

Abstract
A library of 72 quinolones was synthesized from substituted anthranilic acids, using ynone intermediates. These masked β-dicarbonyl synthons allowed cyclization under milder conditions than previously reported quinolone syntheses.
Recently we reported an efficient asymmetric synthesis of cyclic enaminones from ynones derived from saturated β-amino acids (Figure 1).1a We have since expanded the utility of these versatile intermediates by devising novel methods for their functionalization.1b,c Of particular interest was an understanding of the reaction pathway leading to the apparent 6-endo-dig cyclized products. In aliphatic systems our studies suggest that the presence of an external nucleophile facilitates ring formation via an addition-elimination process.1a To gain insight into this reaction pathway we examined anthranilic acid derived ynones as cyclization precursors. The differences in electronic and geometric properties of these systems would test the scope of our reaction. If successful, a route to cyclo-enaminones from anthranilic acid precursors would be of interest since the anticipated 2-phenylquinolones (Figure 1, and Figure 2) have known biologic activity.2-4
Figure 1.
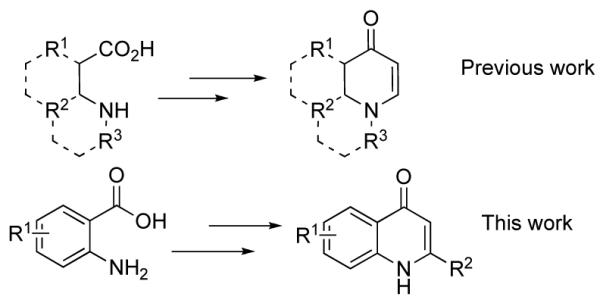
Enaminones from β-amino acids.
Figure 2.
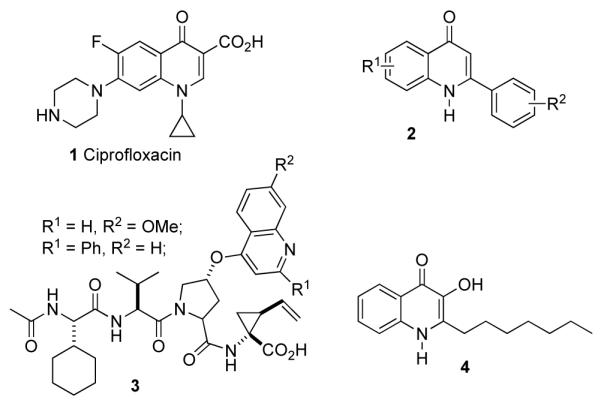
Biologically active quinolones.
2-Phenylquinolones 2 possess antimitotic and cytotoxic properties.2-4 They act as tubulin polymerization inhibitors and are known to compete for the colchicine binding site of tubulin.2-4 Other biologically active quinolones or analogs include the antibacterial agent Ciprofloxacin (1),5 hepatitis C protease inhibitors 3,6 and the P. aeruginosa quorum signaling molecule 4,7 shown in Figure 1. Thus, efficient syntheses of quinolones are of interest in medicinal chemistry and for the synthesis of screening libraries. We hoped to develop a protocol suitable to parallel synthesis within our mission of library development.
Previously reported quinolone syntheses typically employed forcing conditions of elevated temperature and/or strong bases and explicitly employed carbonyl condensation to yield the heterocycle. For example, condensation of ethyl benzoylacetate with anilines has been used with thermal cyclization at 240-250 °C to yield quinolones.2 Alternatively, imine formation from an acetophenone and an anthranilamide followed by ring closure with LDA at −30 °C has produced quinolones.3 Additionally, benzoylation of 2-aminoacetophenones followed by cyclization with t-BuOK at 70 °C for ~1 d yielded quinolones.4 In contrast, the conditions used for the cyclo-enaminone synthesis above1a were much more mild. We were thus interested to see how quinolone synthesis using ynone intermediates as β-diketone synthons would proceed. In addition to using ynone intermediates, we were interested in performing a parallel synthesis of substituted quinolones. The reaction sequence starting with substituted anthranilic acids is shown Scheme 1. This appears to be the first synthesis of quinolones utilizing ynone intermediates.
Scheme 1.
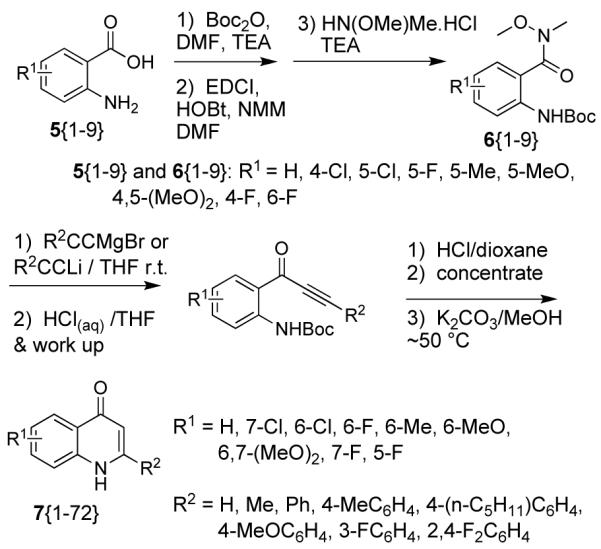
Synthesis of quinolones from ynone intermediates.
We began the synthesis of the quinolone library with the production of Weinreb amides 6{1-9} (Scheme 1). One-pot reactions of the anthranilic acids in the presence of Boc-anhydride, O,N-dimethylhydroxylamine and EDCI/HOBt furnished Boc protected Weinreb amides in yields of 59-80%, after chromatography. It is of note that Boc protection would not occur without added base. The Weinreb amide starting materials were weighed into reaction vials and lyophilized for one day in order to remove adventitious water or solvent traces. Then the metal acetylides were added to the parallel array of Weinreb amides.
Experience in our laboratories with many different types of ynones has shown that the ynone synthesis reaction must be rapidly quenched with excess acid. This prevents Michael adduct formation between the ynones and the methoxymethylamine liberated during the acetylide addition. This efficient acid quench was a key step to our parallel synthesis, since the methoxymethylamine Michael adduct does not allow substantial cyclization to enaminones. Thus, our parallel method also required an extraction step, to separate the ynone products from the N,O-dimethylhydroxylammonium salt. After quenching and work-up, the acetylide adduct samples were concentrated and placed on a lyophilizer to remove excess acetylenes from the ynone products. Samples were examined by NMR to verify ynone formation with characteristic 13C NMR peaks at ~180 ppm, ~104 ppm, and ~98 ppm.
After deprotection of the Boc group with 4M HCl/dioxane and subsequent removal of volatiles, the remaining solids were treated with methanolic K2CO3 at room temperature. Surprisingly, LCMS analysis did not show products of expected mass. However, major products at M+32 were found for the substituted phenylacetylene series, which corresponded to the addition of methanol to the ynone to form the methoxychalcones shown in Scheme 2.
Scheme 2.
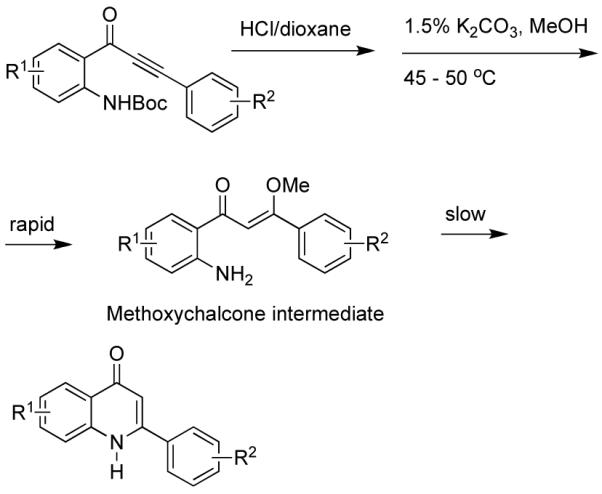
Proposed cyclization pathway for phenylquinolones.
In a related study, the methoxychalcone addition product was isolated (Figure 3). In that case cyclization was presumably prevented because of steric hindrance from the dimethoxybenzyl (DMB) group. Only one isomer of the chalcone was isolated, and there was no evidence for interconversion to other isomers.
Figure 3.
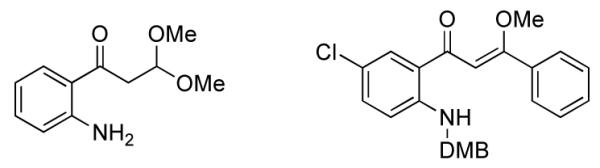
Acetal and methoxychalcone intermediates.
Since quinolone products were not formed, the samples were incubated at 45-50 °C for one day. LCMS showed some products of expected mass were formed, and so incubation was continued for 4 days total. After work up, products were purified by reverse phase chromatography (R2 = H, Me) or fractional crystallization (all phenylquinolones). Characterizations of literature products agreed with literature values, for NMR, MS/HRMS, and IR. Most series of acetylides resulted in useful yields of quinolones (Table 1). Series with R2 = methyl, 4-(n-C5H11)-C6H4 were less efficient at producing quinolones.
Table 1.
Yields (%) and purity (%) of substituted quinolones 7(1-72) produced via Scheme 1.
| Entry/ R1 = |
R2 = H |
R2 = Me |
R2 = Ph |
R2 = 4-(Me)C6H4 |
R2 = 4-(C5H11)C6H4 |
R2 = 4-(MeO)C6H4 |
R2 = 3-FC6H4 |
R2 = 2,4-(F)2C6H3 |
|---|---|---|---|---|---|---|---|---|
| H | 43 / >95 | 14 / >95 | 12 / >95 | 34 / 95 | 7 / >95 | 59 / 95 | 67 / >95 | 67 / 94 |
| 7-Cl | 64 / >95 | 36 / >95 | 81 / >95 | 95 / >95 | 7 / >95 | 30 / 80 | 69 / >95 | 23 / 85 |
| 6-Cl | 79 / >95 | 40 / >95 | 88 / >95 | 63 / >95 | 17 / >95 | 35 / >95 | 73 / 95 | 25 / 95 |
| 6-F | 74 / >95 | 18 / >95 | 63 / >95 | 86 / >95 | 9 / 85 | 90 / >95 | 20 / >95 | 91 / 95 |
| 6-Me | 78 / >95 | 15 / >95 | 35 / >95 | 67 / >95 | 3 / >95 | 60 / >95 | 81 / >95 | 32 / >95 |
| 6-MeO | 38 / >95 | Trace/n.a. | 14 / >95 | 68 / >95 | 8 / >95 | 65 / >95 | 77 / >95 | 87 / >95 |
| 6,7-MeO)2 | 72 / >95 | 8 / >95 | 5 / >95 | 88 / >95 | 6 / 95 | 52 / >95 | 92 / >95 | 36 / >95 |
| 7-F | 11 / >95 | 19 / >95 | 66 / 95 | 74 / >95 | 9 / 95 | 83 / >95 | 93 / >95 | 31 / 95 |
| 5-F | 86 / 90 | Trace/n.a. | 39 / 95 | 57 / >95 | 34 / >95 | 31 / >95 | 68 / 90 | Trace/n.a. |
Table entries are yields%/purity%.8
This synthesis gave us considerable support for our previous inference that an external nucleophile was adding to the ynone followed by cycloenaminone formation, through an addition elimination process. It was previously not possible to ascertain conclusively the intermediate(s) involved in cyclization, as the ring closure was too rapid to analyze for the intermediate by NMR or LCMS. For the anthranilic acid-derived ynones in this study, some intermediates were sufficiently stable to isolate or detect by LCMS. For the anthranilic acid-derived case (Fig. 3) a dimethylacetal was found. For phenylynones as in Scheme 2, methoxychalcones were inferred as intermediates from the LCMS, or isolated as in a related system, shown in Figure 3. Steric and/or conjugative effects would probably stabilize the chalcone in preference to ketal formation. In each case, the intermediates then converted slowly to the corresponding quinolones over the course of several days, at ~50 °C. Higher temperatures (microwave) resulted in appreciable amounts of colored side products. It did not appear that other possible nucleophiles, such as carbonate, were responsible for catalyzing cyclization for the quinolones in this study.
Other nucleophiles could be screened for catalytic activity in the quinolone formation including acetate, thioacetate, methylsulfide, among many others. However, for convenience and ease of removal, methanol is quite useful. In contrast to the related aliphatic case,1 water is disadvantageous as the addition product. Presumably, the β-dicarbonyl, that is formed, deprotonates in weak base to the enolate anion in the anthranilic acid derived case. The enolate anion form does not cyclize.
In conclusion, we have developed a parallel synthesis of quinolones that uses much milder conditions than previously reported methods. No strong bases or high temperatures were required. We have obtained evidence for an addition elimination process to yield the apparent 6-endo-dig products, involving methanol as the catalytic nucleophile. This expands the synthetic utility of ynones as β-carbonyl synthons.
Boc protected Weinreb anthranilamides
Weinreb amide representative procedure: To each substituted anthranilic acid 5{1-9} (6.45 mmol) in a 100 mL round bottom flask with a stirring bar, was added Boc2O (1.48 g, 6.78 mmol). DMF (2.0 mL) was added, a rubber septum cap was attached and the mixture was stirred. When all was dissolved, TEA (0.90 mL) or NMM (0.71 mL) was added by syringe, and a bubbler attached by needle to monitor CO2 evolution. The mixtures were stirred until all gas evolution had ceased, between 3 and 5 days. One mixture was warmed to 50 °C to complete reaction (5-fluoro). (Caution: excessive heating decomposed the Boc2O.) After all gas evolution had ceased, to each flask was added HOBt (0.91 g, 6.5 mmol), DMF (4.5 mL), CH2Cl2 (4.5 mL) and EDCI (1.33 g, 6.94 mmol), and the mixtures were stirred 0.5-1 h. To each flask was added N,O-dimethyl-hydroxylamine hydrochloride (0.76 g, 7.8 mmol), and NMM (0.85 mL) or TEA (1.08 mL), and the mixtures were stirred overnight. Work-up: Each reaction mixture was added to 3:1 hexanes:CH2Cl2 (100 mL) and extracted with 50% brine (7 × 25 mL), and brine (2 × 25 mL). The organics were decanted and concentrated. Crude products 6{1-9} were purified by column chromatography on silica gel (30 mL, 60-200 mesh) using step gradients in 1:1 hexanes:CH2Cl2 with ethyl acetate in proportions of 5%, 10%, 15% as required, collecting 10 mL fractions. Fractions were tested by TLC and appropriate fractions were pooled and concentrated.
Weinreb amide precursors. 6(1)
R1 = H: Yield 75%; 1H NMR (400 MHz, CDCl3) δ = 8.38 (s, 1H), 8.17 (d, J = 8.4 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1.5 Hz, 1H), 7.39 (m, 1H), 7.00 (ddd J = 7.7 Hz, J = 7.5 Hz, J = 1.0 Hz, 1H), 3.57 (s, 3 H), 3.36 (s, 3 H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ = 169.2, 153.1, 138.3, 131.6, 128.9, 121.7, 121.2, 120.6, 80.7, 61.6, 34.7, 28.5; MS (EI): m/z M+H 281; HRMS C14H21N2O4+ calc. 281.1496; found 281.1499; 6(2): R1 = 4-Cl: Yield 72%; 1H NMR (400 MHz, CDCl3) δ = 8.65 (s, 1H), 8.34 (d, J = 1.5 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 6.97 (dd J = 2.1 Hz, J = 8.4 Hz, 1H), 3.55 (s, 3H), 3.38 (s, 3H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ = 168.4, 152.7, 139.9, 137.9, 130.2, 122.2, 121.6, 120.1, 81.1, 61.7, 34.2, 28.5; MS (EI): m/z M+H 315; HRMS C14H20ClN2O4+ calc. 315.1112; found 315.1113; 6(3): R1 = 5-Cl: Yield 70%; 1H NMR (400 MHz, CDCl3) δ = 8.32 (s, 1H), 8.12 (d, J = 9.0 Hz, 1H), 7.43 (d, J = 2.4 Hz, 1H), 7.31 (dd, J = 2.5 Hz, J = 9.0 Hz, 1H), 3.53 (s, 3H), 3.32 (s, 3H), 1.43 (s, 9H); 13C NMR (100 MHz, CDCl3) δ =167.8, 152.9, 137.0, 131.5, 128.6, 126.8, 122.3, 121.9, 81.0, 61.8, 34.2, 28.5; MS (EI): m/z M+H 315; HRMS C14H20ClN2O4+ calc. 315.1112; found 315.1097; 6(4): R1 = 5-F: Yield 69%; 1H NMR (400 MHz, CDCl3) δ = 8.24 (s, 1H), 8.12 (dd, J = 8.6 Hz, J = 4.8 Hz, 1H), 7.21 (dd, J = 8.8 Hz, J = 3.0 Hz, 1H), 7.11 (ddd, J = 9.1 Hz, J = 8.1 Hz, J = 3.0 Hz, 1H), 3.56 (s, 3H), 3.40 (s, 3H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ = 167.7, 158.4, 156.0, 153.2, 134.4, 122.7, 118.5, 118.3, 115.6, 115.3, 80.8, 61.8, 34.1, 28.5; MS (EI): m/z M+H 299; HRMS C14H20FN2O4+ calc. 299.1402; found 299.1383; 6(5): R1 = 5-Me: Yield 67%; 1H NMR (400 MHz, CDCl3) δ = 8.15 (s, 1H), 8.00 (d, J = 8.2 Hz, 1H), 7.25-7.17 (m, 2H), 3.58 (s, 3H), 3.38 (s, 3H), 2.32 (s, 3H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ = 169.3, 153.2, 135.5, 132.2, 131.4, 129.0, 121.8, 120.9, 80.5, 61.5, 34.8, 28.5, 20.8; MS (EI): m/z M+H 295; HRMS C15H23N2O4+ calc. 295.1658; found 295.1656; 6(6): R1 = 5-MeO: Yield 59%; 1H NMR (400 MHz, CDCl3) δ = 8.03-7.85 (m, 2H), 7.00-6.94 (m, 2H), 3.79 (s, 3H), 3.58 (s, 3H), 3.36 (s, 3H), 1.49 (s, 9H); 13C NMR (100 MHz, CDCl3) δ = 168.6, 154.4, 153.4, 130.8, 123.8, 122.9, 116.9, 113.7, 80.3, 61.5, 55.8, 34.4, 28.5; MS (EI): m/z M+H 310; HRMS C15H23N2O5+ calc. 311.1607; found 311.1600; 6(7): R1 = 4,5-(MeO)2 : Yield 62%; 1H NMR (400 MHz, CDCl3) δ = 8.73 (s, 1H), 7.78 (s, 1H), 6.98 (s, 1H), 3.80 (s, 3H), 3.68 (s, 3H), 3.42 (s, 3H), 3.22 (s, 3H), 1.37 (s, 9H); 13C NMR (100 MHz, CDCl3) δ = 168.8, 152.9, 151.4, 142.8, 134.0, 119.3, 111.9, 103.4, 80.0, 61.1, 56.0, 55.8, 34.1, 28.2; MS (EI): m/z M+H 340; HRMS C16H25N2O6+ calc. 341.1713; found 341.1707; 6(8): R1 = 4-F: Yield 80%; 1H NMR (400 MHz, CDCl3) δ = 8.81 (s, 1H), 8.08 (dd, J = 11.9 Hz, J = 2.4 Hz, 1H), 7.55 (dd, J = 8.7 Hz, J = 6.4 Hz, 1H), 6.68 (ddd, J = 8.7 Hz, J = 7.8 Hz, J = 2.5 Hz, 1H), 3.54 (s, 3H), 3.38 (s, 3H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ = 168.6, 165.9, 163.4, 152.8, 141.3, 141.2, 131.2, 131.1, 115.8, 108.6, 108.4, 107.4, 107.1, 81.1, 34.4, 28.5; MS (EI): m/z M+H 299; HRMS C14H20FN2O4+ calc. 299.1402; found 299.1407; 6(9): R1 = 6-F: Yield 59%; two isomers, ~2:1: 1H NMR (400 MHz, CDCl3) δ = 7.75 (d, J = 7.6 Hz, 1H), 7.38 (s, 1H), 7.17 (dd, J = 8.1 Hz, J = 14.9 Hz, 1H), 6.59 (dd, J = 8.8 Hz, J = 8.7Hz, 1H), 3.70-3.31 (m, 3H), 3.22-3.07 (m, 3H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ = 164.5, 161.2, 160.0, 159.6, 157.6, 157.2, 152.2, 138.6, 137.8, 131.9, 131.2, 115.6, 115.5, 112.1, 111.8, 109.2, 109.0, 80.6, 61.5, 60.7, 36.2, 31.9, 28.1, 27.9; MS (EI): m/z M+H 299; HRMS C14H20FN2O4+ calc. 299.1402; found 299.1424.
Quinolone Library Method
Boc protected anthranilic Weinreb amides 6{1-9} (0.150 mmol) were weighed into numbered Mettler-Bohdan Miniblock reaction tubes (11×150 mm) and placed into a lyophilizer overnight, one set of 6{1-9} for each metal acetylide. The tubes were removed from the lyophilizer under argon, and assembled in the Miniblock 24 tube holder with stir bars in each tube. Empty tubes were placed in the remaining positions, the gasket and cover were affixed and the system purged by 4 argon/vacuum cycles and then left under argon. Into each tube containing a Boc protected anthranilic Weinreb amide was added by syringe a solution of metal acetylide. For HCCMgBr and MeCCMgBr, (5.0 mL × 0.5 M in THF) commercial solutions (Aldrich) were used. Lithium phenylacetylides were prepared from each phenylacetylene by deprotonation with methyllithium. Each phenylacetylene (25 mmol) was weighed into a dry 100 mL two-neck flask containing a stir bar under nitrogen. Dry THF (14 mL) was added and stirred to dissolve the acetylene, and the flask was cooled in an ice bath. Methyllithium (16.0 mL × 1.5 M MeLi in ether, Aldrich) was added dropwise (caution: foaming). After addition was complete, cooling was removed, and portions of the lithiumphenylacetylide solution were drawn up into a syringe and 3.0 mL was added to each tube containing a Weinreb amide. Thus each Weinreb amide (0.150 mmol) was allowed to react with an metal acetylide (~2.3-2.5 mmol). Stirring was started and the systems with magnesium acetylides were reacted for 23 h, while lithium phenylacetylides were reacted for 5 h at room temperature. Work-up: The Miniblock was chilled with an ice bath. Each reaction mixture was quenched by rapid injection with chilled 4 M HCl(aq) in THF (1.0 mL) with rapid stirring (prepared from 8.3 mL conc. HCl, water was added to 12 mL, then THF was added to 25 mL). The cover and gasket were then removed and hexane and ether were added to each tube (~2.5 mL each) except for tubes from 6(7), THF and CH2Cl2 was added. Stirring was stopped and the acid layer was removed by pipette. Water (2 mL) was added to each tube with rapid stirring. The stirring was stopped after 2 min, and after settling, the aqueous acid layers removed by pipette. The washing steps were repeated four times total, in the ice bath. Powdered NaHCO3 (~100 mg) was added to each tube and the systems were stirred 15 min. Powdered MgSO4 (100 mg) was added to each tube and stirred for ~15 min (suspended MgSO4 was visible in each tube). After settling, organics were then separately pipetted into individual scintillation vials (20 mL) and the solvents removed in vacuo. For substituted phenylacetylene derivatives, samples were left on high vacuum (lyophilizer) to remove excess phenylacetylenes. Stirring bars were added to each vial, HCl/dioxane (3 mL × 4 M HCl in dioxane) was added and then the vials were capped tightly with a Teflon lined cap. The mixtures were stirred overnight. The HCl/dioxane was then removed under nitrogen. To each vial was added K2CO3 in MeOH (12 mL × 1.5% solution). The vials were tightly capped with Teflon caps, and put on a warm stir plate to react for 4 d at 45-50 °C. The vials were cooled to room temperature and CH2Cl2 (12 mL) used to dilute each reaction mixture, and then the mixtures were filtered through silica gel and flushed with a 1:1 mixture of CH2Cl2 and methanol. The filtrates were concentrated in scintillation vials, and compound series with R2=H and Me were subjected to HPLC purification. The remaining phenylquinolone compounds were not sufficiently soluble to allow efficient LC. All phenylquinolones were individually dissolved in a minimum of TFA and filtered through glass wool to remove silica gel and concentrated. The samples were then washed with ether, CH2Cl2, MeCN and subjected to digestion or fractional crystallization using MeCN and methanol mixtures. Purities were determined by forming solutions in methanol and performing LCMS and HRMS for all compounds 7(1-72). NMR spectra obtained for compounds in d4-methanol or d6-DMSO agreed with literature values.2,3,4
Acknowledgments
Financial support of this work by the National Institute of General Medical Sciences is gratefully acknowledged, P50-GM069663.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1 (a).Turunen BJ, Georg GI. J. Am. Chem. Soc. 2006;128:8702. doi: 10.1021/ja0609046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X, Turunen BJ, Leighty MW, Georg GI. Tetrahedron Lett. 2007;48:8811. doi: 10.1016/j.tetlet.2007.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ge H, Niphakis MJ, Georg GI. J. Am. Chem. Soc. 2008;130:3708. doi: 10.1021/ja710221c. [DOI] [PubMed] [Google Scholar]
- 2.Kuo SC, Lee HZ, Juang JP, Lin YT, Wu TS, Chang JJ, Lednicer D, Paull KD, Lin CM, Hamel E, Lee KH. J. Med. Chem. 1993;36:1146. doi: 10.1021/jm00061a005. [DOI] [PubMed] [Google Scholar]
- 3.Li L, Wang HK, Kuo SC, Wu TS, Lednicer D, Lin CM, Hamel E, Lee KH. J. Med. Chem. 1994;37:1126. doi: 10.1021/jm00034a010. For related syntheses see: Chen BC, Huang X, Wang J. Synthesis. 1987:482. Kasahara A, Izumi T, Watabe H, Takahashi S. Chem. Indust. 1981:121.
- 4.Li L, Wang HK, Kuo SC, Wu TS, Mauger A, Lin CM, Hamel E, Lee KH. J. Med. Chem. 1994;37:3400. doi: 10.1021/jm00046a025. [DOI] [PubMed] [Google Scholar]
- 5.Grohe Klaus, Zeiler Hans Joachim, Metzger Karl. DE 3142854 A1 (19830511) (Bayer A.-G., Fed. Rep. Ger.). Ger. Offen. 1983
- 6.Llinas-Brunet M, Bailey MD, Ghiro E, Gorys V, Halmos T, Poirier M, Rancourt J, Goudreau N. J. Med. Chem. 2004;47:6584. doi: 10.1021/jm0494523. [DOI] [PubMed] [Google Scholar]
- 7.Pesci EC, Milbank JB, Pearson JP, McKnight S, Kende AS, Greenberg EP, Iglewski BH. Proc. Natl. Acad. Sci. USA. 1999;96:11229. doi: 10.1073/pnas.96.20.11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The standards for compound purity and quality are based on those requested for the National Institutes of Health’s Molecular Libraries Small Molecule Repository ( http://mlsmr.glpg.com/MLSMR_HomePage/submitcompounds.html) The compound purities were determined by reverse-phase HPLC with peak area (UV) at 214 nm.