

Introduction
Systolic heart failure is a feed-forward phenomenon with devastating consequences. Impaired cardiac function is the initiating event, but central nervous system mechanisms activated by persistent altered neural and humoral signals from the periphery play an important sustaining role. Animals with experimentally induced heart failure have neurochemical abnormalities in the brain that, when manipulated, profoundly affect sympathetic drive, volume regulation and cardiac remodeling – critical determinants of outcome. This brief review explores recent studies that provide a strong rationale for the development of pharmaceutical agents that target the central nervous system abnormalities in heart failure.
The references S1 - S22 can be found in the online supplement.
Traditional approaches
Standard therapy for systolic heart failure is based largely upon the recognition that neurohumoral systems – particularly the renin-angiotensin-aldosterone system and the sympathetic nervous system - are activated in patients with heart failure and contribute to the progression of the disease (s1). The optimal medical regimen targets the peripheral manifestations of these neurohumoral derangements, counteracting vasoconstriction, volume accumulation, and cardiac and vascular remodeling. Thus, conventional therapy consists of angiotensin converting enzyme (ACE) inhibitors, angiotensin type-1 (AT1) receptor blockers, and mineralocorticoid receptor (MR) antagonists to reduce the effects of angiotensin II (ANG II) and aldosterone (ALDO) on the heart, blood vessels and kidneys; loop diuretics and ultrafiltration to control the effects of ANG II, ALDO, and sympathetic nerve activity (SNA) on renal handling of sodium and water; and beta-blockers to counter the effects of persistent augmented sympathetic drive on cardiac function. Ancillary therapies not specifically based on neurohumoral dysfunction but likely to ameliorate it include inotropic agents (e.g., digoxin, milrinone) and vasodilators (e.g., hydralazine, isosorbide dinitrate, nitroprusside). We also have device therapy with cardiac resynchronization therapy to improve cardiac output, implantable cardioverter defibrillators to protect against sudden death, and, as a last resort for some patients, cardiac transplantation.
Many of the drugs in common usage – e.g., ACE-inhibitors, MR antagonists, beta-blockers - have proven effective in large clinical trials, but their application in clinical practice is frequently limited, especially in the sickest patients who need them most, by serious side effects including hypotension, hyperkalemia, and declining renal function. Some approaches – e.g., aggressive diuresis – may even exacerbate the heart failure syndrome. New ways to intervene in this devastating disease process are clearly needed.
The central nervous system in heart failure
The peripheral manifestations of neurohumoral excitation in heart failure are paralleled by, and in large part facilitated by, excitatory and inflammatory events in the central nervous system. Recent experimental work has focused on how these neurochemical changes in the central nervous system contribute to the pathophysiology of heart failure.
Cardiovascular regulatory regions of the brain
Several regions of the brain (Figure 1) have attracted attention because of their relevance to the physiological derangements observed in heart failure: 1) the circumventricular organs of the lamina terminalis – the subfornical organ (SFO) and the organum vasculosum of the lamina terminalis (OVLT) - that lack a tight blood-brain barrier and can therefore sense the ambient level of circulating hormones like ANG II; 2) the paraventricular nucleus (PVN) of the hypothalamus, a forebrain nucleus that integrates and responds to a variety of neural and humoral signals regulating sympathetic drive and extracellular fluid volume (s2); and 3) the rostral ventrolateral medulla (RVLM), a point of convergence for signals from forebrain and hindbrain centers that determines the intensity of sympathetic activity (s3).
Figure 1.

Schematic depicting selected neural and humoral signals impinging upon the central nervous system (green), key effector nuclei (orange), and behavioral and autonomic responses (red). See text for explanation. Detailed pathways are not shown, but are described in references provided in the online supplement. Abbreviations: SFO, subfornical organ; OVLT, organum vasculosum of the lamina terminalis; PVN, paraventricular nucleus; RVLM, rostral ventrolateral medulla; IML, intermediolateral cell column; NTS, nucleus tractus solitarius; AVP, arginine vasopressin; ACTH, adrenocorticotropic hormone; PIC, pro-inflammatory cytokines.
The SFO and OVLT are concerned primarily with thirst and sodium appetite (s4), behaviors that adversely affect volume regulation in heart failure. The PVN and RVLM contain neurons with long descending axons that innervate the preganglionic sympathetic neurons in the intermediolateral cell column (IML) of the spinal cord (s5), ultimately affecting sympathetic drive to the heart and the vascular tree and, importantly, renal handling of sodium and water and renin release (s6). In addition, the PVN has specialized neurons that regulate the release of adrenocorticotropic hormone (ACTH) and arginine vasopressin (AVP), humoral factors that may contribute to sodium accumulation and vasoconstriction in heart failure. All three regions respond to altered conditions in the periphery in heart failure: the circumventricular organs via humoral cues (e.g., ANG II), the PVN and the RVLM via input from sensory receptors in the heart and vascular tree. In heart failure, the balance of signals from the periphery favors excitation of neurons in all three regions (Figure 2), resulting in volume accumulation and peripheral vasoconstriction. Peripheral consequences of central neural activation in animal models of heart failure include increased sodium consumption and augmented sympathetic drive (1, 2) with decreased renal excretion of sodium and water (1, 2) and worsening left ventricular dysfunction (3).
Figure 2.
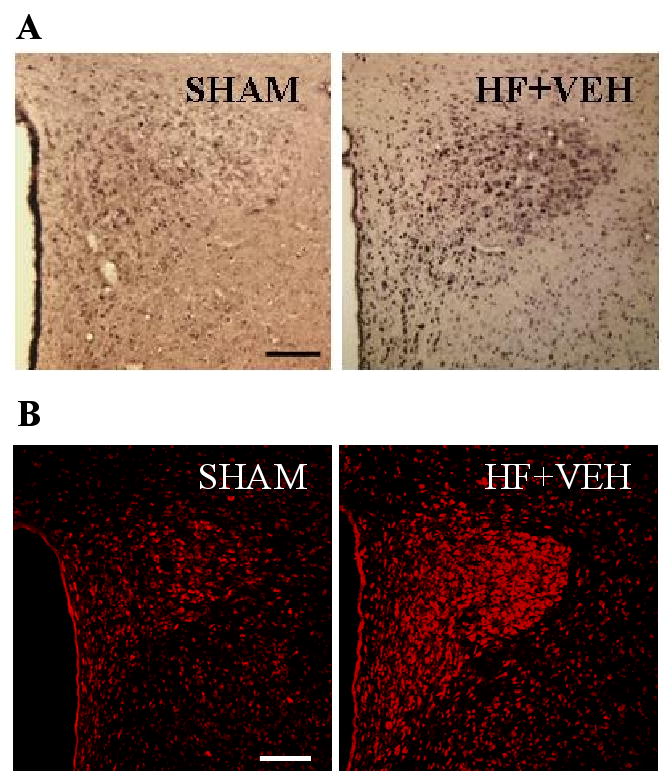
Activation of the central nervous system in heart failure. Coronal (unilateral) sections of the hypothalamic paraventricular nucleus (PVN) from rats with heart failure (HF) of 4 weeks duration and sham-operated controls (SHAM). A) Chronic excitation of PVN neurons is indicated by Fra-Like activity (black dots). B) Superoxide production in PVN neurons is indicated by dihydroethidium fluorescence (red). Superoxide production and neuronal excitation are increased in the PVN and in other cardiovascular regions (not shown) in rats with HF. The HF rats in this study had received chronic intracerebroventricular (ICV) infusions of vehicle (VEH), but no other treatment. In all images, the third ventricle is to the left. Bar=200 microns. Figure adapted from reference (12), and reprinted courtesy of Hypertension.
Angiotensin and aldosterone
In heart failure, the neurochemical milieu in the brain mirrors that in the periphery. Here again, the renin-angiotensin system plays a dominant role. Activity of the intrinsic brain renin-angiotensin system (s7) is exaggerated. In the PVN, angiotensin converting enzyme (ACE) activity, which converts angiotensin I to ANG II, is upregulated.(4) ANG II type-1 (AT1) receptors that mediate the central nervous system effects of ANG II are increased in the PVN (4, 5), the SFO (5) and the RVLM (6). ANG II type-2 (AT2) receptors, that appear to modulate the excitatory effects of ANG II on AT1 receptors (s8), are downregulated in the RVLM in heart failure (7). In acute studies, the abnormalities in central processing of cardiovascular reflexes in animals with heart failure are normalized by blocking brain AT1 receptors (8-10); unfortunately, an effort to stimulate AT2 receptors was not effective (7). Chronic intracerebroventricular (ICV) administration of an AT1 receptor blocker or an ACE inhibitor reduces sympathetic nerve activity (1, 11) and improves volume regulation (1) in animals with heart failure.
ALDO crosses the blood-brain barrier from the circulation, though in restricted quantities, and is also be synthesized in brain tissue (12-14). Like ANG II, ALDO acts within the brain to stimulate sodium appetite (s4) and sympathetic drive (s9). Exactly how ALDO effects these central actions remains under investigation. In peripheral tissues, ALDO has genomic effects mediated by activation of cytosolic MR that are protected from binding with corticosterone by activity of the enzyme 11β-hydroxysteroid dehydrogenase type 2, and non-genomic actions that are partially or completely independent of classical ALDO-sensitive MR (14). ICV administration of an MR antagonist, like ICV administration of an AT1 receptor blocker or an ACE inhibitor, reduces sympathetic activity and improves volume regulation in animals with heart failure (2). ANG II and ALDO may share a common effector mechanism: both can increase NAD(P)H oxidase activity to produce superoxide (6, 15), ALDO likely via a non-genomic mechanism (15). NAD(P)H oxidase-dependent superoxide generation has been implicated as an important factor driving sympathetic activity in animals with heart failure (6, 16).
The pro-inflammatory cytokines
The pro-inflammatory cytokines tumor necrosis factor - alpha (TNF-α) and interleukin-1 beta (IL-1β) also activate the central nervous system to increase sympathetic drive in heart failure, but by a different mechanism (s10, 17). TNF-α and IL-1β induce cyclooxygenase-2 (COX-2) activity in perivascular macrophages of the blood-brain barrier (18), generating prostaglandin E2 (PGE2) which enters the brain (19) and stimulates PVN neurons regulating ACTH release (17) and sympathetic drive (20). TNF-α and IL-1β levels increase in the brain as well as in the periphery in heart failure (21, 22). The increase in pro-inflammatory cytokines may be linked to changes in the brain renin-angiotensin system - in other tissues, activation of AT1 receptors induces and activation of AT2 receptors inhibits the expression of pro-inflammatory cytokines (s11, 23). The presence of pro-inflammatory cytokines in brain tissues is functionally significant, not just a marker of inflammation. Thus, in rats with heart failure, inhibiting brain synthesis of pro-inflammatory cytokines (22), or countering their effects with an anti-inflammatory cytokine (24), reduces COX-2 expression in perivascular macrophages, PGE2 levels in the cerebrospinal fluid and plasma norepinephrine, an indicator of sympathetic nerve activity. It is particularly interesting that the pro-inflammatory cytokines, like ANG II and ALDO, stimulate NAD(P)H oxidase-dependent superoxide production in the brain (22). Thus, the pro-inflammatory cytokines may contribute to sympathetic drive via both PGE2 dependent and NAD(P)H oxidase dependent mechanisms.
Cell signaling
Since NAD(P)H oxidase-dependent superoxide generation is common to several mechanisms leading to sympathetic excitation, it is of interest to consider the downstream molecular events that might ultimately effect an increase in sympathetic drive. Recent data suggest that mitogen-activated protein kinase (MAPK) signaling pathways may play such a role. ANG II, ALDO and the pro-inflammatory cytokines share the ability to stimulate the three major MAPK signaling pathways – p44/42 MAPK, p38 MAPK and JNK – all of which are NAD(P)H oxidase dependent (s12). In heart failure, these three MAPK pathways are activated in the PVN, correlating with superoxide production and neuronal excitation (5). Blocking the p44/42 MAPK pathway substantially reduces sympathetic nerve activity in rats with established HF (Figure 3), but not in sham-operated control rats (25). Notably, MAPK signaling leads to the generation of activator protein-1, which has been implicated as a possible factor in the upregulation of brain AT1 receptors (26).
Figure 3.
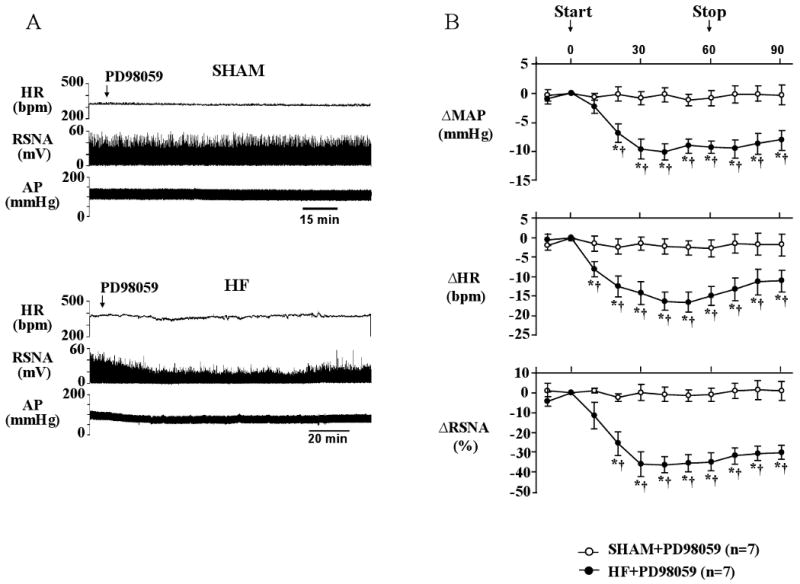
MAPK signaling and sympathetic excitation in heart failure. A) Representative recordings from a sham-operated (SHAM) rat (top panel) and a rat with heart failure (HF) of 4 weeks duration (bottom panel). An intracerebroventricular infusion of the selective p44/42 MAPK inhibitor PD98059 reduced heart rate (HR, beats/min (bpm)), renal sympathetic nerve activity (RSNA, integrated voltage (mV)) and arterial pressure (AP (mmHg)) in the HF rat, but had is effect on the SHAM rat. B) Grouped data, demonstrating that the p44/42 MAPK inhibitor substantially reduced sympathetic drive in rats with established HF. *P_0.05 vs baseline; †P_0.05 vs SHAM. The data suggest that p44/42 MAPK is activated in HF and contributes to the sustained increase in sympathetic nerve activity typical of the HF syndrome. p38 MAPK and JNK inhibitors had no effect. Figure adapted from (25), and reprinted courtesy of Hypertension.
Therapeutic targets in the brain
These experimental studies suggest that interventions at the central nervous system level are beneficial in heart failure. ANG II, AT1 and AT2 receptors, ALDO and ALDO-sensitive MR, the pro-inflammatory cytokines, NAD(P)H oxidase, COX-2, and MAPK all appear to be potential targets for central interventions that might substantially reduce the adverse peripheral effects of sympathetic nerve activity in heart failure. In a direct comparison of chronic peripheral versus central administration of an AT1 receptor blocker in rats with heart failure, the central intervention was actually more effective in reducing left ventricular remodeling following a small myocardial infarction (27). In other studies, chronic central administration of an MR antagonist reduced sodium consumption, normalized renal handling of sodium and water (2), improved baroreceptor regulation of sympathetic nerve activity (2), and reduced circulating levels of pro-inflammatory cytokines (28). Similar effects on sympathetic drive and volume regulation were observed when heart failure rats were treated chronically with an ACE inhibitor (1). Finally, chronic central administration of a cytokine synthesis inhibitor to heart failure rats reduced not only the PVN expression of TNF-α and IL-1β, but also the PVN expression of ACE and AT1 receptors, the superoxide production in the PVN, COX-2 expression in the microvasculature of the PVN, PGE2 in CSF, and plasma ACTH and norepinephrine (22).
Brain mechanisms may be attractive targets for therapeutic intervention in heart failure, but they are not so readily accessible in patients. Systemically administered ACE inhibitors in doses that patients can tolerate are unlikely to penetrate the blood-brain barrier in effective concentrations (s13). In theory, ACE inhibitors may even cause harm, since ANG I levels increase (s14) to provide even more substrate for conversion of ANG I to ANG II in the circumventricular organs where ACE concentrations are very high (s15). AT1 receptor antagonists may cross the blood-brain barrier to varying extents (29), but the doses that can be administered are likely to be limited by effects on blood pressure. Whether the beneficial central effects of MR antagonists observed in animals can be achieved in humans using doses that do not induce hyperkalemia remains to be determined. Though animal studies suggest that inhibiting PGE2 synthesis in the perivascular cells of the blood-brain barrier would have a desirable effect, systemically administered COX-2 inhibitors have adverse effects in heart failure (s16). Intuitively, antioxidants would seem to be a logical approach, since superoxide production is a key factor driving sympathetic activity. Unfortunately, despite the effectiveness of central antioxidant therapy in animal models, systemic antioxidant therapy in cardiovascular disease has not met with clinical success (s17).
At present, the best hope for minimizing the effects of the brain in heart failure may be to reduce the levels of the key offending hormones. Ongoing investigations with renin inhibitors (30) and aldosterone synthase inhibitors (31) will address that hypothesis. Recent data from our laboratory suggest that blood-borne ANG II contributes to upregulation of AT1 receptors inside the blood-brain barrier, in the PVN, and thus predicts a beneficial central effect of lowering circulating ANG II (32). Since ANG II is a primary stimulus for adrenal release of ALDO, a secondary benefit of renin inhibition may be a reduction in plasma ALDO. Efforts to reduce circulating pro-inflammatory cytokines with recombinant human soluble tumor necrosis factor-α receptors have been ineffective (s18) and even potentially hazardous (s19). However, systemic treatment with the cytokine synthesis inhibitor pentoxifylline, which is in common use for other indications with minimal side effects, has been very effective in animal studies (21, 33, 34) and in some (s20, s21) but not all (s22) clinical studies in patients with heart failure. Pentoxifylline readily crosses the blood-brain barrier, so systemically administered drug might reduce both circulating and brain levels of pro-inflammatory cytokines. Finally, in an animal study a systemically administered MR antagonist had the surprising effect of reducing circulating cytokines (33), suggesting a combined approach to mineralocorticoid receptor blockade and anti-cytokine therapy. The general approach of reducing the levels of humoral factors that activate the central nervous system has the obvious advantage of simultaneously treating their adverse effects on peripheral tissues. Moreover, because of the “crosstalk” that has been observed among central neurochemical systems (22, 34), one might anticipate that modifying the activity of one may secondarily mollify another.
In future, the development of drugs that readily cross the blood-brain barrier (35) or preferentially target neural tissues might better address the central nervous system mechanisms contributing to heart failure in humans. Newer technologies - viral vectors (36) or perhaps nanoparticles (37) – hold promise for more specific tissue- or molecule-directed drug therapy. The experimental data presented here suggest that selective treatment of the central nervous system may mimic the improvements now seen with systemically administered drugs, without the associated systemic side effects. However, until new and more selective modes of drug delivery are developed it will not be possible to compare the effects of central versus peripheral interventions, or to evaluate the possible synergistic effects of simultaneous treatment of both facets of the disease.
Supplementary Material
Acknowledgments
This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, NIH RO1s HL HL063915 and HL73986, an American Heart Association Heartland Grant-In-Aid, and funds provided by the University of Iowa.
Footnotes
Disclosures: The authors have nothing to disclose.
References Cited
- 1.Francis J, Wei SG, Weiss RM, Felder RB. Brain angiotensin-converting enzyme activity and autonomic regulation in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H2138–46. doi: 10.1152/ajpheart.00112.2004. [DOI] [PubMed] [Google Scholar]
- 2.Francis J, et al. Central mineralocorticoid receptor blockade improves volume regulation and reduces sympathetic drive in heart failure. Am J Physiol Heart Circ Physiol. 2001;281:H2241–51. doi: 10.1152/ajpheart.2001.281.5.H2241. [DOI] [PubMed] [Google Scholar]
- 3.Huang BS, Leenen FH. Blockade of brain mineralocorticoid receptors or Na+ channels prevents sympathetic hyperactivity and improves cardiac function in rats post-MI. Am J Physiol Heart Circ Physiol. 2005;288:H2491–7. doi: 10.1152/ajpheart.00840.2004. [DOI] [PubMed] [Google Scholar]
- 4.Tan J, Wang H, Leenen FH. Increases in brain and cardiac AT1 receptor and ACE densities after myocardial infarct in rats. Am J Physiol Heart Circ Physiol. 2004;286:H1665–71. doi: 10.1152/ajpheart.00858.2003. [DOI] [PubMed] [Google Scholar]
- 5.Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB. Mitogen-activated protein kinases mediate upregulation of hypothalamic angiotensin II type 1 receptors in heart failure rats. Hypertension. 2008;52:679–86. doi: 10.1161/HYPERTENSIONAHA.108.113639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao L, et al. Superoxide mediates sympathoexcitation in heart failure: roles of angiotensin II and NAD(P)H oxidase. Circ Res. 2004;95:937–44. doi: 10.1161/01.RES.0000146676.04359.64. [DOI] [PubMed] [Google Scholar]
- 7.Gao L, Wang WZ, Wang W, Zucker IH. Imbalance of angiotensin type 1 receptor and angiotensin II type 2 receptor in the rostral ventrolateral medulla: potential mechanism for sympathetic overactivity in heart failure. Hypertension. 2008;52:708–14. doi: 10.1161/HYPERTENSIONAHA.108.116228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang WZ, Gao L, Wang HJ, Zucker IH, Wang W. Interaction between cardiac sympathetic afferent reflex and chemoreflex is mediated by the NTS AT1 receptors in heart failure. Am J Physiol Heart Circ Physiol. 2008;295:H1216–26. doi: 10.1152/ajpheart.00557.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao L, Schultz HD, Patel KP, Zucker IH, Wang W. Augmented input from cardiac sympathetic afferents inhibits baroreflex in rats with heart failure. Hypertension. 2005;45:1173–81. doi: 10.1161/01.HYP.0000168056.66981.c2. [DOI] [PubMed] [Google Scholar]
- 10.Zhu GQ, Gao L, Patel KP, Zucker IH, Wang W. ANG II in the paraventricular nucleus potentiates the cardiac sympathetic afferent reflex in rats with heart failure. J Appl Physiol. 2004;97:1746–54. doi: 10.1152/japplphysiol.00573.2004. [DOI] [PubMed] [Google Scholar]
- 11.Zhang W, Huang BS, Leenen FH. Brain renin-angiotensin system and sympathetic hyperactivity in rats after myocardial infarction. Am J Physiol. 1999;276:H1608–15. doi: 10.1152/ajpheart.1999.276.5.H1608. [DOI] [PubMed] [Google Scholar]
- 12.Yu Y, Wei SG, Zhang ZH, Gomez-Sanchez E, Weiss RM, Felder RB. Does Aldosterone Upregulate the Brain Renin-Angiotensin System in Rats With Heart Failure? Hypertension. 2008;51:727–33. doi: 10.1161/HYPERTENSIONAHA.107.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang BS, White RA, Ahmad M, Tan J, Jeng AY, Leenen FH. Central infusion of aldosterone synthase inhibitor attenuates left ventricular dysfunction and remodelling in rats after myocardial infarction. Cardiovasc Res. 2009;81:574–581. doi: 10.1093/cvr/cvn222. [DOI] [PubMed] [Google Scholar]
- 14.Connell JM, Davies E. The new biology of aldosterone. J Endocrinol. 2005;186:1–20. doi: 10.1677/joe.1.06017. [DOI] [PubMed] [Google Scholar]
- 15.Callera GE, et al. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension. 2005;45:773–9. doi: 10.1161/01.HYP.0000154365.30593.d3. [DOI] [PubMed] [Google Scholar]
- 16.Lindley TE, Doobay MF, Sharma RV, Davisson RL. Superoxide is involved in the central nervous system activation and sympathoexcitation of myocardial infarction-induced heart failure. Circ Res. 2004;94:402–9. doi: 10.1161/01.RES.0000112964.40701.93. [DOI] [PubMed] [Google Scholar]
- 17.Ericsson A, Arias C, Sawchenko PE. Evidence for an intramedullary prostaglandin-dependent mechanism in the activation of stress-related neuroendocrine circuitry by intravenous interleukin-1. J Neurosci. 1997;17:7166–79. doi: 10.1523/JNEUROSCI.17-18-07166.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiltz JC, Sawchenko PE. Signaling the brain in systemic inflammation: the role of perivascular cells. Front Biosci. 2003;8:s1321–9. doi: 10.2741/1211. [DOI] [PubMed] [Google Scholar]
- 19.Engblom D, Ek M, Saha S, Ericsson-Dahlstrand A, Jakobsson PJ, Blomqvist A. Prostaglandins as inflammatory messengers across the blood-brain barrier. J Mol Med. 2002;80:5–15. doi: 10.1007/s00109-001-0289-z. [DOI] [PubMed] [Google Scholar]
- 20.Zhang ZH, Wei SG, Francis J, Felder RB. Cardiovascular and renal sympathetic activation by blood-borne TNF-alpha in rat: the role of central prostaglandins. American journal of physiology. 2003;284:R916–27. doi: 10.1152/ajpregu.00406.2002. [DOI] [PubMed] [Google Scholar]
- 21.Francis J, Chu Y, Johnson AK, Weiss RM, Felder RB. Acute myocardial infarction induces hypothalamic cytokine synthesis. Am J Physiol Heart Circ Physiol. 2004;286:H2264–71. doi: 10.1152/ajpheart.01072.2003. [DOI] [PubMed] [Google Scholar]
- 22.Kang YM, Zhang ZH, Xue B, Weiss RM, Felder RB. Inhibition of brain proinflammatory cytokine synthesis reduces hypothalamic excitation in rats with ischemia-induced heart failure. Am J Physiol Heart Circ Physiol. 2008;295:H227–36. doi: 10.1152/ajpheart.01157.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaschina E, et al. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation. 2008;118:2523–32. doi: 10.1161/CIRCULATIONAHA.108.784868. [DOI] [PubMed] [Google Scholar]
- 24.Yu Y, et al. Central gene transfer of interleukin-10 reduces hypothalamic inflammation and evidence of heart failure in rats after myocardial infarction. Circ Res. 2007;101:304–12. doi: 10.1161/CIRCRESAHA.107.148940. [DOI] [PubMed] [Google Scholar]
- 25.Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB. Angiotensin II-triggered p44/42 mitogen-activated protein kinase mediates sympathetic excitation in heart failure rats. Hypertension. 2008;52:342–50. doi: 10.1161/HYPERTENSIONAHA.108.110445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu D, Gao L, Roy SK, Cornish KG, Zucker IH. Neuronal angiotensin II type 1 receptor upregulation in heart failure: activation of activator protein 1 and Jun N-terminal kinase. Circ Res. 2006;99:1004–11. doi: 10.1161/01.RES.0000247066.19878.93. [DOI] [PubMed] [Google Scholar]
- 27.Huang BS, Ahmad M, Tan J, Leenen FH. Sympathetic hyperactivity and cardiac dysfunction post-MI: Different impact of specific CNS versus general AT(1) receptor blockade. J Mol Cell Cardiol. 2007;43:479–86. doi: 10.1016/j.yjmcc.2007.07.047. [DOI] [PubMed] [Google Scholar]
- 28.Francis J, Weiss RM, Johnson AK, Felder RB. Central mineralocorticoid receptor blockade decreases plasma TNF-alpha after coronary artery ligation in rats. American journal of physiology. 2003;284:R328–35. doi: 10.1152/ajpregu.00376.2002. [DOI] [PubMed] [Google Scholar]
- 29.Wang JM, Tan J, Leenen FH. Central nervous system blockade by peripheral administration of AT1 receptor blockers. J Cardiovasc Pharmacol. 2003;41:593–9. doi: 10.1097/00005344-200304000-00012. [DOI] [PubMed] [Google Scholar]
- 30.Luft FC, Weinberger MH. Antihypertensive therapy with aliskiren. Kidney international. 2008;73:679–83. doi: 10.1038/sj.ki.5002732. [DOI] [PubMed] [Google Scholar]
- 31.Fiebeler A, et al. Aldosterone synthase inhibitor ameliorates angiotensin II-induced organ damage. Circulation. 2005;111:3087–94. doi: 10.1161/CIRCULATIONAHA.104.521625. [DOI] [PubMed] [Google Scholar]
- 32.Wei SG, Yu Y, Zhang ZH, Felder RB. Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol. 2009;296:H1425–33. doi: 10.1152/ajpheart.00942.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang YM, et al. Novel effect of mineralocorticoid receptor antagonism to reduce proinflammatory cytokines and hypothalamic activation in rats with ischemia-induced heart failure. Circ Res. 2006;99:758–66. doi: 10.1161/01.RES.0000244092.95152.86. [DOI] [PubMed] [Google Scholar]
- 34.Guggilam A, Patel KP, Haque M, Ebenezer PJ, Kapusta DR, Francis J. Cytokine blockade attenuates sympathoexcitation in heart failure: Cross-talk between nNOS, AT-1R and cytokines in the hypothalamic paraventricular nucleus. Eur J Heart Fail. 2008;10:625–634. doi: 10.1016/j.ejheart.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munoz L, et al. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. Journal of neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar P, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 37.Silva GA. Nanotechnology approaches to crossing the blood-brain barrier and drug delivery to the CNS. BMC neuroscience. 2008;9 3:S4. doi: 10.1186/1471-2202-9-S3-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.