potentiated cytotoxicity was not better than for low doses (not shown) indicating that it is might not be necessary to use high dose treatment to elicit maximal cytotoxic response. The resulting data were analyzed with a dedicated Calcusyn software to verify potential synergistic interactions between the investigated treatments using Chow and Talalay calculations. In this mathematical model, synergism can be defined when the combination index (CI) is below 1.0 (when CI is below 0.5 the synergism is defined as very strong). Most of the calculated CIs indicate that proteasome inhibitors and PDT exert synergistic and strongly synergistic cytotoxic effects against tumor cells. Incubation of tumor cells with proteasome inhibitors did not increase Photofrin uptake (Supplementary Table 1) indicating that is an unlikely mechanism of potentiated photosensitization.
Figure 5. Proteasome inhibitors potentiate cytotoxic effects of PDT.

EMT6, C-26 and HeLa cells were incubated for 24 hours with 10 μg/ml Photofrin and/or proteasome inhibitors (bortezomib, MG132 or PSI) at indicated concentrations. After 24 hours of incubation, the cells were exposed to different doses of light (as indicated on the right). Following 24 hours of incubation the cytotoxic effects were measured with crystal violet staining. The bars represent percent survival versus untreated controls. Data refer to mean ± SD. Next to each graph showing survival of tumor cells there are results of Chow-Talalay analyses of the combination indices (CI) presented here as a function of inhibition of cell survival in cells treated with proteasome inhibitors and PDT (solid circles). Each numbered circle represents a combination on the survival graph. The straight line at CI = 1 represents the additive effects of both drugs.
To further study the mechanism of potentiated cytotoxicity we incubated HeLa cells with 250 nM MG132 for either 24 or 1 h before illumination. A 24-h exposure to MG132 proteasome inhibitor potentiated cytotoxic effects of PDT to a higher degree than incubation of tumor cells for 1 h before illumination (Fig. 6A and 6B and Supplementary Figure 2). A significant cytotoxicity was observed already at 4 h after illumination with 2.4 J/cm2 in HeLa cells preincubated with MG132, while cytotoxic effects of PDT alone and PDT combined with a 1 h preincubation with MG132 were recorded 24 h after illumination (Figure 6B and Supplementary Figure 2). The higher cytotoxicity was associated with accumulation of higher amounts of carbonylated proteins in cells pre-incubated with 250 nM MG132 for 24 h and treated with PDT (Fig. 6C). Since proteasome inhibitors induce accumulation of proteins that under normal conditions should be degraded (49), including misfolded proteins in the ER, it can be hypothesized that the potentiated cytotoxicity results from accrue of misfolded and/or oxidatively damaged proteins beyond the proteasomal degradative capacity. Indeed, potentiated cytotoxic effects of PDT were also observed in HeLa cells pre-incubated with other ER-stress inducing agents, such as tunicamycin (Fig. 6D, only at 1 μM tunicamycin concentration potentiated cytotoxicity has been observed), or thapsigargin (Fig. 6E), both of which induce a build-up of misfolded proteins within the ER thereby eliciting ER stress. Furthermore, the influence of proteasome inhibitors on the cytotoxic effects of PDT with different photosensitizers was evaluated. For these studies we used photosensitizers that localize predominantly either in mitochondria (Verteporfin, ALA) or in ER (hypericin). The ER-localizing photosensitizer hypericin was found most effective in eliciting potentiated cytotoxic effects in the combined regimen (Fig. 7).
Figure 6. Accumulation of undegraded proteins and ER stress sensitize tumor cells to PDT-mediated cytotoxicity.
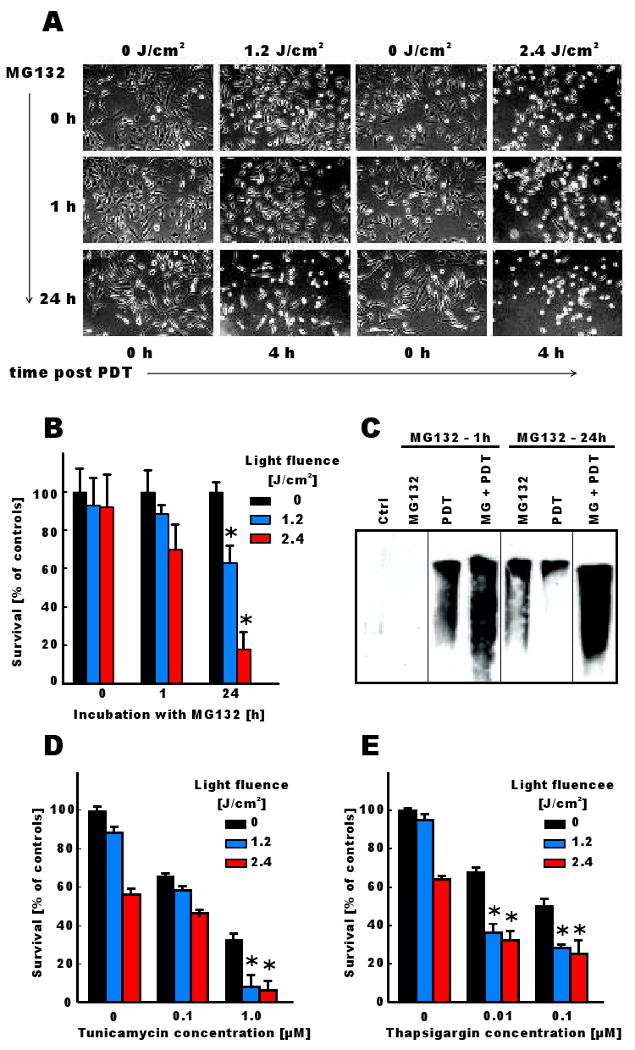
[A] HeLa cells were incubated for 24 hours with 10 μg/ml Photofrin. MG132 at 250 nM concentration was added together with Photofrin or for the last 1 h of incubation. Tumor cells were then illuminated with light at a fluence of 1.2 or 2.4 J/cm2 and photographed under inverted microscope 4 h after illumination.
[B] HeLa cells were incubated for 24 hours with 10 μg/ml Photofrin. MG132 at 250 nM concentration was added together with Photofrin or for the last 1 h of incubation. Tumor cells were then illuminated with light at a fluence of 1.2 or 2.4 J/cm2 and cytotoxic effects were measured with crystal violet staining 8 h after illumination. The bars represent percent survival versus untreated controls. Data refer to mean ± SD. *p<0.05 versus single modality (PDT or MG132) treated cells (Student's t-test).
[C] HeLa cells were incubated for 24 hours with 10 μg/ml Photofrin. MG132 at 250 nM concentration was added together with Photofrin or for the last 1 h of incubation. Tumor cells were then illuminated with light at a fluence of 1.2 J/cm2 and the level of protein carbonylation was measured as described earlier.
[D and E] HeLa cells were incubated for 24 hours with 10 μg/ml Photofrin and/or tunicamycin or thapsigargin at indicated concentrations. Tumor cells were then illuminated with light at a fluence of 1.2 or 2.4 J/cm2 and cytotoxic effects were measured with crystal violet staining 24 h after illumination. The bars represent percent survival versus untreated controls. Data refer to mean ± SD. *p<0.05 versus single modality (PDT or MG132) treated cells (Student's t-test).
Figure 7. Cytotoxic effects of PDT with other photosensitizers combined with proteasome inhibitors.

HeLa cells were incubated for 1 h with 5 μg/ml Verteporfin [A], 6 h with 10 mM ALA [B] or 24 h with 50 nM Hypericin [C] with or without bortezomib. Tumor cells were then illuminated with light at indicated light fluencies. Cytotoxic effects were measured with crystal violet staining 24 h after illumination. The bars represent percent survival versus untreated controls. Data refer to mean ± SD. *p<0.05 versus single modality (PDT or bortezomib) treated cells (Student's t-test).
To gain further insight into mechanisms of potentiated cytotoxicity of the combination treatment electron microscopy studies were done. The most remarkable lesions observed within 8 h of illumination in cells treated with the combination of bortezomib and PDT included robust vacuolization of the cytoplasm, with frequent lysosomal/autophagosomal vesicles and extended ER (Fig. 8).
Figure 8. Electron microscopy of cells treated with PDT and/or bortezomib.
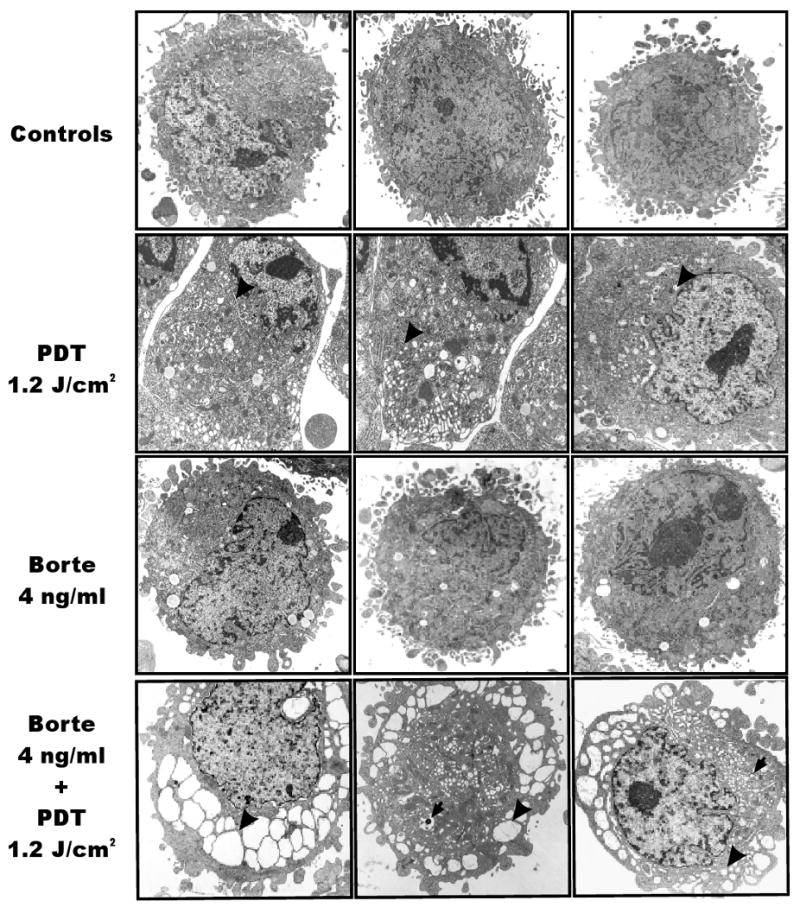
HeLa cells were incubated with 10 μg/ml of Photofrin and/or 4 ng/ml of bortezomib for 24 h and exposed to 1.2 J/cm2 of light. For electron microscopy cells were collected and fixed 8 h after PDT as described under Materials and Methods section. Black arrowheads indicate distended ER, black arrows indicate autophagosomal/lysosomal structures.
Pre-incubation of tumor cells with proteasome inhibitors potentiates antitumor effects of PDT in vivo
The in vitro studies revealed that proteasome inhibitors might effectively sensitize tumor cells to PDT-mediated antitumor effects. Therefore, antitumor effects of the combination treatment with PDT and proteasome inhibitors were evaluated in vivo. In the initial experiment we compared the antitumor effects of the combination treatment with bortezomib administered either before or after suboptimal PDT in an immunogenic murine breast carcinoma (EMT6) model transplanted into syngeneic BALB/c mice (Fig. 9A and B). Interestingly, proteasome inhibition significantly potentiated antitumor effects of PDT in both experimental paradigms, with complete responses observed when bortezomib was administered before or after PDT. Long-term surviving animals were observed in groups (>60% in each group) treated with the combination of bortezomib and PDT in either sequence. Furthermore PSI, another proteasome inhibitor, significantly potentiated antitumor effects of PDT in two different tumor models: a murine colon adenocarcinoma (C-26) as well as EMT6 (Fig. 9C and D). Altogether, in all experiments a marked potentiation of the antitumor effects of PDT in vivo was observed, resulting in retardation of tumor growth, prolongation of the survival time and complete disappearance of tumors in at least 60% of animals (no tumor re-growth for 120 days of observation).
Figure 9. Antitumor effects of the combination treatment with PDT and/or proteasome inhibitors.

BALB/c mice were inoculated with 1 × 105 of EMT6 cells [A-C] or with 2 × 105 of C-26 cells [D] into the right hind limb. Photofrin was administered intraperitoneally at a dose of 10 mg/kg on day 6 [A and B] or 5 [C and D] after inoculation of tumor cells and 24 hours later the tumor site was illuminated with laser light at a dose of 90 J/m2. Bortezomib was administered i.p. on days 5 and 7 [A] or on days 7, 9, 11 and 13 [B] after inoculation of tumor cells at a dose of 1 mg/kg. PSI was administered i.t. on days 6-12 after inoculation of tumor cells at a dose of 20 nmoles/mouse [C and D]. Measurements of tumor diameter were started on day 5 of the experiment and were performed every two days. The graph represents the influence of the PDT treatment on the growth of C-26 tumors. Each group consisted of 5-9 animals. *p<0.05 (Student's t-test), as compared with all other groups. #p<0.05 (Log-rank test), as compared with all other groups.
Discussion
Numerous hydrophobic photosensitizers, including Photofrin, localize to the endoplasmic reticulum, and other membranous structures, such as mitochondria and/or plasma membrane. ER is responsible for the synthesis of the majority of cellular proteins. It contains a number of specialized enzymatic complexes that participate in correct folding and/or glycosylation of cellular proteins. In normal conditions about 30-40% of proteins are incorrectly folded and retrotranslocated from ER to the cytoplasm where they undergo rapid ubiquitination and degradation by proteasomes through ERAD pathway (22). Here we observed that PDT leads to selective retention of ER-membrane associated model proteins (δCD3) but not cytosolic, such as GFP-GV (Fig. 1H), thereby indirectly indicating that PDT might impair ERAD pathway. Similar changes might also occur in other cellular membranes, including mitochondrial or plasma membrane, but were not further studied here. Retrotranslocation and proteasomal degradation of mitochondrial membrane proteins has only recently been suggested (50), and requires further studies. Enlargement of mitochondria in PDT-treated cells (Fig. 2D and Fig. 8) might be an indirect measure of impaired mitochondrial protein retrotranslocation, but this issue requires further studies.
It was previously demonstrated by Magi et al. that PDT with ER- and mitochondria-localizing Purpurin-18 induces protein carbonylation, which is a typical oxidative protein modification (28). Interestingly, many of the carbonylated proteins identified by two-dimensional electrophoresis followed by mass spectrometry are normally functioning within ER and include BiP (GRP78), calreticulin, phosphate disulphide isomerase (PDI) and heat shock protein cognate 71 (HSC71). Additionally, Grabenova et al. reported that PDT downregulates calreticulin, PDI and ERp57 (51). Although carbonylation of these proteins was not studied in this report, it is possible that their decreased amounts in PDT-treated cells result from proteasomal degradation, which follows carbonylation. Indeed, the major mechanism for the elimination of carbonylated proteins is their degradation by the UPS (15, 48). Therefore, we decided to study the influence of Photofrin-mediated PDT on carbonylation and ubiquitination of proteins. Interestingly, although the level of protein carbonylation was maximal at 8 h after PDT, and then slowly decreased (Fig. 1A and C), the amount of polyubiquitinated proteins was still increasing until 24 h after light exposure (Fig. 1B and D). There are several potential mechanisms for this apparent discrepancy. It is possible that not only carbonylated proteins are ubiquitinated, and these are continuously accumulating in PDT-treated cells. It is also possible that heavily carbonylated proteins are preferential substrates for proteasomes, thereby resulting in accumulation of other proteins destined for degradation. Accumulation of polyubiquitinated proteins might also be caused by impaired proteasomal degradation, as has been observed with radiotherapy, another cytotoxic and therapeutic procedure that generates ROS (52). However, the fact that neither the function of purified 20S proteasome subunits after direct photosensitization, nor the proteasome activity in PDT-treated cells were impaired, indicates that PDT does not damage UPS (Fig. 1E-G). Immunofluorescence studies of HeLa cells expressing model substrates of the UPS (29) indicated that PDT leads to selective accumulation of ER-associated reporter protein (δCD3), but not a cytosolic one (GFP-GV), although both undergo carbonylation after PDT (Fig. 1H, 4C). This observation indicates that PDT might lead to a selective impairment of ERAD pathway, which is responsible for retrotranslocation of damaged or misfolded proteins from the ER for their cytoplasmic degradation within proteasomes (53). While components of the ERAD machinery have not been studied in this report, we and others have shown that PDT leads to a rapid ROS-mediated loss-of-function of proteins, enzymes and transporters co-localized in various cellular compartments with photosensitizers (54-58). For example, the ER-localizing hypericin leads to a rapid (within minutes) disappearance of SERCA2 Ca2+ transporters, which is associated with increased cytoplasmic Ca2+ levels (54). Rapid disappearance of SERCA2 might be caused by a number of mechanisms such as fragmentation or crosslinking, but might also be caused by its carbonylation resulting in change of the antibody binding sites (epitopes). The possibility that PDT leads to ROS-mediated damage to the components of ERAD machinery was not addressed here, and will require further studies. It is also possible that elimination of carbonylated proteins observable within 24 h results from engagement of other mechanisms such as autophagy. Indeed, PDT was shown to induce autophagy in tumor cells (59). Electron microscopy studies revealed that there are lysosomal/autophagosomal vesicles forming in PDT-treated cells. The potential role of autophagy in elimination of carbonylated proteins remains to be elucidated. It was recently observed that thioreoxin participates in decarbonylation of cellular proteins (60). We observed that PDT leads to an increased thioredoxin activity (unpublished results), but the role of this enzyme in protein decarbonylation, especially in the setting of PDT, is still unexplored. Robust accumulation of carbonylated proteins in tumor cells incubated with proteasome inhibitors and exposed to PDT confirms that UPS is a significant mechanism responsible for the removal of carbonylated proteins. In addition to our findings that PDT affects ER membrane-associated proteins, it is possible that similar events take place in other membranous cellular compartments (such as mitochondria or plasma membrane), which are sites of Photofrin localization. The possibility of retrotranslocation of proteins from the mitochondrial membrane have only recently been observed (50), and it will be of utmost importance to study carbonylation and retrotranslocation of mitochondrial membrane proteins in the setting of PDT.
Accumulation of carbonylated proteins within ER is accompanied by induction of ER stress and UPR after PDT (Fig. 2). Although it was previously shown, that PDT induces damage to ER (51, 59, 61), and increases expression of ER chaperones (62-64), the signaling pathways associated with UPR have not been studied. We observed that PDT leads to cytoplasmic splicing of XBP1 mRNA (Fig. 2C), a transcription factor associated with UPR. Together, these observations indicate that PDT-induced damage leads to aberrant protein folding within ER. The balance between the extent of ER stress and the adaptive UPR response can dictate cell fate (22). Excessive loading of ER with unfolded proteins that results in extensive ER stress can overwhelm the capacity of UPR. In such conditions UPR triggers cell death (22, 46). Indeed, pre-incubation of tumor cells with thapsigargin and tunicamycin that lead to accumulation of aberrantly folded proteins within ER prior to illumination significantly potentiates PDT-mediated cytotoxicity (Fig. 6D and 6E). All these observations indicate that therapeutic procedures that would elicit ER stress might effectively potentiate cytotoxic effects of PDT.
There is inhomogeneous light distribution during PDT of solid tumors. While well illuminated superficial areas of the tumors receive optimal fluence to trigger robust ROS production and damage to cellular macromolecules, the deeper regions frequently receive an insufficient light dose. UPR would elicit protective responses that will ultimately rescue some tumor cells and compromise the therapeutic efficacy of PDT. Therefore, we decided to study the cytotoxicity of a combined regimen consisting of pre-incubation of tumor cells with proteasome inhibitors followed by PDT. Such combinations would be particularly attractive from the translational perspective as bortezomib, a proteasome inhibitor is a clinically approved drug that undergoes clinical testing in the treatment of solid tumors (65). For the combination studies we used three different proteasome inhibitors (bortezomib, MG132 and PSI) and three cell lines (EMT6, C-26 and HeLa). In all cell lines and all combinations studied we have observed strongly potentiated cytotoxicity as compared to either PDT- or proteasome inhibitor-alone groups (Fig. 5). It is highly possible that the potentiated cytotoxicity results from accumulation of proteins within ER membrane at the time of illumination and not from any direct interaction of PDT and proteasome inhibitors as only 24 h, but not 1 h, preincubation of HeLa cells with MG132 significantly sensitized HeLa cells to PDT (Fig. 6A and 6B). Combined treatment led to accumulation of more carbonylated and polyubiquitinated proteins in tumor cells as compared with single modality-treated cells (Fig. 6C), and the proteins that preferentially accumulate were associated with ER membrane (δCD3 and αTCR), but not with ER lumen (α1-AT) or cytoplasm (GFP-GV) (Fig. 4A). These observations indicate that the most severe damage results from the accumulation of Photofrin in ER membrane. Indeed, TEM studies revealed that there is a marked vacuolization of the cytoplasm in bortezomib + PDT treated cells (Fig. 8), which might be caused by robust extension of the ER lumen. However, it cannot be excluded that large vacuoles observed in tumor cells treated with bortezomib and PDT are of autophagic origin. In PDT-treated cells we observed numerous swollen mitochondria (which also accumulate Photofrin), but these structures were not present in tumor cells after combination treatment. It is likely, although not addressed further, that severely damaged mitochondria were removed in a process of mitophagy.
The most important observation is that proteasome inhibitors significantly potentiate antitumor effects of Photofrin-mediated PDT in vivo (Fig. 9). There was a complete tumor regression in all mice treated with the combined regimen, and after 120 days of observation tumors relapsed in only up to 40% of animals. Intriguingly, potentiated antitumor effects were observed independently of the timing of proteasome inhibitor administration, and occurred when bortezomib was administered either before or after PDT. Since PDT exerts potent antivascular effects it is possible that PDT-treated tumors undergo ischemia-reperfusion episodes that lead to prolonged oxidative stress. Therefore, administration of proteasome inhibitors before or after PDT might elicit potentiated antitumor effects that would result from either sensitization to photooxidation or post-illumination oxidative stress caused by ischemia-reperfusion.
In conclusion, we observed that Photofrin-mediated PDT leads to carbonylation and ubiquitination of ER membrane-associated proteins, accompanied by induction of ER stress and unfolded protein response. Compounds that induce ER stress, such as proteasome inhibitors, can effectively sensitize tumor cells to PDT-mediated cytotoxicity. These observations are of immediate clinical application as proteasome inhibitors are successful drugs approved for use in oncology.
Supplementary Material
Acknowledgments
This work was supported in part by grants: N401 123 31/2736 (J.G.) and N401 3240 33 (D.N.) from Ministry of Science and Higher Education in Poland, 1M19/N (M.J.), 1M19/WB1/07 (M.J.), 1M19/WB2/07 (D.N.), and 1M19/NM2/07 (P.S.) from the Medical University of Warsaw, OT/06/49 from the K.U.Leuven (P.A.), G.0492.05 from the “Fonds voor Wetenschappelijk Onderzoek (FWO)-Vlanderen (P.A.), R01-CA/AI838801 (MRH and PM) from the US National Institutes of Health, as well as an institutional appropriation of the American Cancer Society grant IRG-84-002-22 (C.W.). Tadeusz Issat is a recipient of the START stipend from the Foundation for Polish Science. We would like to thank Elzbieta Gutowska and Anna Czerepinska for excellent technical assistance.
References
- 1.Weishaupt KR, Gomer CJ, Dougherty TJ. Identification of singlet oxygen as the cytotoxic agent in photoinactivation of a murine tumor. Cancer Res. 1976;36:2326–9. [PubMed] [Google Scholar]
- 2.Juzenas P, Moan J. Singlet oxygen in photosensitization. J Environ Pathol Toxicol Oncol. 2006;25:29–50. doi: 10.1615/jenvironpatholtoxicoloncol.v25.i1-2.30. [DOI] [PubMed] [Google Scholar]
- 3.Bachowski GJ, Pintar TJ, Girotti AW. Photosensitized lipid peroxidation and enzyme inactivation by membrane-bound merocyanine 540: reaction mechanisms in the absence and presence of ascorbate. Photochem Photobiol. 1991;53:481–91. doi: 10.1111/j.1751-1097.1991.tb03660.x. [DOI] [PubMed] [Google Scholar]
- 4.Girotti AW. Photosensitized oxidation of cholesterol in biological systems: reaction pathways, cytotoxic effects and defense mechanisms. J Photochem Photobiol B. 1992;13:105–18. doi: 10.1016/1011-1344(92)85050-5. [DOI] [PubMed] [Google Scholar]
- 5.Davies MJ. Singlet oxygen-mediated damage to proteins and its consequences. Biochem Biophys Res Commun. 2003;305:761–70. doi: 10.1016/s0006-291x(03)00817-9. [DOI] [PubMed] [Google Scholar]
- 6.Cadet J, Ravanat JL, Martinez GR, Medeiros MH, Di Mascio P. Singlet oxygen oxidation of isolated and cellular DNA: product formation and mechanistic insights. Photochem Photobiol. 2006;82:1219–25. doi: 10.1562/2006-06-09-IR-914. [DOI] [PubMed] [Google Scholar]
- 7.Klotz LO, Kroncke KD, Sies H. Singlet oxygen-induced signaling effects in mammalian cells. Photochem Photobiol Sci. 2003;2:88–94. doi: 10.1039/b210750c. [DOI] [PubMed] [Google Scholar]
- 8.Nowis D, Golab J. Photodynamic therapy and oxidative stress. In: Hamblin MR, Mroz P, editors. Advances in photodynamic therapy: basic, translational, and clinical. Boston, Mass.; London: Artech House; 2008. pp. 151–78. [Google Scholar]
- 9.Petropoulos I, Friguet B. Protein maintenance in aging and replicative senescence: a role for the peptide methionine sulfoxide reductases. Biochim Biophys Acta. 2005;1703:261–6. doi: 10.1016/j.bbapap.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 10.Wang HP, Hanlon JG, Rainbow AJ, Espiritu M, Singh G. Up-regulation of Hsp27 plays a role in the resistance of human colon carcinoma HT29 cells to photooxidative stress. Photochem Photobiol. 2002;76:98–104. doi: 10.1562/0031-8655(2002)076<0098:urohpa>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 11.Jalili A, Makowski M, Switaj T, et al. Effective photoimmunotherapy of murine colon carcinoma induced by the combination of photodynamic therapy and dendritic cells. Clin Cancer Res. 2004;10:4498–508. doi: 10.1158/1078-0432.CCR-04-0367. [DOI] [PubMed] [Google Scholar]
- 12.Xue LY, He J, Oleinick NL. Rapid tyrosine phosphorylation of HS1 in the response of mouse lymphoma L5178Y-R cells to photodynamic treatment sensitized by the phthalocyanine Pc 4. Photochem Photobiol. 1997;66:105–13. doi: 10.1111/j.1751-1097.1997.tb03145.x. [DOI] [PubMed] [Google Scholar]
- 13.Korbelik M, Sun J, Cecic I. Photodynamic therapy-induced cell surface expression and release of heat shock proteins: relevance for tumor response. Cancer Res. 2005;65:1018–26. [PubMed] [Google Scholar]
- 14.Mitra S, Goren EM, Frelinger JG, Foster TH. Activation of heat shock protein 70 promoter with meso-tetrahydroxyphenyl chlorin photodynamic therapy reported by green fluorescent protein in vitro and in vivo. Photochem Photobiol. 2003;78:615–22. doi: 10.1562/0031-8655(2003)078<0615:aohspp>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 15.Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. Embo J. 2005;24:1311–7. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunlop RA, Rodgers KJ, Dean RT. Recent developments in the intracellular degradation of oxidized proteins. Free Radic Biol Med. 2002;33:894–906. doi: 10.1016/s0891-5849(02)00958-9. [DOI] [PubMed] [Google Scholar]
- 17.Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA. Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem. 2008;283:21837–41. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grune T, Merker K, Sandig G, Davies KJ. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem Biophys Res Commun. 2003;305:709–18. doi: 10.1016/s0006-291x(03)00809-x. [DOI] [PubMed] [Google Scholar]
- 19.Duarte TL, Lunec J. Review: When is an antioxidant not an antioxidant? A review of novel actions and reactions of vitamin C. Free Radic Res. 2005;39:671–86. doi: 10.1080/10715760500104025. [DOI] [PubMed] [Google Scholar]
- 20.Wojcik C, Di Napoli M. Ubiquitin-proteasome system and proteasome inhibition: new strategies in stroke therapy. Stroke. 2004;35:1506–18. doi: 10.1161/01.STR.0000126891.93919.4e. [DOI] [PubMed] [Google Scholar]
- 21.Golab J, Bauer TM, Daniel V, Naujokat C. Role of the ubiquitin-proteasome pathway in the diagnosis of human diseases. Clin Chim Acta. 2004;340:27–40. doi: 10.1016/j.cccn.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 22.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 23.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 24.Romisch K. Endoplasmic reticulum-associated degradation. Annu Rev Cell Dev Biol. 2005;21:435–56. doi: 10.1146/annurev.cellbio.21.012704.133250. [DOI] [PubMed] [Google Scholar]
- 25.Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519–30. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 26.Nowis D, McConnell EJ, Dierlam L, Palamarchuk A, Lass A, Wojcik C. TNF potentiates anticancer activity of bortezomib (Velcade) through reduced expression of proteasome subunits and dysregulation of unfolded protein response. Int J Cancer. 2007;121:431–41. doi: 10.1002/ijc.22695. [DOI] [PubMed] [Google Scholar]
- 27.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100:9946–51. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magi B, Ettorre A, Liberatori S, et al. Selectivity of protein carbonylation in the apoptotic response to oxidative stress associated with photodynamic therapy: a cell biochemical and proteomic investigation. Cell Death Differ. 2004;11:842–52. doi: 10.1038/sj.cdd.4401427. [DOI] [PubMed] [Google Scholar]
- 29.Wojcik C, Rowicka M, Kudlicki A, et al. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol Biol Cell. 2006;17:4606–18. doi: 10.1091/mbc.E06-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen B, Roskams T, Xu Y, Agostinis P, de Witte PA. Photodynamic therapy with hypericin induces vascular damage and apoptosis in the RIF-1 mouse tumor model. Int J Cancer. 2002;98:284–90. doi: 10.1002/ijc.10175. [DOI] [PubMed] [Google Scholar]
- 31.Golab J, Nowis D, Skrzycki M, et al. Antitumor effects of photodynamic therapy are potentiated by 2-methoxyestradiol. A superoxide dismutase inhibitor. J Biol Chem. 2003;278:407–14. doi: 10.1074/jbc.M209125200. [DOI] [PubMed] [Google Scholar]
- 32.Nowis D, Legat M, Grzela T, et al. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene. 2006;25:3365–74. doi: 10.1038/sj.onc.1209378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hendrickx N, Volanti C, Moens U, et al. Up-regulation of cyclooxygenase-2 and apoptosis resistance by p38 MAPK in hypericin-mediated photodynamic therapy of human cancer cells. J Biol Chem. 2003;278:52231–9. doi: 10.1074/jbc.M307591200. [DOI] [PubMed] [Google Scholar]
- 34.Makowski M, Grzela T, Niderla J, et al. Inhibition of cyclooxygenase-2 indirectly potentiates antitumor effects of photodynamic therapy in mice. Clin Cancer Res. 2003;9:5417–22. [PubMed] [Google Scholar]
- 35.Nakamura A, Goto S. Analysis of protein carbonyls with 2,4-dinitrophenyl hydrazine and its antibodies by immunoblot in two-dimensional gel electrophoresis. J Biochem. 1996;119:768–74. doi: 10.1093/oxfordjournals.jbchem.a021306. [DOI] [PubMed] [Google Scholar]
- 36.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 37.Nowis D, McConnell E, Wojcik C. Destabilization of the VCP-Ufd1-Npl4 complex is associated with decreased levels of ERAD substrates. Exp Cell Res. 2006;312:2921–32. doi: 10.1016/j.yexcr.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 39.Mlynarczuk-Bialy I, Roeckmann H, Kuckelkorn U, et al. Combined effect of proteasome and calpain inhibition on cisplatin-resistant human melanoma cells. Cancer Res. 2006;66:7598–605. doi: 10.1158/0008-5472.CAN-05-2614. [DOI] [PubMed] [Google Scholar]
- 40.Golab J, Olszewska D, Mroz P, et al. Erythropoietin restores the antitumor effectiveness of photodynamic therapy in mice with chemotherapy-induced anemia. Clin Cancer Res. 2002;8:1265–70. [PubMed] [Google Scholar]
- 41.Golab J, Wilczynski G, Zagozdzon R, et al. Potentiation of the anti-tumour effects of Photofrin-based photodynamic therapy by localized treatment with G-CSF. Br J Cancer. 2000;82:1485–91. doi: 10.1054/bjoc.1999.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golab J, Kozar K, Kaminski R, et al. Interleukin 12 and indomethacin exert a synergistic, angiogenesis-dependent antitumor activity in mice. Life Sci. 2000;66:1223–30. doi: 10.1016/s0024-3205(00)00427-6. [DOI] [PubMed] [Google Scholar]
- 43.Kozar K, Kaminski R, Switaj T, et al. Interleukin 12-based immunotherapy improves the antitumor effectiveness of a low-dose 5-Aza-2′-deoxycitidine treatment in L1210 leukemia and B16F10 melanoma models in mice. Clin Cancer Res. 2003;9:3124–33. [PubMed] [Google Scholar]
- 44.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 45.Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: Part three-Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagn Photodyn Ther. 2005;2:91–106. doi: 10.1016/S1572-1000(05)00060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends Mol Med. 2003;9:169–76. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 48.Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Divald A, Powell SR. Proteasome mediates removal of proteins oxidized during myocardial ischemia. Free Radic Biol Med. 2006;40:156–64. doi: 10.1016/j.freeradbiomed.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 50.Margineantu DH, Emerson CB, Diaz D, Hockenbery DM. Hsp90 inhibition decreases mitochondrial protein turnover. PLoS ONE. 2007;2:e1066. doi: 10.1371/journal.pone.0001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grebenova D, Kuzelova K, Smetana K, et al. Mitochondrial and endoplasmic reticulum stress-induced apoptotic pathways are activated by 5-aminolevulinic acid-based photodynamic therapy in HL60 leukemia cells. J Photochem Photobiol B. 2003;69:71–85. doi: 10.1016/s1011-1344(02)00410-4. [DOI] [PubMed] [Google Scholar]
- 52.Pajonk F, Pajonk K, McBride WH. Apoptosis and radiosensitization of hodgkin cells by proteasome inhibition. Int J Radiat Oncol Biol Phys. 2000;47:1025–32. doi: 10.1016/s0360-3016(00)00516-2. [DOI] [PubMed] [Google Scholar]
- 53.Ahner A, Brodsky JL. Checkpoints in ER-associated degradation: excuse me, which way to the proteasome? Trends Cell Biol. 2004;14:474–8. doi: 10.1016/j.tcb.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 54.Buytaert E, Callewaert G, Hendrickx N, et al. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. Faseb J. 2006;20:756–8. doi: 10.1096/fj.05-4305fje. [DOI] [PubMed] [Google Scholar]
- 55.Petrat F, Pindiur S, Kirsch M, de Groot H. NAD(P)H, a primary target of 1O2 in mitochondria of intact cells. J Biol Chem. 2003;278:3298–307. doi: 10.1074/jbc.M204230200. [DOI] [PubMed] [Google Scholar]
- 56.Usuda J, Chiu SM, Murphy ES, Lam M, Nieminen AL, Oleinick NL. Domain-dependent photodamage to Bcl-2. A membrane anchorage region is needed to form the target of phthalocyanine photosensitization. J Biol Chem. 2003;278:2021–9. doi: 10.1074/jbc.M205219200. [DOI] [PubMed] [Google Scholar]
- 57.Xue LY, Chiu SM, Oleinick NL. Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene. 2001;20:3420–7. doi: 10.1038/sj.onc.1204441. [DOI] [PubMed] [Google Scholar]
- 58.Salet C, Moreno G, Ricchelli F, Bernardi P. Singlet oxygen produced by photodynamic action causes inactivation of the mitochondrial permeability transition pore. J Biol Chem. 1997;272:21938–43. doi: 10.1074/jbc.272.35.21938. [DOI] [PubMed] [Google Scholar]
- 59.Buytaert E, Callewaert G, Vandenheede JR, Agostinis P. Deficiency in apoptotic effectors Bax and Bak reveals an autophagic cell death pathway initiated by photodamage to the endoplasmic reticulum. Autophagy. 2006;2:238–40. doi: 10.4161/auto.2730. [DOI] [PubMed] [Google Scholar]
- 60.Wong CM, Cheema AK, Zhang L, Suzuki YJ. Protein carbonylation as a novel mechanism in redox signaling. Circ Res. 2008;102:310–8. doi: 10.1161/CIRCRESAHA.107.159814. [DOI] [PubMed] [Google Scholar]
- 61.Kessel D, Castelli M, Reiners JJ. Ruthenium red-mediated suppression of Bcl-2 loss and Ca(2+) release initiated by photodamage to the endoplasmic reticulum: scavenging of reactive oxygen species. Cell Death Differ. 2005;12:502–11. doi: 10.1038/sj.cdd.4401579. [DOI] [PubMed] [Google Scholar]
- 62.Gomer CJ, Ferrario A, Rucker N, Wong S, Lee AS. Glucose regulated protein induction and cellular resistance to oxidative stress mediated by porphyrin photosensitization. Cancer Res. 1991;51:6574–9. [PubMed] [Google Scholar]
- 63.Mak NK, Li KM, Leung WN, et al. Involvement of both endoplasmic reticulum and mitochondria in photokilling of nasopharyngeal carcinoma cells by the photosensitizer Zn-BC-AM. Biochem Pharmacol. 2004;68:2387–96. doi: 10.1016/j.bcp.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 64.Buytaert E, Matroule JY, Durinck S, et al. Molecular effectors and modulators of hypericin-mediated cell death in bladder cancer cells. Oncogene. 2008;27:1916–29. doi: 10.1038/sj.onc.1210825. [DOI] [PubMed] [Google Scholar]
- 65.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14:1649–57. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.