

Abstract
This review integrates recent understanding of a novel role for NDPK-A in two related directions: Firstly, its role in an airway epithelial cell when bound to the luminal (apical) membrane and secondly in the cytosol of many different cells (epithelial and non-epithelial) where an isoform-specific interaction occurs with a regulatory partner, AMPKα1. Thus NDPK-A is present in both a membrane and cytosolic environment but in the apical membrane, its roles are not understood in detail; preliminary data suggest that it co-localises with the cystic fibrosis protein (CFTR). In cytosol, we find that NDPK-A is coupled to the catalytic alpha1 isoform of the AMP-activated protein kinase (AMPKα subunit), which is part of a heterotrimeric protein complex that responds to cellular energy status by switching off ATP-consuming pathways and switching on ATP-generating pathways when ATP is limiting. We find that ATP is located within this complex and ‘fed’ from NDPK to AMPK without ever ‘seeing’ bulk solution. Importantly, the reverse can also happen such that AMPK activity can be made to decline when NDPK-A ‘steals’ ATP from AMPK. Thus we propose a novel paradigm in NDPK-A function by suggesting that AMP-kinase can be regulated by NDPK-A, independently of AMP.
Keywords: AMP kinase, Trinucleotide, Energy metabolism, ATP, GTP, Chloride, CFTR, AMP
Introduction
Epithelial cells transduce energy from ATP to create bidirectional ion gradients across the plasma membrane to energetically drive numerous functions, the details of which are reviewed elsewhere (Mehta, 2005; Mehta and Bush, 2005). This process is often deemed ‘vectorial’ because transport is directional and depends on three related functions. First, that for any transported ion (chloride is a good example), ion influx and exit occur across different faces of the cell (apical, outside facing and basolateral, blood-capillary facing); second, that the transported ion uses different proteins (in the two membranes) to generate differential accumulation (and exit) of ions and third, that proteins mediating ion influx and exit can somehow co-ordinate their functions with cellular energy supply, but without signal confusion. Thus, the difference between say a muscle cell or a fat cell and an epithelial cell is that the ‘non-blood facing’ or apical epithelial membrane contains different transporters and channels compared to the capillary facing basolateral membrane—hence, vectorial transport. This review is focused on potential ‘signals’ that integrate energy with ions and asks how an epithelial cell membrane senses energy availability (Treharne et al., 2001). We reveal the crucial role of NDPK-A in this process.
The general idea that the apical and basolateral signals are different is well illustrated by comparing the movement of chloride across epithelial membranes. Two excellent paradigms are revealed by comparing the differential regulatory mechanisms driving apical, G-protein regulated, CFTR-dependent, chloride transport as described by Schweibert et al. (Schwiebert et al., 1994) relative to that process in the basolateral membrane where Dowd and Forbush find that intracellular chloride concentration provides a feedback signal to control accumulative chloride co-transport via the activity of a protein kinase (Dowd and Forbush, 2003). The details are described elsewhere (Mehta, 2005).
Vectorial transport
The observed direction of chloride flux cannot be deduced intuitively based on the known intra- versus extracellular chloride concentrations found in vivo (40–50 and 90–110 mM respectively). The net direction of chloride flux (inside-apical to outside-apical say) is principally driven by the magnitude of the (negative-inside) transmembrane potential difference set up by the outward movement of potassium ions from cell interior to extracellular space. Calculations show the counter-intuitive result that despite having up to twice the chloride concentration outside than inside an epithelial cell, chloride may nevertheless move out provided a protein gate is open. The cystic fibrosis transmembrane conductance regulator (CFTR) provides one of a number of such gates located in the apical membrane of many epithelial cells (Gabriel et al., 1994). Note the contrast with the transmembrane sodium gradient of 10–20 mM (inside) versus 130–140 mM (in the blood) where concentration difference and potential difference are vectorially-additive (i.e. both are driven inward into the cell). Thus, sodium always moves into a cell when a gate is open (typically through apical sodium channels). These facts have been understood for many years but the regulation of underlying proteins remain problematic.
What did we know between 1990 and 2000?
The history of the work prior to 1990 on the role of chloride concentration and membrane protein function is described elsewhere (Treharne et al., 2001).Briefly, Anil Mehta (AM) was investigating the notion that chloride concentration could act as a signal to the apical membrane using enriched apical human nasal epithelial membranes biopsied from normal human airways in vitro (Treharne et al., 1994). Between 1989 (when CFTR was cloned) and 1994, Kate Treharne (KT) and AM found that chloride principally regulated the steady state intensity of phosphorylation of many (unknown) apical membrane proteins via membrane-associated protein kinase(s). These were unusual kinases that could not be affected by broad spectrum inhibitors such as staurosporine suggesting that they also did not belong to the classical PKA/C family (Fig. 1). That some novel kinase(s) was present was also likely because when this membrane-delimited kinase(s) preparation utilised GTP as a phosphate donor (radio-labelled gamma phosphate), a different pattern of membrane phosphoproteins was generated compared to ATP (compare upper and lower panels in Fig. 1). The evidence for signalling was consistent with the finding that when GTP was replaced with ATP not only was a different chloride-dependent profile of phosphorylated membrane proteins generated, but the chloride-dependence of different membrane-associated phosphoproteins changed dependent on the anion chosen to replace the chloride (gluconate ⋘ nitrate ⋙ sulphate). That these two different nucleotide species differentially altered the net phosphorylation state of different apical membrane proteins suggested two possible explanations. Firstly, that different ion-regulated membrane-bound kinases were present, or alternatively, there existed differential regulation (by ions) of a kinase(s) capable of using either nucleotide. The idea of differential regulation was not confined to kinases because of our related finding as described in Treharne et al. (1994), that phosphatases could also play discrete roles. Thus phosphatase inhibition with broadly acting phosphothiorate nucleotide analogues also (further differentially) changed the profile of apical membrane phosphoproteins. Once again, conventional phosphatase inhibitors such as okadaic acid were ineffective, adding to the novelty. Additionally, the rank order of the anion-dependent intensity of labelling disappeared when the phosphothiorate-containing hydrolysis resistant ATP was present suggesting a complex role for dephosphorylation. However, chloride-dependent regulation was preserved when hydrolysis resistant GTP was added, but this changed the anion rank order further (Treharne et al., 1994). Crucially, the whole apical system was also differentially sensitive to cation species, but this cation acted principally via the ATP pathway, such that sodium concentrations as low as 10 mM were inhibitory (Marshall et al., 1999).
Fig. 1.

Autoradiographs of SDS-PAGE separated apically enriched human airway membranes which had been incubated with thiosulphate (T), sulphate (S), nitrate (N), chloride (C), bromide (B), acetate (A), iodide (I) or gluconate (G) and γ [32P]-ATP/GTP (see Treharne et al., 1994 for details). Each salt contains 50 mM Mg2+. Figure reproduced with permission from AJP.
The key discovery
The pH of the solutions was germane with significantly less phosphotransfer to membrane proteins below pH 7 (unpublished data). This observation was wholly unexpected and suggested that either the process of phosphotransfer was pH-dependent as a physiological phenomenon per se (cellular pH lies just below 7, close to the pK for histidine) or that the membrane protein phosphate species we were studying was not present on phosphoserine/threonine or phosphotyrosine because both are acid-stable. We investigated the latter idea first. Between 1996–1998, Richmond Muimo (RM) made the seminal observation that the ion-sensitive phosphorylation in membrane of airway epithelium occurred on (acid-labile) histidine residues (Muimo et al., 1998, 2000). RM observed loss of phosphate label from a number of chloridesensitive membrane phosphoproteins (including a 37 kDa and a 19/21 kDa doublet bands) on SDS gels de-stained with acetic acid containing solutions. In addition, phosphoamino acid analysis procedures that included acid hydrolysis (6 N HCl for 2 h at 105°C) of phosphoproteins (labeled with [γ-32P]GTP or [γ-32P]ATP for 5 min) removed >95% of the radioactivity from the phosphoproteins. Further analysis showed that the airway membrane chloride sensitive proteins, phosphorylated with either [γ-32P]GTP or [γ-32P]ATP, were not only acid- and hydroxylamine acetatelabile, but also base-stable and inhibitable following preincubation with diethyl pyrocarbonate. Pre-treatment with diethyl pyrocarbonate ethoxycarbonylates the histidine residues on proteins and inhibits histidine phosphorylation (Miles, 1977; Wieland et al., 1993). RM then travelled to the laboratory of the father of the field, Harry Matthews (now retired) at the University of California, Davis to learn the complex HPLC-based phosphoamino acid analysis procedures that would distinguish phosphohistidine from other alkali-stable and acid labile phosphoamino acids (Muimo et al., 2000). The procedures included alkali hydrolysis (6 N KOH for 5 h at 105°C) of phosphoproteins (labeled with [γ-32P]GTP or [γ-32P]ATP for 5 min), which retained the alkali-stable 32P-labelled phosphoamino acids from the above chloride-dependent phosphoproteins. The alkaline hydrolysate was then spiked with non-radiolabelled standards and analysed by anion exchange chromatography coupled with on-line fluorescence detection. Fractions were collected and subjected to Cerencov counting to determine the elution profile of the radioactivity. Thus, ultimately, the final proof was only obtained when RM found, that the radioactivity co-eluted with the appropriate phosphohistidine standards. Taken together, the above results suggested that the chloride-dependent, acid- and hydroxylamine acetate-labile but base-stable phosphoramidate linkage was likely to occur on histidine (and not arginine or lysine, the other amino acids that can be phosphoramidated).
In order to characterise the putative protein kinases involved in generating phosphohistidine inmembrane of ovine airway epithelium, we decided to analyse and identify autophosphorylating proteins. Membrane proteins were separated by SDS-PAGE, renatured and the protein kinases allowed to autophosphorylate “in-gel.“ A number of autophosphorylating bands were observed including a 19 and 21 kDa doublet, which was identified as nucleoside diphosphate kinase by protein sequencing (see Fig. 2).
Fig. 2.
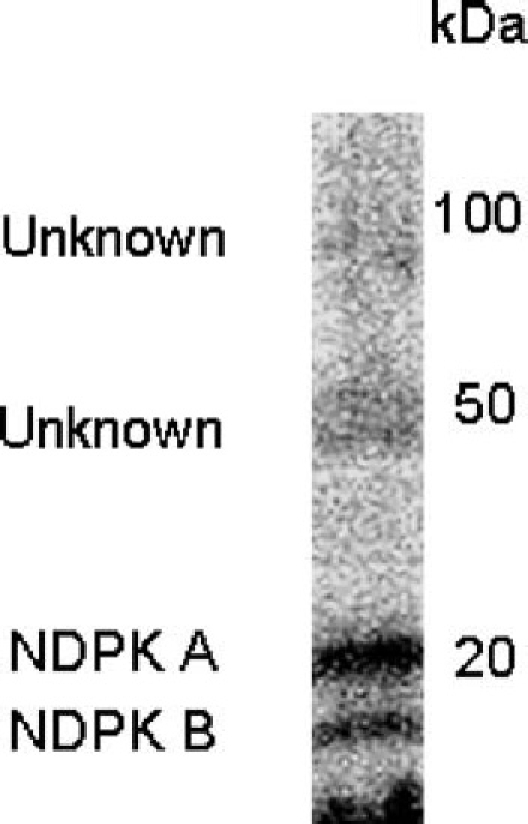
“In-gel“ autophosphorylation of proteins in membrane of ovine airway epithelium. Apical membrane proteins from sheep tracheal epithelia were prepared by sucrose gradient technique and separated by SDS-PAGE (12.5%). Following re-naturation “in-gel“ (Muimo et al., 1998), proteins were autophosphorylated by incubating the gel with 9.25 MBq γ[32P]-ATP (16 nm, specific activity 6000 Ci/mmol) in 50 ml of 25 mM MOPS pH 7.9, 10 mM MnCl2 and 0.05% Triton X-100. Labelled proteins were then detected by electronic autoradiography. Result is representative of at least three separate experiments. This figure has been adapted and reproduced with permission from Am J Resp Cell Mol Biol.
Further work using isoform-specific antibodies suggested that NDPK-A and NDPK-B were present bound to the airway epithelial membranes. RM hypothesized that NDPK was being regulated by another protein because the phosphohistidine content of purified membrane-bound NDPK from epithelial cells was chloride-insensitive. An experiment was devised whereby apical membranes were stripped of peripheral membrane proteins and then added back to purified NDPK in vitro (Fig. 3).
Fig. 3.

Panel (A) Western blot of NDPK A from ovine airway membrane fractions prepared in two sequential stages, each involving two different methods of ‘Membrane Solubilisation’ and ‘Column Elution’. Stage I Membrane Solubilisation: Lanes (a, b, c, d) contain proteins extracted from post-nuclear ovine airway membranes using either 0.1% Chaps (a & c) or 5 mM EDTA (b & d). Stage II Protein Isolation: Solubilised proteins from stage I were applied to a cAMP-POROS 20 EP affinity column (generated according to manufacturer’s instruction). After low salt washes, column-retained proteins were eluted with either 2 mM cAMP (fractions a, b) or 2M salt (fractions c, d). The fractions were run on SDS-PAGE, blotted onto PVDF and probed with an anti-NDPK A antibody. NDPK A was only found in lanes b & d. Panel (B)Western blot of NDPK A shows that overlay of the blot (panel A) with a solution containing NDPK A (lane b, 25 (g) results in NDPK A binding to a 62 kDa protein only in lane a (eluted with cAMP from the cAMP-affinity column). Panel (C) Enhanced phosphorylation of NDPK by eluate from cAMP-affinity column. Incubation of fractions containing NDPK A (Panel A, lane b, 1.78 µg protein) with fractions containing the 62 kDa protein (Panel B, lane a, 10× dilution), in the presence of γ[32P]-ATP and Mn2+, results in a four fold enhancement in NDPK phosphorylation. Quantitation is shown in panel D (n = 3).
This data suggested that a 62 kDa protein was able to increase the phosphohistidine content of apical membrane-purified NDPK-A. At that time, this approach was rather novel because NDPK regulation was not understood and further investigation was supported by the Wellcome Trust for KT to work on the project. Thus we had set out to prove the notion that protein-protein interactions modulate the specific molecular actions of NDPK, a notion that remains current with new binding partners of NDPK being identified at an increasing rate (Roymans et al., 2002). This idea was later to be shown to be consistent with the finding that despite sharing 88% amino acid sequence identity, isoforms NDPK-A (Nm23-H1) and NDPK-B (Nm23-H2) have distinctive cellular functions (Bosnar et al., 2004).
AMP-activated kinase
Unknown to us, Hallows et al. had found that in yeast two-hybrid experiments, apical membrane containing CFTR bound the 63 kDa AMPKα (Hallows et al., 2003). AMPK was first discovered as an activity that inhibited preparations of acetyl-CoA carboxylase (ACC1), a regulator of cellular fatty acid synthesis (Winder et al., 1997). This protein was almost exactly the same molecular weight as that found in our studies and since fatty acid metabolism goes awry in CF (Strandvik, 2004), KT speculated that this kinase was the latent binding partner for NDPK. Her guess proved to be inspired. The AMP-activated protein kinase (AMPK) is a heterotrimeric protein with a 63 kDa catalytic α subunit and two regulatory β and γ subunits (38 and 36 kDa respectively), each of which is encoded by distinct genes (α1, α2; β1, β2; γ 1, γ 2, γ 3) (Stapleton et al., 1996; Nielsen et al., 2003). AMPK is reported to be a sensor of cellular energy status, responding to the cytosolic AMP:ATP ratio that itself varies as the square of the ADP:ATP ratio, the whole process being equilibrated by the adenylate kinase reaction (Hardie and Carling, 1997). Once activated by a rise in cellular AMP concentration, AMPK phosphorylates several downstream substrates, the overall effect of which is to switch off ATP-consuming pathways (e.g. fatty acid synthesis and cholesterol synthesis) and switch on ATP-generating pathways (e.g. fatty acid oxidation and glycolysis) (Hardie and Hawley, 2001). AMPK has other longer-term effects in addition to the more short-term conservation of ATP, by altering both protein and gene expression, although the physiological consequences of these longer term effects are not fully understood (Barnes et al., 2002; Holmes and Dohm, 2004). Recent results have indicated that AMPK is able to respond to stimuli that do not cause a detectable change in the AMP:ATP ratio, suggesting that other signals can feed into the AMPK system allowing for a more complex mechanism of cellular energy control (Hawley et al., 2002).
We recently demonstrated that the AMPKα1 (but not α2) catalytic subunit is associated with NDPK-A such that the local supply of ATP synthesised by NDPK-A (from GTP plus ADP) is capable of altering the activity of AMPK as determined by specific AMPK assay (Crawford et al., 2005). As shown in Fig. 4, we also found that NDPK-catalysed production of ATP can be used by AMPK to phosphorylate a known in vivo target, acetyl CoA carboxylase (ACC), a key regulator of fat metabolism. These data suggest a functional association between two enzymes critical for cellular energy supply such that NDPK is acting as a novel regulator to maintain AMPK activity towards at least one metabolically important target in fatty acid synthesis—the complex of NDPK-A, AMPK α1 and ACC creating a novel ‘fat controller’. We therefore propose that a functional association exists between these two critical protein kinases that could allow both adenine and guanine nucleotide energy levels to be efficiently sensed within a novel protein complex. We propose that firstly, the NDPK-A/AMPK α1 interaction is capable of providing AMPK with NDPK-synthesised substrate ATP even under conditions where cellular ATP might be scarce. Relative to AMPK, NDPK is a rapidly acting nucleotide-balancing enzyme with turnover rates in three figures, efficiently maintaining ATP/GTP levels. That NDPK-A (but not NDPK-B) can undertake this role supports NDPK isoform specificity of function. Secondly, we suggest that the proximity of these enzymes could facilitate efficient nucleotide availability to both GTP- and ATP-utilizing enzymes within the same protein complex, depending on the immediate needs of the cell. In Fig. 4, for illustration purposes alone, NDPK is shown utilising either: GDP and ATP to form GTP, or ADP and GTP to form ATP. Under conditions where ATP is limited (e.g., under conditions of cellular stress), the AMP-activated protein kinase is activated (Hardie and Carling, 1997; Hardie and Hawley, 2001) and is known to phosphorylate a range of downstream targets (ACC, HMG-CoA reducatase, HSL and the glucose transporter, Glut4 etc.) with the end-effect of decreasing cellular ATP consumption and increasing cellular ATP synthesis (Barnes et al., 2002; Fryer et al., 2000; Hardie, 1999; Hardie and Pan, 2002). Our model shows one example of the effects of this process towards fatty acid metabolism. Conversely, we also find that the basal, AMP-independent AMPK activity can be decreased by a further 50% when ATP is “stolen“ by adding GDP to the complex (Crawford et al., 2005).
Fig. 4.
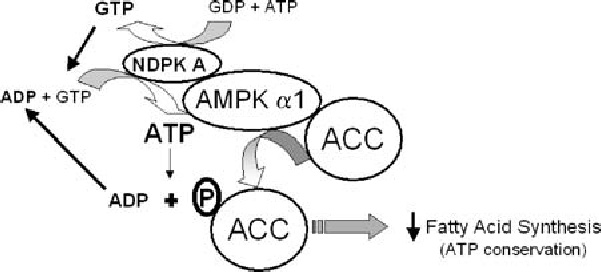
NDPK substrate channelling hypothesis: ATP produced by nucleoside diphosphate kinase (NDPK), from GTP and ADP, is used by the AMP-activated protein kinase (AMPK) as substrate in the AMPK-mediated phospho-transfer reaction. AMPK then uses this NDPK-mediated substrate to phosphorylate its downstream targets, one example of which is acetyl-CoA carboxylase 1 (ACC1). Accumulation of ADP as a result of AMPK phospho-transfer could potentially feed back to NDPK in the microenvironment and fuel the generation of more ATP. By this rationale, even under extremely limiting ATP concentrations, NDPK would provide substrate ATP to AMPK by proxy of their tight association, allowing AMPK to remain functional despite a general shortage of cellular ATP. Thus, NDPK has a role to play in the AMPK signalling cascade. Schematic diagram is based on the observations in Figs. 4 and 5, hypothesising the interaction of NDPK with AMPK taking into account the fates of ADP, GTP and ATP pertaining to the ability of AMPK to phosphorylate ACC1, a major downstream components of the AMPK signalling cascade. Reproduced with permission from Biochem. J.
In conclusion, our approach has illuminated the cellular importance of cytosolic NDPK in unexpected ways, contributing to the evolution of the perception that this enzyme is no longer a mere housekeeping mainstay but can act in an isoform specific manner as a metabolic sensor and more recently, as a key component in a multi-molecular stress response complex. At the beginning of this review we alluded to the role of NDPK-A with respect to CFTR binding. Our preliminary data (Treharne et al., 2001) support the idea that NDPK-A binds CFTR in vitro. This work is in press (Crawford et al., Cell Signalling). Current work in progress aims to understand where the two proteins interact and seeks to elucidate the role of AMPK in the CFTR interaction (Crawford et al. Molecular and Cellular Biology in press).
Acknowledgment
We wish to acknowledge support from the Cystic Fibrosis Trust and the Wellcome Trust (Ref: 075237/Z/04/Z and 069150/Z/02/Z). We thank the Anonymous Trust for recent support.
References
- Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, Foufelle F, Carling D, Hardie DG, Baldwin SA (2002) Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci 115:2433–2442 [DOI] [PubMed]
- Bosnar MH, De Gunzburg J, Bago R, Brecevic L, Weber I, Pavelic J (2004) Subcellular localization of A and B Nm23/NDPK subunits. Exp Cell Res 298:275–284 [DOI] [PubMed]
- Crawford RM, Treharne KJ, Best OG, Muimo R, Riemen CE, Mehta A (2005) A novel physical and functional association between nucleoside diphosphate kinase A and AMP-activated protein kinase alpha1 in liver and lung. Biochem J 392:201–209 [DOI] [PMC free article] [PubMed] [Retracted]
- Dowd BF, Forbush B (2003) PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1). J Biol Chem 278:27347–27353 [DOI] [PubMed]
- Fryer LG, Hajduch E, Rencurel F, Salt IP, Hundal HS, Hardie DG, Carling D (2000) Activation of glucose transport by AMP-activated protein kinase via stimulation of nitric oxide synthase. Diabetes 49:1978–1985 [DOI] [PubMed]
- Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ (1994) Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 266:107–109 [DOI] [PubMed]
- Hallows KR, Kobinger GP, Wilson JM, Witters LA, Foskett JK (2003) Physiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cells. Am J Physiol Cell Physiol 284:C1297–C1308 [DOI] [PubMed]
- Hardie DG (1999) Roles of the AMP-activated/SNF1 protein kinase family in the response to cellular stress. Biochem Soc Symp 64:13–27 [PubMed]
- Hardie DG, Carling D (1997) The AMP-activated protein kinase—fuel gauge of the mammalian cell? Eur J Biochem 246:259–273 [DOI] [PubMed]
- Hardie DG, Hawley SA (2001) AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays 23:1112–1119 [DOI] [PubMed]
- Hardie DG, Pan DA (2002) Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans 30:1064–1070 [DOI] [PubMed]
- Hawley SA, Gadalla AE, Olsen GS, Hardie DG (2002) The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 51:2420–2425 [DOI] [PubMed]
- Holmes B, Dohm GL (2004) Regulation of GLUT4 gene expression during exercise. Med Sci Sports Exerc 36:1202–1206 [DOI] [PubMed]
- Marshall LJ, Muimo R, Riemen CE, Mehta A (1999) Na+ and K+ regulate the phosphorylation state of nucleoside diphosphate kinase in human airway epithelium. Am J Physiol 276:C109–C119 [DOI] [PubMed]
- Mehta A (2005) CFTR: more than just a chloride channel. Pediatr Pulmonol 39:292–298 [DOI] [PubMed]
- Mehta A, Bush A (2005) Beyond chloride transport: CFTR in the 21st century-introductory remarks to a new state of the art series. Pediatr Pulmonol 39:289–291 [DOI] [PubMed]
- Miles EW (1977) Modification of histidyl residues in proteins by diethylpyrocarbonate. Methods Enzymol 47:431–442 [DOI] [PubMed]
- Muimo R, Banner SJ, Marshall LJ, Mehta A (1998) Nucleoside diphosphate kinase and Cl(,)-sensitive protein phosphorylation in apical membranes from ovine airway epithelium. Am J Respir Cell Mol Biol 18:270–278 [DOI] [PubMed]
- Muimo R, Hornickova Z, Riemen CE, Gerke V, Matthews H, Mehta A (2000) Histidine phosphorylation of annexin I in airway epithelia. J Biol Chem 275:36632–36636 [DOI] [PubMed]
- Nielsen JN, Mustard KJ, Graham DA, Yu H, MacDonald CS, Pilegaard H, Goodyear LJ, Hardie DG, Richter EA, Wojtaszewski JF (2003) 5′ AMP-activated protein kinase activity and subunit expression in exercise-trained human skeletal muscle. J Appl Physiol 94:631–641 [DOI] [PubMed]
- Roymans D, Willems R, Van Blockstaele DR, Slegers H (2002) Nucleoside diphosphate kinase (NDPK/NM23) and the waltz with multiple partners: possible consequences in tumor metastasis. Clin Exp Metastasis 19:465–476 [DOI] [PubMed]
- Schwiebert EM, Gesek F, Ercolani L, Wjasow C, Gruenert DC, Karlson K, Stanton BA (1994) Heterotrimeric G proteins, vesicle trafficking, and CFTR Cl- channels. Am J Physiol 267:C272–C281 [DOI] [PubMed]
- Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, House CM, Fernandez CS, Cox T, Witters LA, Kemp BE (1996) Mammalian AMP-activated protein kinase subfamily. J Biol Chem 271:611–614 [DOI] [PubMed]
- Strandvik B (2004) Fatty acid metabolism in cystic fibrosis. N Engl J Med 350:605–607 [DOI] [PubMed]
- Treharne KJ, Marshall LJ, Mehta A (1994) A novel chloride-dependent GTP-utilizing protein kinase in plasma membranes from human respiratory epithelium. Am J Physiol 267:L592–L601 [DOI] [PubMed]
- Treharne KJ, Mehta A, Muimo R (2001) NDPK, a protein kinase defective in CF, binds to and is regulated by AMPK, a CFTR-associated protein. Pediatr Pulmonol 22(Suppl):195
- Wieland T, Nurnberg B, Ulibarri I, Kaldenberg-Stasch S, Schultz G, Jakobs KH (1993) Guanine nucleotide-specific phosphate transfer by guanine nucleotide-binding regulatory protein beta-subunits. Characterization of the phosphorylated amino acid. J Biol Chem 268:18111–18118 [PubMed]
- Winder WW, Wilson HA, Hardie DG, Rasmussen BB, Hutber CA, Call GB, Clayton RD, Conley LM, Yoon S, Zhou B (1997) Phosphorylation of rat muscle acetyl-CoA carboxylase by AMP-activated protein kinase and protein kinase A. J Appl Physiol 82: 219–225 [DOI] [PubMed]