

Summary
Mitochondrial homeostasis reflects a dynamic balance between membrane fission and fusion events thought essential for mitochondrial function. We report here that altered expression of the C. elegans BCL2 homolog CED-9 affects both mitochondrial fission and fusion. Although striated muscle cells lacking CED-9 have no alteration in mitochondrial size or ultrastructure, these cells appear more sensitive to mitochondrial fragmentation. By contrast, increased CED-9 expression in these cells produces highly interconnected mitochondria. This mitochondrial phenotype is partially suppressed by increased expression of the dynamin-related GTPase DRP-1, with suppression dependent on the BH3 binding pocket of CED-9. This suppression suggests that CED-9 directly regulates DRP-1, a model supported by our finding that CED-9 activates the GTPase activity of human DRP1. Thus, CED-9 is capable of regulating the mitochondrial fission-fusion cycle but is not essential for either fission or fusion.
Keywords: Bcl-2, BCL2, CED-9, DRP-1, Mitochondria, Morphology, Dynamics, Homeostasis, C. elegans, Transmembrane domain
Introduction
Bcl-2 proteins regulate apoptosis through a mechanism that is sensitive to their expression levels, which vary among Bcl-2 homologs and different tumor isolates (Findley et al., 1997; Krajewska et al., 1996; Martin et al., 2001; Oltvai et al., 1993; Reed, 2008; Reed et al., 1991). Indeed, Bcl-2 expression levels are thought to accelerate or delay the progression of many diseases including cancer and neurodegenerative diseases (Adams and Cory, 2007; Ay et al., 2001; Kostic et al., 1997; Merry and Korsmeyer, 1997; Thompson, 1995). Although the effects of different expression levels has been intensively studied with respect to apoptosis, their impact on other cellular processes are only beginning to be elucidated. One such process is mitochondrial homeostasis, where increased or decreased expression affects mitochondrial morphology, resulting in either fragmented or interconnected mitochondria (Brooks et al., 2007; Delivani et al., 2006; Frank et al., 2001; Jagasia et al., 2005; Karbowski et al., 2006; Li et al., 2008).
Alterations in mitochondrial morphology can impact mitochondrial function and can perturb various signaling pathways (Detmer and Chan, 2007; McBride et al., 2006). A dramatic example of the correlation between mitochondrial morphology and function occurs during apoptosis, where mitochondrial membranes remodel and fragment near the time that cytochrome c is released from the inner membrane space (Martinou and Youle, 2006). Indeed, perturbation of the mitochondrial fission and fusion machinery affects apoptosis efficiency (Cassidy-Stone et al., 2008; Estaquier and Arnoult, 2007; Fannjiang et al., 2004; Frank et al., 2001; Jagasia et al., 2005; James et al., 2003; Lee et al., 2004; Parone et al., 2006; Sugioka et al., 2004). Conversely, perturbation of Bcl-2 proteins that regulate apoptosis has been found to alter mitochondrial homeostasis (Brooks et al., 2007; Delivani et al., 2006; Karbowski et al., 2006; Li et al., 2008). Intriguingly, the Bcl-2 protein BAX, the mitochondrial fission protein DRP1 (DNML1), and the mitochondrial fusion protein MFN2 cluster at foci on mitochondrial surfaces, further suggesting connections between mitochondrial homeostasis and Bcl-2 proteins (Karbowski et al., 2002).
This connection may occur by Bcl-2 proteins regulating one or more of the large GTPases involved in mitochondrial fission and fusion. The large GTPases involved in mitochondrial fusion are known as mitofusins: FZO-1 in yeast and C. elegans, MFN1 and MFN2 in mammals (Detmer and Chan, 2007). MFN2 constitutively localizes at mitochondrial outer membranes in two populations that are influenced by the Bcl-2 proteins BAX and BAK (Karbowski et al., 2002; Karbowski et al., 2006). Strikingly, mitochondria in cells lacking both BAX and BAK are highly fragmented compared with wild-type cells (Karbowski et al., 2006). This mitochondrial fragmentation is reversed upon increased expression of the mitofusin protein MFN2, consistent with a model whereby the observed mitochondrial defects arise from insufficient mitochondrial fusion. As MFN2 appears mislocalized and does not form higher order structures in the absence of BAX and BAK, these results suggest that Bcl-2 proteins are important for MFN2 activity, and may in turn regulate mitochondrial homeostasis by affecting fusion. Supporting this idea are studies in which either increased BCL-XL (BCL2L1) or CED-9 expression results in interconnected mitochondria in HeLa cells (Delivani et al., 2006). Though these findings and others suggest a tight interplay between Bcl-2 proteins, mitofusins and mitochondrial fusion (Brooks et al., 2007; Delivani et al., 2006; Karbowski et al., 2006), other mechanisms exist such as direct interactions between Bcl-2 proteins and membranes (Leber et al., 2007; Thuduppathy et al., 2006a; Thuduppathy et al., 2006b).
In addition to regulating mitochondrial fusion, Bcl-2 proteins have also been implicated in regulating the large GTPase involved in mitochondrial fission, the dynamin-related protein Drp1. Upon recruitment to sites of future fission, this large GTPase is proposed to polymerize around mitochondria and promote fission (Ingerman et al., 2005; Smirnova et al., 2001). This fission process can be activated by BAX in a DRP1-dependent manner (Frank et al., 2001). Thus, BAX-induced mitochondrial fission appears to depend upon DRP1. Whether DRP1 itself requires Bcl-2 proteins for fission has been difficult to ascertain, due partly to the presence of multiple mammalian Bcl-2 homologs. This difficulty is underscored by a recent study suggesting that BCL-XL also promotes fission by increasing the GTPase activity of DRP1 (Li et al., 2008).
In C. elegans, mitochondrial fission induced by DRP-1 expression in embryos appears to require CED-9, the only Bcl-2 homolog identified to date (Jagasia et al., 2005). We report here that DRP-1-induced fission can occur in the absence of CED-9 activity in differentiated body wall muscle. Despite the lack of a CED-9 requirement for either mitochondrial fission or fusion in body wall muscle, we find that altered CED-9 expression can affect both processes.
Results
Mitochondria in animals carrying ced-9 loss-of-function or gain-of-function mutations appear similar to mitochondria in wild-type animals
To investigate the effects of altered CED-9 expression on C. elegans mitochondrial homeostasis, we examined mitochondrial morphology in the body wall muscle of young adult animals. These muscle cells abut the outer body surface and are relatively large and flat, making it possible to evaluate mitochondria size, shape and distribution (Labrousse et al., 1999). We created multiple independent transgenic lines, each expressing GFP targeted to the mitochondrial matrix (mitoGFP). These lines express mitoGFP using a copy of the myo-3 promoter, which drives expression in striated body wall muscle cells. In muscle cells from wild-type ced-9(+) adult animals, we generally observed tubular mitochondria that were thin, elongated tubules, arranged in roughly parallel rows (Fig. 1A). There was some degree of cell to cell variability, which when quantified was found to be about 25% of the muscle cells that had shorter or fragmented mitochondria (Fig. 2G). This variability was higher in older animals (data not shown), and may be related to stress, as previously observed (Labrousse et al., 1999).
Fig. 1.

Analysis of mitochondria in wild-type, loss-of-function ced-9(n2812lf); ced-3(n717lf) and gain-of-function ced-9(n1950gf) animals.
(A–C)Mitochondrial morphology in wild-type, loss-of-function ced-9(n2812lf); ced-3(n717lf) and gain-of-function ced-9(n1950sd) genetic backgrounds was examined by creating multiple independently generated transgenic lines that expressed GFP targeted to the mitochondrial matrix (mitoGFP). Wide-field epifluorescene images are of body wall muscle cells from young adult animals. n, nucleus. Bar, 10 μm. (D)The average mitochondrial length in a given muscle cell was estimated using ImageJ (see Materials and Methods), and plotted as an individual point [wild-type n=16, ced-9(n2812lf); ced-3(n717lf) n=19, ced-9(n1950gf) n=14]; the mean value is indicated by a horizontal bar.
(E)Mitochondrial ultrastructure in wild-type, ced-9(n2812lf); ced-3(n717lf) and ced-9(n1950sd) animals was examined in both body wall muscle and hypodermis by transmission electron microscopy. m, mitochondria. Bar, 500 nm.
Fig. 2.

Mitochondrial fission and fusion occur in the absence of CED-9. (A-F) Wild-type and dominant negative forms of DRP-1 were expressed using a myo-3 promoter in wild-type ced-9(+) and ced-9(n2812lf); ced-3(n717lf) animals. Similar mitochondrial morphologies were observed in both genetic backgrounds when body wall muscle cells were labeled with mitoGFP. n, nucleus. Bar, 10 μm. (G,H) Wild-type DRP-1 expressed using a myo-3 promoter in ced-9(n1950gf) animals resulted in a similar phenotype. (I) Increased DRP-1 expression induced mitochondrial fragmentation in wild-type, ced-9(n2812lf); ced-3(n717lf), and ced-9(n1950sd) backgrounds. Each point represents the proportion of cells with fragmented mitochondria from an independently generated transgenic line; the mean value is indicated by a horizontal bar, and error bars represent the standard error. The differences between wild-type and ced-9(n2812lf); ced-3(n717lf) backgrounds is significant, z=2.42, P<0.05; raw data is included as Table S1 in supplementary material. (J) Inactivating DRP-1 with dominant negative DRP-1(K40A) resulted in interconnected mitochondria in both wild-type and ced-9(n2812lf); ced-3(n717lf) backgrounds.
To test whether loss of CED-9 expression alters mitochondrial homeostasis, we first examined mitochondrial morphology in ced-9(n2812lf); ced-3(n717lf) animals. We chose these animals, which lack the CED-3 caspase required for almost all apoptosis (Hengartner and Horvitz, 1994b), because loss-of-function ced-9(n2812lf) is lethal due to excessive ectopic apoptosis (Hengartner et al., 1992). We expected to find fragmented mitochondria in these animals based on mammalian studies where mitochondria from BAX and BAK double knockout cells are fragmented (Karbowski et al., 2006). However, muscle cells from ced-9(n2812lf); ced-3(n717lf) animals had mitochondria that were grossly similar to those in wild-type animals (Fig. 1B). Similar results were obtained using the mitochondrial stain MitoTracker Red CMXRos (M-7512, Molecular Probes) when we examined cells from ced-9(n2812lf); ced-3(n717lf) animals, ced-3(n717lf) single-mutant animals, and maternally rescued ced-9(n1950n2161lf) animals harboring a different ced-9 mutant allele (Fig. S1 in supplementary material). Measurement of the mean mitochondrial length (see Materials and Methods) in cells with tubular mitochondrial morphology was also similar: ced-9(+) animals had a mean mitochondrial length of 1.62 μm (±0.24 μm; s.d.), while ced-9(n2812lf); ced-3(n717lf) animals had a mean mitochondrial length of 1.64 μm (±0.35 μm; Fig. 1D). These results suggest that CED-9 is not essential for mitochondrial homeostasis in C. elegans muscle.
We also examined mitochondria in the gain-of-function mutant ced-9(n1950gf) that is deficient in apoptosis activation due to a point mutation in the BH3 binding pocket of CED-9 (Hengartner and Horvitz, 1994a). We reasoned that if CED-9 is not essential for maintaining mitochondrial homeostasis, then this mutant allele should also show little effect on mitochondrial morphology. Indeed, the results were similar with a mean mitochondrial length of 1.66 μm (±0.27 μm; Fig. 1C,D), although we note that the diameter of these mitochondria occasionally appeared slightly larger than those of the other genotypes.
Finally, we analyzed mitochondrial ultrastructure by transmission electron microscopy. This analysis did not reveal any gross mitochondrial abnormalities in either body wall muscle or hypodermis of wild-type, ced-9 loss-of-function and ced-9 gain-of-function animals (Fig. 1E). These results extend previous reports of normal mitochondrial morphology in ced-9 mutant embryos (Jagasia et al., 2005), suggesting that CED-9 is not essential for maintaining mitochondrial homeostasis in C. elegans.
Increased DRP-1 expression induces mitochondrial fragmentation in the absence of CED-9
Even though there were no gross defects in mitochondrial morphology in animals lacking CED-9, it is possible that loss of CED-9 expression primarily affects mitochondrial homeostasis under conditions not assayed in our steady-state experiments. One such condition has been reported in a study examining the role of the mitochondrial fission protein DRP-1: heat-shock expression of DRP-1 in embryos results in mitochondrial fragmentation, which is dependent upon the presence of wild-type CED-9 (Jagasia et al., 2005). We asked whether CED-9 is also required for DRP-1-induced fragmentation of mitochondria in body wall muscle cells by increasing DRP-1 expression in ced-9(+) animals using the myo-3 promoter. Consistent with previous results (Labrousse et al., 1999), increased DRP-1 expression in animals with wild-type CED-9 activity increased the proportion of cells with fragmented mitochondria that were small and vesicular (Fig. 2B,I). When we increased DRP-1 expression in animals lacking CED-9, the proportion of cells with fragmented mitochondria also increased, suggesting that DRP-1 does not always require CED-9 for mitochondrial fragmentation (Fig. 2E,I). Similarly, increased DRP-1 expression in ced-9(n1950gf) animals increased the proportion of cells with fragmented mitochondria (Fig. 2H,I). These results differ from previous results that suggested that CED-9 was required for DRP-1-induced fragmentation (Jagasia et al., 2005). These differences may arise from either a differential sensitivity to mitochondrial fragmentation in the developmental stage or cell-type examined (embryonic cells versus adult muscle cells), or the length and degree of expression (heat-shock versus myo-3 promoter).
Interestingly, increased DRP-1 expression resulted in a higher proportion of cells with fragmented mitochondria in animals lacking CED-9 when compared to animals with wild-type CED-9 (0.84 versus 0.66, z=2.42, P<0.05, Fig. 2I). This difference suggests that CED-9 may be present and has an active role in adult muscle cells. This increased fragmentation could either indicate increased fission or decreased fusion in the absence of CED-9. Increased fission would be somewhat surprising given that the activity of DRP-1 was decreased, not increased, in embryos lacking CED-9 (Jagasia et al., 2005). Similarly, human BCL-XL has recently been reported to increase human DRP1 GTPase activity (Li et al., 2008). We also note that decreased mitochondrial fusion is observed in mammalian cells lacking BAX and BAK (Karbowski et al., 2006).
Inhibiting mitochondrial fission with dominant negative DRP-1(K40A) results in interconnected mitochondria even in the absence of CED-9
These considerations prompted us to test whether altered CED-9 expression affects mitochondrial fusion. We first tested this indirectly by examining the effects of a dominant negative form of DRP-1 in the presence and absence of CED-9. This experiment was expected to produce interconnected mitochondria because of a lack of fission and unchecked fusion, unless mitochondrial fusion is impaired (Bleazard et al., 1999; Karbowski et al., 2006; Labrousse et al., 1999; Sesaki and Jensen, 1999; Smirnova et al., 2001). Upon expression of dominant negative DRP-1(K40A) in the presence of CED-9, we observed two classes of cells with distinct mitochondria as expected from previous studies (Labrousse et al., 1999). The first class of cells, approximately 76% (n=58), contained dilated segments of mitochondria that were spatially separated (Fig. 2C). These mitochondrial structures have previously been reported as clumps of mitochondrial inner membrane-bound segments that have divided, but are still interconnected by thin tubules of mitochondrial outer membrane (Labrousse et al., 1999). These thin tubules were visible in the second class of cells (24%), where spatially separated clumps of mitochondria were connected by thin tubules of mitochondrial inner membrane. Since all of the cells we observed had one of these two phenotypes, DRP-1(K40A) must significantly perturb mitochondrial homeostasis. Given the role of DRP-1 in the fission process, it seems most likely that this mutant protein blocks fission, allowing unchecked mitochondrial fusion to convert a tubular mitochondrial morphology into an interconnected mitochondrial morphology (Fig. 2J).
We then tested the effects of dominant negative DRP-1(K40A) in the absence of CED-9. Upon expression of DRP-1(K40A) in ced-9(n2812lf); ced-3(n717lf) animals, we found that all cells contained partially interconnected, dilated mitochondria (Fig. 2F,J). Approximately 31% of these cells (n=96) contained mitochondrial segments connected by thin tubules. This finding suggests that mitochondrial fusion is still able to proceed in the absence of CED-9. Although subtle fusion defects may not be detectable in this assay, our conclusion is consistent with the generally tubular mitochondria and normal mitochondrial ultrastructure found in muscle cells from ced-9(n2812lf); ced-3(n717lf) animals (Fig. 1). A further understanding of the role for CED-9 in mitochondrial fusion may benefit from direct analysis of fusion rates (Berman et al., 2008).
Increased CED-9 expression alters mitochondrial morphology in muscle cells
A complementary approach to testing Bcl-2 function in mitochondrial homeostasis is through expression studies: increased BCL-XL or CED-9 expression in HeLa cells results in highly interconnected mitochondria (Delivani et al., 2006). This phenotype has been attributed to excess fusion, in part because both BCL-XL and CED-9 are thought to interact with and activate the mitofusin Mfn-2. For these reasons, we asked whether increased CED-9 expression had a similar effect on C. elegans mitochondria.
Upon increased CED-9 expression in both ced-9(+) and ced-9(n2812lf); ced-3(n717lf) animals, mitochondria appeared dilated and often interconnected by thin tubular structures when visualized with either matrix GFP or GFP localized to mitochondrial outer membranes (Fig. 3B,F, and Fig. S1B in supplementary material). This effect was dose dependent, as a lower proportion of cells possessed interconnected mitochondria in transgenic lines created using lower concentrations of the ced-9 expression construct (Table S1 in supplementary material). Further analysis of these animals by electron microscopy revealed clusters of mitochondria that sometimes formed multiple concentric layers (Fig. 3H). We rule out the possibility that the effect of CED-9 on mitochondria is simply a consequence of targeting excess bulk protein to mitochondrial outer membranes, as mitochondria appeared normal when we expressed either TOM70::GFP alone (Fig. 3G), or a CED-9 construct lacking a core structural helix (Fig. 3D), both of which accumulate at mitochondrial outer membranes (Fig. S2 in supplementary material). Additionally, expression of a GFP construct containing the C-terminal transmembrane domain of CED-9 targets efficiently to mitochondrial outer membranes, yet does not alter mitochondrial morphology (Tan et al., 2007).
Fig. 3.

Increased CED-9 expression alters mitochondrial morphology. (A,B) Increased CED-9 expression resulted in dilated, interconnected mitochondria. This effect is similar to inactivation of mitochondrial fission by expression of dominant negative DRP-1(K40A) (Fig. 2C). However, we note that DRP-1(K40A)-induced mitochondria are more rounded, and only a fraction of the cells possess thin tubules interconnecting various mitochondria. (C) Removing the N-terminal 67 residues of CED-9 did not abolish the ability of CED-9 to alter mitochondria. However, removing either (D) a core structural helix or (E) the C-terminal transmembrane domain prevented CED-9 from altering mitochondria. Increased CED-9 expression also altered mitochondrial outer membrane morphology as judged by mitochondria labeled with TOM70::GFP, a mitochondrial outer membrane tethered GFP (Labrousse et al., 1999). (F) In otherwise wild-type animals, the mitochondria had a fairly stereotypical tubular morphology. (G) By contrast, transgenic animals with increased CED-9 expression displayed altered outer membrane morphologies. n, nucleus. Bar, 10 μm. (H) Mitochondrial morphology in the body wall muscle of adult animals with increased CED-9 expression was further examined by transmission electron microscopy. Two animals were found that possessed markedly distinct mitochondrial morphologies; shown are two cells from one of the animals. Bar, 500 nm.
This interconnected mitochondria phenotype presumably reflects excessive mitochondrial fusion due to either an increase in fusion or an inhibition of fission. Since Bcl-2 proteins are thought to activate mitofusins (Brooks et al., 2007; Delivani et al., 2006; Karbowski et al., 2006), and interconnected mitochondria can result from an activation of mitofusins (Santel and Fuller, 2001), we attempted to produce a similar phenotype by increasing the expression of the C. elegans mitofusin FZO-1. Interestingly, increased FZO-1 expression did not produce interconnected mitochondria in either ced-9(+) or ced-9(n2812lf); ced-3(n717lf) animals, instead producing a vesicular morphology distinct from that resulting from CED-9 or DRP-1(K40A) expression (Fig. S3A in supplementary material). This phenotype is consistent with increased MFN2 expression (Santel and Fuller, 2001) and may represent an intermediate stage of mitochondrial fusion or a dominant negative effect of increased FZO-1 expression. A dominant negative effect is supported by comparisons with mitochondrial morphology in cells from loss-of-function fzo-1(tm1133lf) animals that show similar phenotypes (Fig. S3B,C in supplementary material). If increased FZO-1 expression results in a dominant negative effect as our data suggests, then a dependence on CED-9 expression may be obscured.
The interconnected mitochondria phenotype produced upon increased CED-9 expression might also arise from an inhibition of fission by CED-9. If this were the case, we expect the morphology would be similar to the dominant negative DRP-1(K40A) phenotype discussed above. Although similarities exist, increased CED-9 expression produced less rounded, more elongated mitochondria than DRP-1(K40A; Fig. 2C and Fig. 3B). More strikingly, increased CED-9 expression resulted in nearly all of the analyzable cells having thin tubules of mitochondrial inner membrane connecting various mitochondrial segments, as compared to 24% with DRP-1(K40A). We interpret these data to suggest a mechanism whereby increased CED-9 expression does not simply inhibit mitochondrial fission, but may instead increase fusion of both mitochondrial inner and outer membranes (Fig. 3B,F).
To determine what structural features of CED-9 are required to alter mitochondria, we analyzed a number of mutant forms of CED-9. The ability of CED-9 to alter mitochondria does not require the N-terminal 67 residues, a feature not conserved among mammalian Bcl-2 homologs (Fig. 3C). However, deletion of the sixth α-helix in CED-9, which forms the hydrophobic core of the protein and is required to regulate apoptosis (Tan et al., 2007), does prevent CED-9 from altering mitochondria (Fig. 3D) even though this construct is able to accumulate at mitochondria (Fig. S2 in supplementary material). As this construct is predicted to result in a misfolded structure, these findings suggest that targeting the CED-9 transmembrane domain to mitochondria is not sufficient to alter mitochondrial morphology, and that this alteration requires a folded Bcl-2 domain.
Mitochondria remained tubular after expression of CED-9 lacking the C-terminal transmembrane domain (Fig. 3D), a construct previously shown not to accumulate at mitochondria (Tan et al., 2007). These findings, and the decreased activity of several CED-9 constructs with point mutations in the Bcl-2 domain of CED-9 predicted to affect its ability to interact with mitochondrial membranes (Table S1 in supplementary material), suggests that the Bcl-2 domain of CED-9 requires direct contact with mitochondrial membranes to alter mitochondria. This requirement may reflect a close membrane interaction necessary for CED-9 modulation of mitochondrial fusion proteins such as the mitofusin FZO-1 (Delivani et al., 2006). Intriguingly, this requirement may also reflect a direct remodeling of membranes by CED-9, which is capable of promoting vesicle fusion in vitro (F.J.T. and J. E. Zuckerman, unpublished).
Effects of CED-9 on mitochondria are partially suppressed by co-expressing DRP-1
To further characterize the interconnected mitochondria seen upon increased CED-9 expression, we tested whether or not the effects of CED-9 on mitochondria could be suppressed by concurrently increasing the expression of the wild-type form of the mitochondrial fission protein DRP-1. Such interconnected mitochondria can sometimes be converted to tubular or fragmented mitochondria by activating the mitochondrial fission machinery (James et al., 2003; Otsuga et al., 1998; Tieu et al., 2002; Yoon et al., 2003).
When DRP-1 was co-expressed using a copy of the myo-3 promoter on the same transgene array as myo-3::ced-9, the interconnected mitochondrial phenotype was partially rescued, suggesting that CED-9-altered mitochondria are still structurally intact and responsive to remodeling by components of the mitochondrial fission machinery. We observed fragmented, tubular and interconnected morphologies that probably represent different degrees of DRP-1-mediated suppression of the CED-9 phenotype, and not lack of CED-9 expression (Fig. S4 in supplementary material). Our data cannot exclude that these morphologies represent CED-9 rescuing DRP-1-fragmented mitochondria. We conclude that the mitochondrial morphology in a given cell is probably the consequence of both the relative expression level between the two genes and the particular environmental conditions. Additionally, CED-9 and DRP-1 may directly regulate each other’s activity, rather than primarily acting on mitochondria.
The ability of a protein to regulate the CED-9 effect on mitochondrial homeostasis has been previously shown when the pro-apoptotic BH3-only protein EGL-1 was co-expressed with CED-9 in HeLa cells, which resulted in fewer cells with interconnected mitochondria (Delivani et al., 2006). As EGL-1 binds to the BH3 binding pocket of CED-9 and causes a structural rearrangement (Yan et al., 2004), this result suggests that the effect of CED-9 on mitochondria may be regulated via interactions with the BH3 binding pocket of CED-9. To test whether co-expression of DRP-1 modulates CED-9 via interactions with the BH3 binding pocket, we first created a construct corresponding to the ced-9(n1950gf) allele. This allele encodes a mutation where glycine 169 in the BH3 binding pocket is replaced with glutamate (Fig. 4C) (Hengartner and Horvitz, 1994a), which inhibits EGL-1 from binding and triggering a conformational change in CED-9 (del Peso et al., 2000; Yan et al., 2004).
Fig. 4.

CED-9-altered mitochondria can be partially suppressed by co-expression of DRP-1. (A) Co-expression of DRP-1 and CED-9 from the same transgene resulted in three classes of mitochondrial morphology: tubular, fragmented and interconnected (percentages represent the mean value of three independently generated transgenic lines; raw data is presented in C and Table S1 in supplementary material). These three phenotypes suggest that increased DRP-1 expression is able to partially suppress the effects of CED-9 on mitochondria, or vice-versa. n, nucleus. Bar, 10 μm. (B) Structure of CED-9 (yellow surface) bound to a portion of the BH3 containing protein EGL-1 (blue ribbon) (1TY4.pdb). In the gain-of-function ced-9(n1950sd) allele, glycine 169, which resides in the CED-9 BH3 binding pocket, is mutated to glutamate (G169E). This mutation has been reported to decrease EGL-1 binding to CED-9 (del Peso et al., 2000). (C) Co-expression of DRP-1 and CED-9(G169E) from the same transgene also resulted in three classes of mitochondrial morphology. (D) Each point represents the proportion of cells with interconnected mitochondria from an independently generated transgenic line; the mean value is indicated by a horizontal bar, and error bars represent the standard error. No mean was calculated for the CED-9(G169E) construct because of the apparent bimodal distribution. This variation among lines expressing CED-9(G169E) was examined by western blot analysis (Fig. S4 in supplementary material). The difference in proportions between co-expression of DRP-1 with CED-9 and co-expression of DRP-1 with CED-9(G169E) is significant, z=2.59, P<0.01. (E) The GTPase activity of 2.5 μM human DRP1 was assayed using a continuous assay coupled to a regenerative system. (F) The amount of GTP hydrolyzed by 2.5 μM human DRP1 with 12.5 μM CED-9 after 40 minutes (296±23 μM) was significantly higher than the amount of GTP hydrolyzed by 2.5 μM human DRP1 alone after 40 minutes (221±17 μM), t(4)=4.56, P<0.025.
When CED-9(G169E) was expressed alone, we still observed interconnected mitochondria indistinguishable from wild-type CED-9 except for its penetrance (Fig. S1C in supplementary material). In contrast to increased expression of wild-type CED-9, where most cells had interconnected mitochondria, CED-9(G169E) expression resulted in cell-to-cell and line-to-line variability where only a portion of the cells in any given animal contained interconnected mitochondria (Fig. 4D). Analysis of these lines by western blot analysis using an α-CED-9 antibody indicated that a variable amount of protein was present in different CED-9(G169E) transgenic lines (Fig. S4 in supplementary material), where even lines with low CED-9 protein abundance had a portion of interconnected mitochondria. We interpret these results to suggest that CED-9(G169E) retains most of the ability to alter mitochondrial homeostasis.
To further explore CED-9(G169E) and mitochondrial homeostasis, we tested whether the effect of CED-9(G169E) on mitochondria could still be suppressed by increasing DRP-1 expression. In transgenic animals expressing both CED-9(G169E) and DRP-1, the proportion of cells with interconnected mitochondria was higher (0.73) than in animals co-expressing wild-type CED-9 and DRP-1 (0.52; Fig. 4B,D). The difference in proportions, which is significant (z=2.59, P<0.01), appears to result from a difference in CED-9 activity rather than differences in expression or protein levels (Fig. S4 in supplementary material). This difference in the proportion of cells with interconnected mitochondria is consistent with the G169E mutation affecting an interaction between CED-9 and DRP-1, perhaps one that occurs via the BH3 binding pocket.
To test whether CED-9 is capable of interacting with DRP-1, we asked whether or not CED-9 affects the GTPase activity of human DRP1 (which is 59% identical to C. elegans DRP-1 at the amino acid level) using bacterially expressed, recombinant proteins. Using a continuous GTPase assay coupled to a regenerative system (Ingerman et al., 2005), 2.5 μM human DRP1 alone hydrolyzed 221±17 μM (mean ± s.d.) GTP after 40 minutes (Fig. 4E,F). In the presence of 12.5 μM CED-9, the same amount of human DRP1 hydrolyzed 296±23 μM GTP. This activation of human DRP1 by CED-9 is significant [t(4)=4.56, P<0.025], and appears specific as BSA did not alter the GTPase activity of DRP1. These findings suggest that CED-9 may regulate mitochondrial fission by directly modulating DRP-1. Similar tests with recombinant S. cerevisiae Dnm1 did not show a difference in the presence or absence of CED-9 (data not shown).
Discussion
In summary, animals lacking CED-9 do not have gross mitochondrial defects (Fig. 1). This finding in adult animals, along with studies in embryos (Jagasia et al., 2005), suggests that either CED-9 does not regulate mitochondrial homeostasis, or that CED-9 plays a more complex role regulating fission and fusion under special circumstances such as stress or apoptosis. Support of a role for CED-9 in mitochondrial homeostasis comes from the finding that there is increased fragmentation upon increased DRP-1 expression in ced-9(n2812lf); ced-3(n717lf) animals (Fig. 2I). This phenotype may arise from decreased mitochondrial fusion in the absence of CED-9. This hypothesis is supported by the finding that CED-9 interacts with human MFN2, a homolog of FZO-1 (Delivani et al., 2006). Alternatively, this phenotype may arise from increased DRP-1 activity in the absence of CED-9, implying that CED-9 inhibits DRP-1, although our data (and that of others) do not support such inhibition (Fig. 4D,E).
If CED-9 inhibits DRP-1, then we would expect that upon increased CED-9 expression interconnected mitochondria would appear similar to interconnected mitochondria upon dominant negative DRP-1(K40A) expression, which is not the case (Figs 2 and 3). We further tested whether CED-9 inhibits DRP-1 by co-expressing DRP-1 with either wild-type CED-9 or CED-9(G169E). If CED-9 did inhibit DRP-1 via the BH3 binding pocket of CED-9, then the G169E mutation would disrupt DRP-1 binding, causing less interconnected mitochondria; instead, we see more interconnected mitochondria (Fig. 4D). These results suggest that interconnected mitochondria upon increased CED-9 expression do not result from decreased fission, but rather increased fusion. Furthermore, the ability of DRP-1 to better suppress wild-type CED-9 expression than CED-9(G169E) (Fig. 4D) suggests that either CED-9 activates DRP-1 activity, or DRP-1 inhibits a CED-9 fusogenic activity. The CED-9 activation of the GTPase activity of human DRP1 (Fig. 4E) is consistent with other studies suggesting that Bcl-2 proteins potentiate Drp-1 activity (Jagasia et al., 2005; Li et al., 2008). However, these studies do not exclude the possibility that DRP-1 inhibits CED-9-induced mitochondrial fusion.
These observations extend previous reports on the role of CED-9 in mitochondrial homeostasis. Similar to previous experiments in HeLa cells (Delivani et al., 2006), we found that increased CED-9 expression in C. elegans results in interconnected mitochondria (Fig. 3). In earlier HeLa cell experiments, co-expression of EGL-1 suppressed this CED-9 fusogenic activity (Delivani et al., 2006). In our C. elegans experiments, co-expression of DRP-1 also suppressed CED-9 fusogenic activity (Fig. 4), but suppressed CED-9 (G169E) to a lesser extent, suggesting that the BH3 binding pocket may be a general point of regulation for CED-9 fusogenic activity. Furthermore, as increased CED-9 expression in ced-4(n1162lf) loss-of-function animals also resulted in interconnected mitochondria, this finding suggests that CED-4, which binds to a different interface on CED-9, may not play a major role in the mitochondrial homeostasis of adult C. elegans muscle (Table S1 in supplementary material).
In a previous study of C. elegans embryos, mitochondria appeared fragmented in animals harboring a temperature-sensitive ced-9(n1653ts) mutation (Delivani et al., 2006). As this previous study examined embryos at a restrictive temperature in the context of functional CED-3, at least some of the cells are probably undergoing apoptosis, which might result in mitochondrial fragmentation (Jagasia et al., 2005), and may explain the observed differences.
Previously, DRP-1 was found to be dependent upon CED-9 for mitochondrial fragmentation (Jagasia et al., 2005), which at first glance appears to contradict the present study. Our data show that increased DRP-1 expression fragment mitochondria in animals that lack CED-9 (Fig. 2E); in addition, a higher proportion of cells contain fragmented mitochondria compared with animals that do express CED-9 (Fig. 2G). However, the apparent contradiction may be resolved when considering that fragmented phenotypes can arise from inhibition of mitochondrial fusion as well as activation of fission. Thus, we interpret the differences between the studies to reflect decreased mitochondrial fusion in animals lacking CED-9, a decrease that is only unmasked when mitochondrial fission is sufficiently activated.
A model for Bcl-2 activity in mitochondrial homeostasis
Our data are consistent with current models in which Bcl-2 proteins potentiate both mitochondrial fusion and fission. For mitochondrial fusion, these models propose that Bcl-2 proteins activate mitofusins, such as FZO-1, resulting in interconnected mitochondria (Brooks et al., 2007; Delivani et al., 2006; Karbowski et al., 2006). Our data further these models by indicating that in C. elegans, mitochondrial fusion does not strictly require CED-9 (Figs 1 and 2), but CED-9 may still potentiate FZO-1 activity under certain conditions. For mitochondrial fission, current models propose that Bcl-2 proteins activate dynamin-related GTPases, such as DRP-1, resulting in fragmented mitochondria (Frank et al., 2001; Li et al., 2008). Our data further these models by indicating that in C. elegans, mitochondrial fission does not strictly require CED-9 (Figs 1, 2 and 4), but CED-9 may still potentiate DRP-1 activity under certain conditions. We note, however, that our data is also consistent with a model whereby DRP-1 inhibits a CED-9 activity that promotes mitochondrial fusion, which would provide a second mechanism causing mitochondrial fragmentation (Fig. 5). The cellular signals that regulate whether Bcl-2 proteins, such as CED-9, activate mitochondrial fission or fusion processes remain to be identified.
Fig. 5.
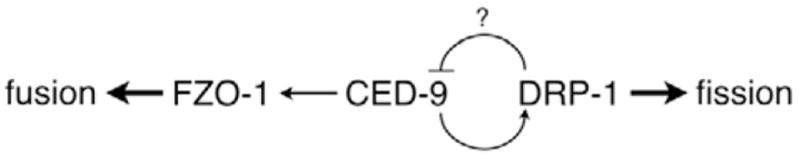
A model for CED-9 function in mitochondrial homeostasis. In C. elegans, CED-9 does not appear to be essential for mitochondrial homeostasis. Mitochondrial fusion, which probably involves the mitofusin FZO-1, can occur in the absence of CED-9. Similarly, mitochondrial fission, which involves DRP-1 can also occur in the absence of CED-9. This and other studies (Delivani et al., 2006; Jagasia et al., 2005), however, suggest that CED-9 may play a role in potentiating the activities of both DRP-1 and FZO-1. Additionally, CED-9 may itself be regulated by the mitochondrial fission protein DRP-1. If true, regulation of CED-9 by DRP-1 would provide a mechanism for cross-regulation between the mitochondrial fission and fusion machineries. Such a pathway would allow DRP-1 to promote mitochondrial fission both directly, and through downregulation of FZO-1 by way of inhibiting CED-9.
The multifunctional nature of CED-9 suggests that Bcl-2 proteins may provide a point of regulation through which the amount of fission and fusion is tuned in response to cellular requirements. Accordingly, this study suggests that Bcl-2 proteins may provide a link between components of the mitochondrial fission and fusion machinery, and may provide insight into the effects of altered Bcl-2 expression such as are found in many human cancers.
Materials and Methods
Genetic methods and strains
C. elegans were cultured and maintained as described previously on modified Youngren’s, only Bacto-peptone agar at 20°C (Tan et al., 2007). The complete genotype for ced-9 gain of function is ced-9(n1950sd) (Hengartner and Horvitz, 1994a); for loss-of-function ced-9(lf) is ced-9(n2812lf), ced-3(n717lf) (Hengartner and Horvitz, 1994b). The fzo-1(tm1133) deletion strain, a generous gift from S. Mitani (National Bioresource Project for the nematode, Japan), exhibited delayed growth, decreased brood sizes and uncoordinated locomotion.
Germline transformations were performed as described previously (Tan et al., 2007). We generally injected constructs (ced-9 being approximately 7.1 kb, drp-1 being approximately 8.4 kb) at 16.7 μg/ml, along with the pSAK4 plasmid (7.0 kb) containing the mitoGFP marker at 15.6 μg/ml, and the pRF4 plasmid (7.3 kb) containing the rol-6(su1006) gene at 109.4 μg/ml as the co-injection marker. For injection mixes with both ced-9 and drp-1 constructs, the amount of pRF4 was adjusted to maintain a constant DNA concentration. Transgenic lines expressing a variety of constructs had growth rates, brood sizes and locomotion similar to control transgenic lines expressing only mitoGFP and rol-6 markers.
Plasmid constructs
During previous experiments in which we studied a CED-9 construct with GFP at the N terminus (Tan et al., 2007), we occasionally noticed subtle alterations in mitochondrial morphology. Therefore, we created transgenic lines expressing CED-9 without GFP at the N terminus. These lines expressed CED-9 at higher levels than GFP::CED-9 when compared by western blot analysis (data not shown).
The myo-3::ced-9 plasmid was constructed by removing gfp from the myo-3::gfp::ced-9 plasmid previously described (Tan et al., 2007). All other ced-9 variants were constructed in a similar fashion. The myo-3::drp-1(K40A) plasmid was a generous gift from A. van der Bliek (UCLA, Los Angeles, CA). Standard site-directed mutagenesis was performed to revert this plasmid back to a wild-type copy of drp-1. The DNA sequences of all coding regions amplified by PCR were determined to ensure that no point mutations were introduced by PCR.
Recombinant CED-9 was bacterially expressed using a pHis-Gβ1 fusion construct (pFT30.31), which was made by PCR amplification of ced-9 from the pB30 cDNA clone, kindly provided by Bob Horvitz (MIT, Boston, MA). A 753 nt PCR product lacking the transmembrane domain (residues 252–280) was cloned into the pHis-Gβ1 vector (Amezcua et al., 2002). This vector provides an N-terminal 6xHis tag, the β1 domain from streptococcal protein G, and a recognition site for cleavage by TEV protease. Cleavage by 6xHis-TEV protease results in CED-9ΔTM with the amino acid sequence GEF at the N terminus as a cloning artifact.
Light microscopy and image analysis
To image body wall muscle cells, young adult animals were anesthetized in 0.5% 1-phenoxy-2-propanol for ~25 minutes, and then mounted onto a microscope slide containing a 4% agarose pad. Images were taken on an Olympus BX51 microscope using an Olympus UPlanFl 100× 1.30 NA oil immersion objective and equipped with an Optronics MagnaFire camera. Images were rotated at most once, cropped, and rescaled without resampling. Imaging of mitochondria in body wall muscle by confocal microscopy revealed that these mitochondria generally lie in a 3–4 μm thick layer; analysis of the various phenotypes revealed little difference between confocal and widefield fluorescence microscopy.
The number of GFP-positive body wall muscle cells, as well as the intensity of GFP expression varied from animal to animal; variation in expression did not appear to correlate with altered morphology (data not shown). In addition, mitochondria from animals with increased CED-9 expression may be less efficient in protein import, since mitoGFP often appeared diffuse throughout the cell. Therefore, only GFP-positive body wall muscle cells with discernable mitochondria were examined; cells where GFP was found to be too diffuse in fluorescence to the point of not being able to discern mitochondria were not scored. In general, anywhere from one to five GFP-positive cells per animals were suitable for imaging.
Individual body wall muscle cells were examined by eye and categorized on the overall mitochondrial morphology: ‘fragmented’ if more than half of the mitochondria by area had a length:width ratio <2; ‘interconnected’ if any of the mitochondria were dilated or had thin tubules interconnecting them (often crossing multiple parallel rows); or ‘tubular’ if otherwise.
Analysis of mitochondria lengths was performed using ImageJ v1.39p. Individual mitochondria were identified using an adaptive thresholding algorithm (median, radius = 4.0 pixels). The major axis fit for an individual particle by the built-in Analyze Particles routine was interpreted as the length of an individual mitochondrion. Manual inspection of processed images revealed that this mitochondria identification procedure had an accuracy of approximately 85%. In general, this procedure had a tendency to break longer mitochondria into shorter fragments. The results from a hand measurement of a small sample resulted in comparable results. A double-blind test where participants visually scored mitochondria lengths also concluded that there was no significant difference between ced-9(+) and ced-9(n2812lf); ced-3(n717lf) animals.
Electron microscopy
In preparation for fixation, young adult animals were anesthetized in 0.5% 1-phenoxy-2-propanol for ~25 minutes and then bisected. Animals were fixed overnight at 4°C with 3.0% paraformaldehyde, 1.5% glutaraldehyde in 0.1 M sodium cacodylate pH 7.4, 2.5% sucrose, supplemented with 1.0 mM MgCl2 and 0.5 mM CaCl2. These samples were post-fixed with Palade’s OsO4, stained overnight with Kellenberger uranyl acetate, dehydrated in graded ethanols and propylene oxide, and embedded in Spurr’s resin. Sections were examined on either a Philips EM 410 or FEI Tecnai 12 Twin, and images were collected using a Soft Imaging System Megaview III digital camera. We note that because of the sickness of fzo-1(tm1133) animals, the staging as young adults may not be precise.
Statistical analysis
Statistical data are presented as the mean proportion ± standard error. As the number of cells imaged and analyzed typically varied for each independently generated transgenic line, a weighted mean was calculated for each construct to allow comparison between constructs. In general, the proportion for each line of a given construct was not significantly different when compared with other lines of the same construct; a notable exception is with +CED-9(G169E), as discussed in Results. When comparing the mean proportion between constructs, z scores were calculated for the difference between proportions. Raw data for each line is given in Table S1 in the supplementary material.
Protein expression and purification
pHis-Gβ1-CED-9ΔTM fusion protein was expressed and purified from Escherichia coli Rosetta cells (Novagen) in LB medium containing 50 μg/ml carbenicillin and 34μg/ml chloramphenicol. Cells were grown at 37°C to an OD600 of ~0.2 while shaking at 300 r.p.m., chilled to 18°C until OD600 0.6–0.8, and induced by addition of 0.5 mM IPTG for 12–16 hours. The resulting cells were collected by centrifugation and resuspended in buffer A (20 mM Tris, 0.5 mM NaCl, 1 mM DTT at pH 8.0) containing protease inhibitors (Complete, EDTA-free Protease Inhibitor Cocktail Tablets; Roche Applied Science), lysed by three passes through an EmulsiFlex C3 (Avestin, Ottawa, Canada), supplemented with 2 mM MgCl2 and treated with 1 μg/ml DNase I (Sigma), and clarified at 30,000 r.c.f. for 45 minutes at 4°C. The fusion protein in the resulting supernatant was bound to a Ni2+ chelating column (Ni Sepharose 6 Fast Flow; Amersham) and eluted with imidazole. Concurrent with dialysis into buffer A, the desired CED-9 construct was obtained by cleavage with recombinant 6xHis-TEV protease (1:100 molar ratio) for 4 hours at 23°C. The reaction mixture was loaded onto the Ni2+ chelating column, and the flow-through containing CED-9ΔTM was collected and concentrated. The concentrate was further purified over a HiLoad 16/60 Superdex 75 gel filtration column (Amersham), quantified using UV absorbance (ε280=30,680 M/cm in 6 M GuHCl), and judged to be >95% pure by Coomassie-Blue-stained gel electrophoresis. The final protein stock was stored at a concentration of 40 μM in 20 mM Tris (pH 8.0), 150 mM NaCl, 1 mM DTT, and 1 mM EDTA at 4°C until used.
Human DRP1 with a calmodulin-binding peptide (CBP) at the N terminus was expressed and purified as previously described (Zhu et al., 2004). The final protein stock was stored at 23 μM in 20 mM Tris (pH 7.4), 150 mM NaCl, 10 mM EDTA, flash frozen in liquid nitrogen, and stored at −80°C until used.
GTPase assay
A continuous GTPase assay was used whereby the rate of GTP hydrolysis was determined through coupling to a GTP regeneration system (Ingerman et al., 2005). The activity of 2.5 μM human DRP1 was assayed in 200 μl of GTPase reaction buffer (final 200 mM NaCl, 600 μM NADH, 1 mM GTP). Modulation of GTPase activity was assayed on a Molecular Devices SpectraMAX 250 at 23.5°C by the addition of BSA (Sigma P5369), CED-9, or buffer control.
Supplementary Material
Acknowledgments
We thank Barbara Conradt and Stephane Rolland (Dartmouth Medical School, Hanover, NH) for sharing their results on CED-9 in mitochondrial fusion before publication. The authors also thank Joohong Ahnn, Rosa Alcazar, Michael Edidin, Jamie Fleenor, J. Marie Hardwick, Edward Hedgecock, Chaya Krishna, J. Michael McCaffery, Ned Perkins, Rui Proenca, SiQun Xu, Judy Yanowitz and Jonathan Zuckerman for resources and suggestions. Alexander van der Bliek generously provided the myo-3::drp-1(K40A) plasmid, Sergei Morozov helped with statistical analysis, H. Robert Horvitz provided the ced-9 cDNA, Craig Blackstone provided the human DRP1 plasmid, and Santa Cruz Biotech, Inc. provided sample α-CED-9 antibodies. Finally, we thank Shoehei Mitani (National Bioresource Project for the nematode, Japan) for kindly providing fzo-1(tm1133), and the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources, for some nematode strains used in this work. This work was supported by NIH grants R01GM067180 (R.B.H.) and R01GM37706 (A.Z.F.), American Cancer Society award IRG-58-005-41 (R.B.H.), a training grant from NIH (2T32GM007231) and startup funds provided by JHU (R.B.H.). F.J.T. was supported in part by a Department of Defense NDSEG fellowship, and a Millipore Foundation Dimitri V. d’Arbeloff award.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/121/20/3373/DC1
References
- Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amezcua CA, Harper SM, Rutter J, Gardner KH. Structure and interactions of PAS kinase N-terminal PAS domain: model for intramolecular kinase regulation. Structure. 2002;10:1349–1361. doi: 10.1016/s0969-2126(02)00857-2. [DOI] [PubMed] [Google Scholar]
- Ay I, Sugimori H, Finklestein SP. Intravenous basic fibroblast growth factor (bFGF) decreases DNA fragmentation and prevents downregulation of Bcl-2 expression in the ischemic brain following middle cerebral artery occlusion in rats. Brain Res Mol Brain Res. 2001;87:71–80. doi: 10.1016/s0169-328x(00)00285-0. [DOI] [PubMed] [Google Scholar]
- Berman SB, Pineda FJ, Hardwick JM. Mitochondrial fission and fusion dynamics: the long and short of it. Cell Death Differ. 2008;15:1147–1152. doi: 10.1038/cdd.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie ZJ, Dong Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in bax/bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Peso L, Gonzalez VM, Inohara N, Ellis RE, Nunez G. Disruption of the CED-9. CED-4 complex by EGL-1 is a critical step for programmed cell death in Caenorhabditis elegans. J Biol Chem. 2000;275:27205–27211. doi: 10.1074/jbc.M000858200. [DOI] [PubMed] [Google Scholar]
- Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–773. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–1094. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- Fannjiang Y, Cheng WC, Lee SJ, Qi B, Pevsner J, McCaffery JM, Hill RB, Basanez G, Hardwick JM. Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev. 2004;18:2785–2797. doi: 10.1101/gad.1247904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley HW, Gu L, Yeager AM, Zhou M. Expression and regulation of Bcl-2, Bcl-xl, and Bax correlate with p53 status and sensitivity to apoptosis in childhood acute lymphoblastic leukemia. Blood. 1997;89:2986–2993. [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR. Activation of C. elegans cell death protein CED-9 by an amino-acid substitution in a domain conserved in Bcl-2. Nature. 1994a;369:318–320. doi: 10.1038/369318a0. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994b;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, Song K, Kitada S, Reed JC. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am J Pathol. 1996;148:1567–1576. [PMC free article] [PubMed] [Google Scholar]
- Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- Leber B, Lin J, Andrews DW. Embedded together: The life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12:897–911. doi: 10.1007/s10495-007-0746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci USA. 2008;105:2169–2174. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Toquet C, Oliver L, Cartron PF, Perrin P, Meflah K, Cuillere P, Vallette FM. Expression of bcl-2, bax and bcl-xl in human gliomas: a re-appraisal. J Neurooncol. 2001;52:129–139. doi: 10.1023/a:1010689121904. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Youle RJ. Which came first, the cytochrome c release or the mitochondrial fission? Cell Death Differ. 2006;13:1291–1295. doi: 10.1038/sj.cdd.4401985. [DOI] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–R560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–267. doi: 10.1146/annurev.neuro.20.1.245. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Otsuga D, Keegan BR, Brisch E, Thatcher JW, Hermann GJ, Bleazard W, Shaw JM. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J Cell Biol. 1998;143:333–349. doi: 10.1083/jcb.143.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111:3322–3330. doi: 10.1182/blood-2007-09-078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC, Meister L, Tanaka S, Cuddy M, Yum S, Geyer C, Pleasure D. Differential expression of bcl2 protooncogene in neuroblastoma and other human tumor cell lines of neural origin. Cancer Res. 1991;51:6529–6538. [PubMed] [Google Scholar]
- Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114:867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JA, Mindell JA, Finkelstein A, Shen WH, Collier RJ. Mutational analysis of the helical hairpin region of diphtheria toxin transmembrane domain. J Biol Chem. 1994;269:22524–22532. [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugioka R, Shimizu S, Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem. 2004;279:52726–52734. doi: 10.1074/jbc.M408910200. [DOI] [PubMed] [Google Scholar]
- Tan FJ, Fire AZ, Hill RB. Regulation of apoptosis by C. elegans CED-9 in the absence of the C-terminal transmembrane domain. Cell Death Differ. 2007;14:1925–1935. doi: 10.1038/sj.cdd.4402215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Thuduppathy GR, Craig JW, Kholodenko V, Schon A, Hill RB. Evidence that membrane insertion of the cytosolic domain of Bcl-x(L) is governed by an electrostatic mechanism. J Mol Biol. 2006a;359:1045–1058. doi: 10.1016/j.jmb.2006.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuduppathy GR, Terrones O, Craig JW, Basanez G, Hill RB. The N-terminal domain of Bcl-xL reversibly binds membranes in a pH-dependent manner. Biochemistry. 2006b;45:14533–14542. doi: 10.1021/bi0616652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu Q, Okreglak V, Naylor K, Nunnari J. The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J Cell Biol. 2002;158:445–452. doi: 10.1083/jcb.200205031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Gu L, Kokel D, Chai J, Li W, Han A, Chen L, Xue D, Shi Y. Structural, biochemical, and functional analyses of CED-9 recognition by the proapoptotic proteins EGL-1 and CED-4. Mol Cell. 2004;15:999–1006. doi: 10.1016/j.molcel.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem. 2004;279:35967–35974. doi: 10.1074/jbc.M404105200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.