

Abstract
Chronic granulomatous disease (CGD) is characterized by recurrent infections and granuloma formation. In addition, we have observed a number of diverse autoimmune conditions in our CGD population, suggesting that patients with CGD are at an elevated risk for development of autoimmune (AI) disorders. In this report, we describe antiphospholipid syndrome (aPL), recurrent pericardial effusion, juvenile idiopathic arthritis (JIA), IgA nephropathy, cutaneous lupus erythematosus, and autoimmune pulmonary disease in the setting of CGD. The presence and type of autoimmune disease has important treatment implications for patients with CGD.
Keywords: Chronic granulomatous disease, autoimmune, antiphospholipid syndrome, IgA nephropathy, lupus, juvenile idiopathic nephropathy
Chronic granulomatous disease (CGD) is a primary immunodeficiency (PID) resulting from a defect in the multicomponent nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, which is responsible for production of bactericidal reactive oxygen species (ROS) in phagocytes. As a result, CGD patients are at increased susceptibility to certain catalase-positive bacteria and fungi, and Aspergillus species1. The primary clinical features of CGD are recurrent infections and granuloma formation. However, reports of sarcoidosis2, JIA3, IgA nephropathy4, pericardial effusion5, and severe Crohn’s-like inflammatory bowel disease6 suggest that the breadth of altered immune regulation extends beyond recurrent infections and granulomas. We propose, in addition, that CGD patients are at significant risk for development of autoimmune disease (AI), and provide a series of case reports and a review of the literature to support that hypothesis. Specifically we report here the following AI in our CGD patients; antiphospholipid syndrome (aPL), juvenile idiopathic arthritis (JIA), IgA nephropathy; steroid-responsive recurrent pericardial effusions, cutaneous lupus erythematosus, and lastly, a case with ‘geographic pulmonary lesions’, a finding we have observed in four other CGD patients.
CASE PRESENTATIONS
CASE ONE: Antiphospholipid Syndrome
A 14.5 year-old Caucasian male was diagnosed with X-linked (gp91phox-deficient) chronic granulomatous disease (CGD) following the development of Serratia marcescens abscesses of his neck and mesentery at 3 years of age. Long-term prophylaxis consisting of trimethoprim-sulfamethoxazole, itraconazole and interferon-γ (IFN-γ) was commenced. Of interest, his mother (a CGD carrier) and maternal aunt (not a carrier) were both diagnosed with discoid lupus. At 10 years, he developed cellulitis of his arm following a minor skin abrasion. Treatment with intravenous antibiotics was complicated by venous thrombosis in his affected arm, which was treated with a 3-month course of warfarin. At age 14, he described acute swelling, pain and redness of the left thigh, with no other associated symptoms, fever, or history of trauma. His laboratory tests were unremarkable, other than an ESR of 50 mm/hr (NIH range 0–25mm/hr). Doppler ultrasound revealed a left femoral deep venous thrombosis and he was treated initially with heparin, followed by warfarin anticoagulation. Four months later he was seen at our clinic with fever, bullous otitis media and severe headaches. On physical examination, he was normotensive, and his fundoscopy was unremarkable. He had a leukocytosis with a total white blood cell (WBC) of 16×109/l (neutrophils 81.4%), and ESR of 101 mm/hr. CT imaging of his head at this time was unremarkable. He was treated with levofloxacin for bullous otitis media, and maintained on warfarin 3.5mg daily with a goal INR of 3. His frontal and ocular headaches persisted, and ophthalmologic examination one month later revealed flame hemorrhages and papilledema due to raised intracranial pressure. A venous MR-angiography revealed extensive old cerebral sinus venous thrombosis involving the right sigmoid, transverse and superior sagittal sinuses (Figure 1). The patient’s headaches resolved, and he resumed normal activities. He returned two months later with extensive new clots in the left common femoral, superficial femoral, popliteal veins and a superficial vein in the left calf resulting in near-complete veno-occlusion. Extensive hematologic evaluation ruled out an underlying hereditary hypercoagulability including the absence of anti-clotting factor antibodies. Negative evaluations included APC resistance, protein S and protein C deficiency, ATIII deficiency, prothrombin G20210A mutation, methylentetrahydrofolate reductase C677T polymorphism, homocysteinemia, and β2-glycoprotein antibodies. Screening was also negative for rheumatoid factor and the following autoantibodies: anti-cardiolipin antibodies (IgG and IgM), anti-ENA, anti-dsDNA, c-ANCA, and p-ANCA. However, lupus anticoagulant, not uncommonly detected in asymptomatic CGD patients, was present, and ANA was positive. A trial of oral prednisone at 0.5 mg/kg/day (30 mg/day) was commenced. Within 24 hours, his leg swelling and pain improved dramatically. His prednisone dose was slowly tapered over several weeks with recurrence of symptoms (leg swelling, erythema, and pain on ambulation) at a dose of 5 mg daily. His prednisone dose was increased to 15mg daily with resolution of his symptoms. Repeat evaluation of his autoantibodies and inflammatory markers revealed elevated ESR and CRP (101 mm/hr and 13.6 mg/dL), and again, positive lupus anticoagulant. Thus he fulfilled the recently revised criteria for the diagnosis of aPL (2006 International Consensus Statement on an update of the classification criteria for definite antiphospholipid syndrome7. Methotrexate was instituted as a steroid-sparing agent and has allowed tapering of prednisone to 7.5mg (current weight 57.8kg) on alternate days while continuing warfarin 3.0 mg daily (INR 2.6) without further progression of thrombosis to date. Follow up imaging exams have demonstrated slow resolution of the thromboses with venous recanalization in the head and leg.
Figure 1.
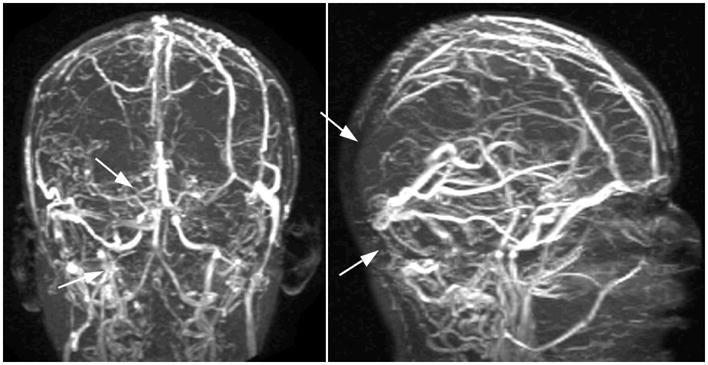
A magnetic resonance angiogram of the brain revealing filling defects (white arrows) due to extensive intracerebral venous thrombosis.
CASE TWO: Recurrent extensive pericardial effusion
Patient #2 is a 13-year-old white male diagnosed with X-linked CGD in infancy. Trimethoprim-sulfamethoxazole and itraconazole prophylaxis without IFNγ was commenced upon diagnosis. At 9 months of age, he presented with respiratory distress without fevers or any obvious sites of infection. A significant pericardial effusion was identified and pericardiocentesis performed for both diagnostic and therapeutic purposes. No etiology for the effusion could be determined and the patient was given an empiric trial of prednisone, which was effective and then tapered over several weeks. However, the pericardial effusion recurred approximately every 6 months over the next three years with similar workup, treatment and outcome. At age 7, he developed pulmonary Cryptococcus, which responded to fluconazole. A month later, he returned with fevers, malaise and dysphagia. His laboratory parameters (WBC count, electrolytes, liver, kidney and thyroid function tests) were unremarkable except for an elevated ESR (108 mm/hr) and CRP (6.7 mg/dL). A chest X-ray showed an enlarged cardiac shadow and a large pericardial effusion was confirmed by chest CT (Figure 2). An echocardiogram revealed normal cardiac structure and function. Bacterial, fungal, and viral cultures from the effusion fluid obtained from pericardiocenteses were also negative, as were cultures from his blood. The pericardial effusion resolved promptly upon recommencement of methylprednisolone over the next 5 days, and empiric antibiotics were discontinued (all cultures were negative). The patient was discharged a week later with resolution of the effusion by echocardiogram and sent home on a tapering dose of oral prednisone. On low dose maintenance alternate day prednisone he has not had any recurrences to date over more than 4 years of follow up.
Figure 2.
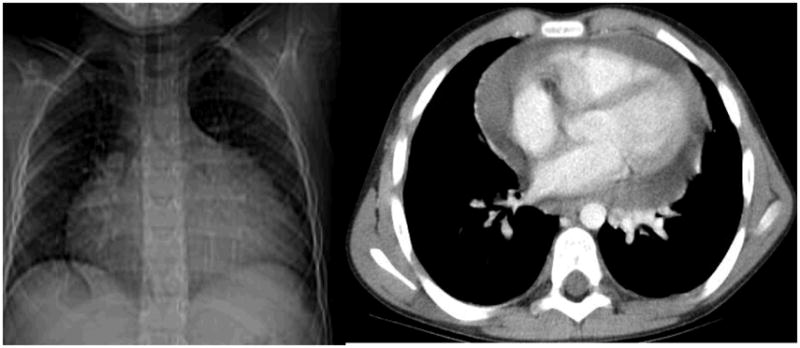
An enlarged cardiac silhouette on chest X-ray due to massive pericardial effusion confirmed on chest CT.
CASE THREE: IgA nephropathy
Patient #3 is a 17-year-old white male with X-linked CGD diagnosed at 3 years of age. Prophylaxis included trimethoprim-sulfamethoxazole and itraconazole, without IFN-γ. His past history is significant for recurrent cystic acne, Staphylococcus aureus liver abscesses, lymphadenitis, and multiple pulmonary infections (including Aspergillus, Nocardia). At age 13, during an admission to the NIH for a fungal pneumonia (treated with voriconazole), isolated hematuria and trace proteinuria were noted with a serum creatinine of 1.0 mg/dl. Significant proteinuria, 2+ blood cells in the urine, and granular and hyaline casts persisted and over the next month. His creatinine (which had a historical baseline of 0.8 mg/dl) increased to 1.9 mg/dl and he became oliguric. An extensive renal assessment was performed to evaluate for post-infectious glomerulonephritis, IgA nephropathy, vasculitis, or connective tissue disorders. ANA, anti-ENA, anti-dsDNA, p-ANCA, c-ANCA, anti-myeloperoxidase, anti-proteinase-3, RF, serum complement C3 and C4 were all within normal limits. Urinary excretion of calcium and phosphorus were within normal range. A renal biopsy revealed a focal segmental proliferative and necrotizing crescentic glomerulonephritis and mesangial IgA deposits on immunofluorescence consistent with IgA nephropathy. He was treated with prednisone 30 mg twice daily, and his creatinine rapidly improved to to 1.1 mg/dl over the next week, allowing a slow taper of oral prednisone. Two months later his creatinine had stabilized at his baseline level of 0.8 mg/dl. Over the following years, his renal function has been normal on continued low-dose, alternate-day oral prednisone 2.5 mg. We have in our cohort another patient with similar steroid-responsive renal impairment due to IgA nephropathy. In both patients, there was no family history of autoimmune diseases.
CASE FOUR: Juvenile Idiopathic Arthritis
Patient #4 is a 26-year-old white female diagnosed with p47phox-deficient CGD at birth. Her early course was remarkable for recurrent sinusitis, otitis media, and pulmonary infections. At age 15, she developed arthralgias of her elbows and hands, which progressed to bilateral involvement of her elbows, knees, and hands. Physical examination revealed erythematous macules and papules on the arms and forehead, bilateral swelling of the metacarpal and proximal interphalangeal joints, as well as numerous rheumatoid nodules. ANA, ENA, and dsDNA were unremarkable, as were cultures of blood and synovial fluid, except for an elevated RF (97 IU/mL). A biopsy of the joint showed perivascular synovitis without lymphoid aggregates. X-rays of the hands, wrists, elbows, feet and ankles revealed mild subchondral lucency at the right third metacarpal head, but no joint space loss or bone destruction. She was treated with non-steroidal anti-inflammatory medications and minocycline with only transient improvement. Etanercept 25 mg subcutaneously bi-weekly was commenced which led to resolution of her symptoms. When etanercept was discontinued for a poorly healing wound, her symptoms recurred. Treatment with naproxen and tramadol gave only transient improvement, so Etanercept was reinstituted with good response. This case is representative of one other patient with JIA in our cohort, who required gold injections in addition to plaquenil and prednisone for maintenance of disease remission.
CASE FIVE: Cutaneous Lupus Erythematosus
Patient #5 is a 21-year old white male diagnosed with X-linked CGD at 18 months of age. Long term antibiotic and antifungal prophylaxis, as well as IFNγ were initiated. At age 12, he was diagnosed with cutaneous lupus erythematosus (LE) following development of a rash on his chest. His rash improved with residual scarring after treatment with topical corticosteroid therapy. Of note, his mother, a CGD-carrier, was diagnosed with cutaneous LE. At the time of this presentation, he reported a two-month history of large annular, erythematous plaques on the chest, malar rash (Figure 3), and erythematous plaques on the scalp with associated alopecia. He denied symptoms of other organ involvement, significant diarrhoea or abdominal pain. Other than the rash, his physical exam was unremarkable. Laboratory evaluation revealed negative antibodies to cardiolipin, ds DNA, ENA, ANA, anti-Smith, anti-histone, RF, anti-Ro and –La, and lupus anticoagulant. His ESR was not elevated. A diagnostic skin biopsy again confirmed cutaneous LE. Following a week of clobetasol ointment 0.05% without significant improvement, a tapering dose of oral prednisone was commenced which resulted in marked improvement. He continues on low dose alternate day-5mg daily prednisone.
Figure 3.
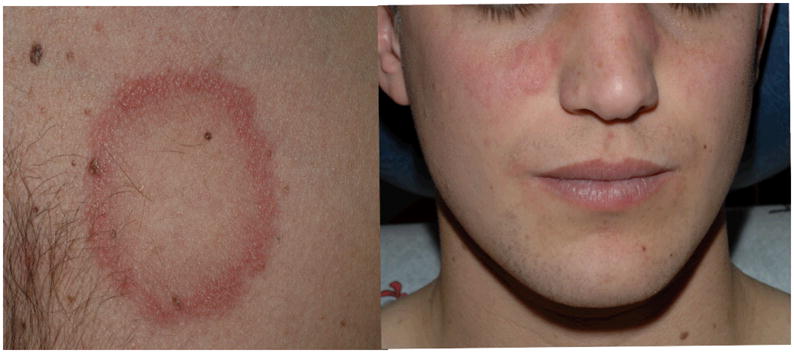
Arcuate and annular plaques with central clearing on the chest of patient 5; malar erythema and focal plaque on the right cheek.
CASE SIX: Recurrent aphthous stomatitis and pulmonary infiltrates
Patient #6 is a 14-year-old Caucasian male with p47phox-deficient CGD diagnosed at 16 months of age, following a history of lymphadenitis and staphylococcal skin abscesses. Standard CGD prophylaxis consisting of sulfamethoxazole/trimethoprim, itraconazole and IFNγ was commenced. He had a history of multiple dermatologic problems, including photosensitivity (face, arms, trunk), eczematous skin lesions, guttate psoriasis, and impetigo. Photosensitivity symptoms did not improve following cessation of trimethoprim-sulfamethoxazole, a known photosensitizer. At age 10, he developed persistent cheilitis and multiple oral ulcers. He had no evidence of active infection, malnutrition, or associated gastrointestinal complaints that can sometimes be associated with aphthous ulcers or cheilitis. Over the past 2 years, he had also developed a fluctuating, rim-enhancing, multifocal fluffy pulmonary infiltrate on chest CTs (Figure 4) without associated fevers, respiratory symptoms, or other constitutional complaints. Empiric antibacterial (levofloxacin) and antifungal therapy (itraconazole, voriconazole) had no effect on the infiltrates on repeat CT. Laboratory investigations revealed absence of leukocytosis, ESR 22–43mm/hr, and angiotensin converting enzyme (ACE) level 45–49 IU/L (16–52 IU/L). His ANA was strongly positive at 5.5 EU (0.0–0.9 EU), as was anti-cyclic citrullinated peptide (CCP) of 33 U (0–20 U). Other normal laboratory tests included CRP 0.49mg/dL, proteinase-3, anti-dsDNA, ENA, p-and c-ANCAs. We have followed three other CGD patients with similar appearance of wax-and-wane type geographic pulmonary lesions on CT (Figure 4). Biopsies from these other three CGD patients have only revealed interstitial granulomatous inflammation and lymphocytic infiltrates, and all without any evidence of infection. In our experience, treatment with empiric methotrexate in these 3 patients resulted in improvement of pulmonary disease. As patient #6 remains asymptomatic, no lung biopsies have been performed on him, nor has any specific immunosuppressive therapy been instituted. The oral ulcers are controlled on oral dexamethasone rinse twice daily.
Figure 4.
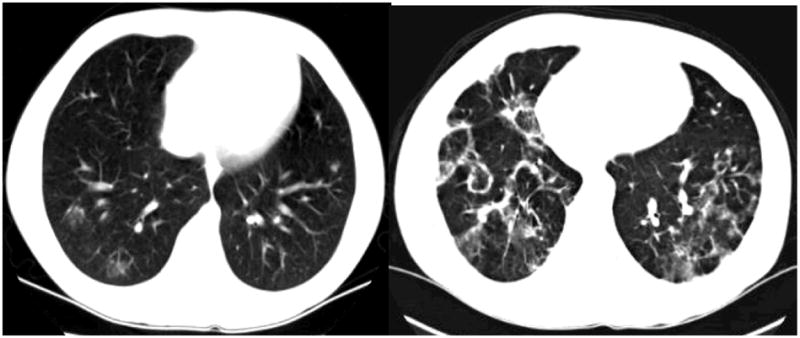
Chest CTs illustrating the fluctuating, circumscribed, rim-enhancing pulmonary infiltrates seen in Patient 6 and others.
DIFFERENTIAL DIAGNOSIS
In cases 1, 3, 4, and 5, presenting signs and symptoms suggested specific autoimmune conditions (aPL, IgA nephropathy, JIA, and LE). However, in cases 2, and 6 (pericardial effusions and fluctuating pulmonary infiltrates with oral aphthous ulcers), a clear autoimmune diagnosis could not be identified, despite the presence of some features suggestive of autoimmunity.
Venous thromboembolism in childhood is rare, and can occur in the presence of endothelial damage, blood stasis or a hypercoagulable state8. Although the first episode of thrombosis in patient #1 occurred in the setting of a venous catheter, the subsequent episodes were not related to any identified triggering events. Lupus anticoagulant (LA), a known risk factor for thrombosis, was present on repeated evaluations of this patient. Although LA has been reported in CGD patients as well as carriers9, recurrent thrombosis has not been previously described in CGD patients. The 2006 revised aPL classification criteria requires the fulfillment of at least one clinical feature (vascular thrombosis or pregnancy morbidity) and one laboratory parameter (positive anticardiolipin antibodies and lupus anticoagulant) on equal or more than two occasions at least 12 weeks apart7.
Pericardial effusion is also uncommon in the pediatric population. Known causes include previous cardiac surgery, infections, connective tissue disorders and malignancies10. Patient #2’s rapid response to prednisone is consistent with pericardial effusions of “sterile” inflammatory origin in childhood in contrast to a more prolonged course in bacterial pericarditis10. Rare cases of cardiac and pericardial involvement in CGD with granulomatous infiltration of the epicardium and myocardium and development of pericardial effusion have previously been reported5, 11, 12.
IgA nephropathy is an immune-complex-mediated glomerulonephritis (GN) defined histopathologically by mesangial IgA deposit13. To our knowledge, there is one previously reported case of IgA nephropathy in a CGD patient, who concurrently had multiple soft tissue abscesses with Staphylococcus aureus, thus postulating the role of infections triggering the IgA immune complex formation14. Sato et al. demonstrated abnormal phagocytic activity in polymorphonuclear leukocytes in 72% of patients with IgA nephropathy, implicating the role of neutrophils in the clearance of IgA-containing immune complexes and therefore preventing their accumulation in the glomeruli 15.
The differential diagnosis of new onset polyarthritis includes post-infectious (Reiter’s syndrome), rheumatic fever, in association with inflammatory bowel disease, and psoriatic arthritis. However, the association of symmetrical polyarthritis with multiple large and small joints, the presence of rheumatoid nodules, and rheumatoid factor (RF) seropositivity is consistent with JIA16. JIA was previously reported in one CGD patient, whose clinical course was similar to that of patient #4 reported here4. Cutaneous LE is well described in CGD carriers and more recently, in CGD patients9, 17, 18. Of note, the diagnosis of cutaneous lupus in his mother suggests a shared genetic susceptibility.
The differential diagnosis of pulmonary infiltrates in the context of CGD includes bacterial, fungal or viral pathogens. However, patient #6’s characteristic CT pulmonary appearance or pattern, which did not respond to, or have any correlations with empiric antibiotic and antifungal treatment or leukocytosis, suggested a non-infective etiology. Of note, similar rim-enhancing lesions with a characteristic wax-and-wane pattern over a period of years, have been observed in 3 other CGD patients. Over time, these patients reported dyspnea on exertion and demonstrated oxygen desaturation after exercise. They were without leukocytosis, fevers or chills, and lung biopsies failed to demonstrate any infectious pathogens. Treatment with empiric methotrexate in these 3 patients resulted in improvement of pulmonary disease. This constellation of photosensitive rash, oral aphthous ulcers sensitive to oral dexamethasone elixir, and distinct ‘geographic’ pulmonary lesions in patient #6, in the absence of autoantibodies or other organ involvement, do not fit within the criteria for any known autoimmune diseases.
LABORATORY AND OTHER TESTING/PROCEDURES
The diagnosis of CGD in each case was confirmed by dihydrorhodamine 123 (DHR) assay, Western blot, and in all but patient #4 (p47 deficiency), confirmed with genomic sequencing mutation analysis. Laboratory and other testing specific for each case has been described above.
DISCUSSION OF PATHOGENESIS
There is an increasing awareness of autoimmune phenomena in primary immunodeficiencies (PID), particularly PID associated with lymphoid and humoral defects19. The most frequently described immune-related inflammatory disorders in CGD are inflammatory bowel disease and lupus erythematosus. CGD-associated colitis is clinically and histologically similar to Crohn’s disease, but appears distinct from the typical granulomatous lesions causing obstruction in the gastrointestinal and urogenital system6. Discoid lupus is most commonly reported in female carriers of X-linked CGD, although it has been described in CGD patients as well9, 20. Isolated case reports of other immune-mediated phenomena in CGD patients include systemic lupus erythematosus (SLE) 4, 21, autoimmune thrombocytopenia22, idiopathic thrombocytopenic purpura23, rheumatoid arthritis3, eosinophilic cystitis24, IgA nephropathy14, sarcoidosis2 and celiac disease coexisting with pulmonary hemosiderosis 25.
Classically, the definition of an autoimmune disease requires the detection of an autoimmune response (autoantibody or cell-mediated), the identification of a corresponding autoantigen, and induction of such response and disease in experimental animals26. However, more recently, there has been a broadening list of parameters for diagnosis of an autoimmune disorder 27, in particular, the inclusion of clinical response to immunosuppression as a diagnostic criterion. The most consistent feature of the cases presented here, with the exception of case #6, is the remarkable response to immunosuppressive therapy. Also striking, is the relative rarity of these conditions in the general population, particularly in childhood. In CGD patients followed by our clinic, thirteen have an AI disorder, comprising over 10% of our cohort. This does not include patients who have inflammatory bowel disease, which is not uncommon in CGD patients, occurring in some 30 percent of the patients followed at NIH6. Given that our program receives referrals of patients with more complex problems associated with their CGD, it is possible that our patient population may not be representative of the general clinical immunology community. It would be of great value to ascertain the prevalence of autoimmune manifestations in CGD from all available registry databases that track CGD patients.
We believe that these autoimmune disorders are not simply part of the inflammatory ‘spectrum’ seen in CGD, as these unusual presentations are outside the range of what is commonly seen in this disease. Nor are they isolated phenomena coincidentally observed in CGD patients, but rather, the new hypothesis that we propose is that patients with CGD are at increased risk of developing any of a large spectrum of known and unknown types of specific autoimmune diseases. The type of autoimmune disease that develops likely has a specific genetic basis or susceptibility risk, and/or environmental triggers. Iatrogenic factors that could impact on development or exacerbation of autoimmunity include trimethoprim-sulfamethoxazole, itraconazole, or IFN-γ. In this context, we propose that CGD patients who satisfy established diagnostic criteria for a specific autoimmune disease such as IgA nephropathy, sarcoidosis, lupus erythematosus, or juvenile idiopathic arthritis actually have these disorders and are not just a mirror phenotype that represents part of the spectrum of CGD inflammation. Furthermore, when viewed from this paradigm it provides a basis for viewing the recurrent thrombosis or antiphospholipid syndrome (patient #1), the recurrent pericardial effusion syndrome (patient #2), or the recurrent aphthous ulcers with fluctuating pulmonary infiltrates (patient #6) as autoimmune diseases which respond to immunosuppressive therapy.
FINAL DIAGNOSIS AND TREATMENT-MANAGEMENT PLAN
Chronic granulomatous disease acquired its original nomenclature from the signature pathognomonic clinical feature of this disorder, the widespread multi-organ occurrence of characteristic multinucleated giant cell granulomas. When these are manifested in and around hollow organs such as in the gastrointestinal or genitourinary systems, it causes pyloric/small bowel obstruction and ureter/bladder outlet obstruction which are rapidly steroid responsive. Another generalized manifestation of this exuberant granuloma forming process includes impairment of wound healing that paradoxically is improved with steroids. Furthermore, studies in CGD knockout mice of both the gp91phox deficient X-linked or p47phox-deficient autosomal recessive varieties, have demonstrated that inhalation of sterilized non-viable Aspergillus spores can cause acute pulmonary decompensation from rapid granuloma formation that is not seen in normal mice28, 29, and injection of a sterile stimulus of inflammation into the peritoneum of CGD mice elicits a robust cellular inflammation response that is an order of magnitude greater than seen with normal mice1. Recent studies have demonstrated that CGD patient neutrophils have delayed apoptosis associated with diminished production of anti-inflammatory mediators relative to normal neutrophils, providing another mechanism by which inflammation in CGD is prolonged with impaired resolution30. Heretofore, the prevailing paradigm has been that all manifestations of inflammation in CGD are just extremes in the spectrum of CGD granuloma formation, including uncommon presentations as well as those which fulfill specific diagnostic criteria for well characterized autoimmune disorders. Some types of inflammation, such as those characteristic of Crohn’s-like inflammatory bowel disease in its most mild and low-dose steroid responsive form, occur with such high frequency in CGD patients (more than 20–30% of patients) that under the prevailing paradigm this inflammatory bowel disease has been viewed as part of the CGD granuloma spectrum. However, a subset of CGD patients with inflammatory bowel disease have severe Crohn’s-like colonic disease associated with extensive bloody diarrhea, strictures and fistula formation. It is not clear whether these patients actually have Crohn’s disease triggered by CGD, or have a CGD-specific process.
Recently, it has been shown that T lymphocytes exhibit a very small burst production of reactive oxygen products in response to mitogen stimulus, and that a portion of this burst is missing in mouse and human CGD T lymphocytes31. In the murine CGD model, CD4+ T lymphocytes have a strong bias toward a Th1 pattern of cytokine production when differentiated to an effector phenotype31, and when challenged with pulmonary aspergillosis, led to excessive inflammation and a dominant production of IL1732. This further raises the possibility that CGD is a risk factor in the development of Th1 types of autoimmune diseases. While some long term medications that CGD patients receive, in particular IFNγ, may influence this risk, data from CGD mouse models not on medications has undeniably demonstrated that CGD is associated with excessive inflammation. Our current study lacks the statistical power to address the potential contribution of any particular medication on the development of autoimmunity in CGD. We hypothesize that all CGD patients are at significantly increased risk of developing autoimmune disease, but that the actual expression of a particular autoimmune disease is likely due to a combination of familial genotype and environmental exposure factors.
The new paradigm we propose is that manifestations of autoimmune diseases in CGD which meet established diagnostic criteria for a particular autoimmune disease, be regarded as such and not solely regarded as being only part of a broad spectrum of granuloma formation associated with CGD. This has important therapeutic implications in that, in addition to corticosteroid therapy, appropriate steroid-sparing therapies for that autoimmune disease should also be initiated to treat the disease pathology. Without this paradigm shift there seem to be significant barriers to consideration of the use of potent steroids and steroid-sparing immunosuppressant therapy such as anti-TNFα agents, methotrexate, azathioprine or even cyclosporine A or tacrolimus in CGD because of the fear that the increased risk of infection in a CGD patient would outweigh the potential benefits. While there probably is an increased infection risk associated with use of potent non-steroidal anti-inflammation agents in CGD patients, those CGD patients with autoimmune disease that do not respond to prednisone or who require more than low dose alternate day prednisone for symptom control should be considered for treatment with non-steroidal agents with established and known efficacy in treatment of the specific autoimmune disorder, albeit with close clinical monitoring.
SUMMARY
Although infections and granuloma formation are undoubtedly the most common manifestations in CGD patients, there is a significant subset of CGD patients who experience a broad variety of autoimmune diseases, which respond well to immunosuppressive therapy. To explain the diversity of autoimmune disorders observed in CGD patients, we suspect that CGD might itself serve as a genetic cofactor, which lowers the threshold for overt phenotypic development of autoimmunity, and where the particular type of autoimmune disease manifested is dictated by the genetic and/or environmental background. This is a new paradigm that will require additional observational input with statistical assessment, but it has specific implications for clinical management. Maintaining a high index of suspicion for autoimmune disorders in patients with CGD will minimize unnecessary invasive procedures, and allow early institution of appropriate therapy to control inflammation and reduce risks of long-term complications. Furthermore, reporting of additional cases will allow for identification of common patterns of disease and will held direct appropriate studies and eventually lead to a better understanding of the pathogenesis of autoimmune disorders in patients with CGD.
CLNICAL PEARLS
Autoimmune diseases should be considered early in the differential diagnosis of CGD patients, particularly in the absence of identifiable microbial pathogens
Supportive laboratory parameters should be actively sought for, for example, serological markers of autoantibodies, RF, and serum angiotensin converting enzyme
Despite the immune-compromised state of CGD patients, though it may appear paradoxical, prednisone and/or other immunosuppressive drugs are often beneficial or necessary in these autoimmune conditions and appropriate therapy should be not be withheld because of concern over risk of infection.
Abbreviations
- CGD
chronic granulomatous disease
- AI
autoimmune
- aPL
antiphospholipid syndrome
- JIA
juvenile idiopathic arthritis
- IFN-γ
interferon gamma
- ANA
anti-nuclear antibody
- RF
Rheumatoid factor
References
- 1.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 2.De Ravin SS, Naumann N, Robinson MR, Barron KS, Kleiner DE, Ulrick J, et al. Sarcoidosis in chronic granulomatous disease. Pediatrics. 2006;117:e590–5. doi: 10.1542/peds.2005-1349. [DOI] [PubMed] [Google Scholar]
- 3.Lee BW, Yap HK. Polyarthritis resembling juvenile rheumatoid arthritis in a girl with chronic granulomatous disease. Arthritis Rheum. 1994;37:773–6. doi: 10.1002/art.1780370524. [DOI] [PubMed] [Google Scholar]
- 4.Schmitt CP, Scharer K, Waldherr R, Seger RA, Debatin KM. Glomerulonephritis associated with chronic granulomatous disease and systemic lupus erythematosus. Nephrol Dial Transplant. 1995;10:891–5. [PubMed] [Google Scholar]
- 5.Macedo F, McHugh K, Goldblatt D. Pericardial effusions in two boys with chronic granulomatous disease. Pediatr Radiol. 1999;29:820–2. doi: 10.1007/s002470050704. [DOI] [PubMed] [Google Scholar]
- 6.Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O’Brien S, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114:462–8. doi: 10.1542/peds.114.2.462. [DOI] [PubMed] [Google Scholar]
- 7.Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS) J Thromb Haemost. 2006;4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x. [DOI] [PubMed] [Google Scholar]
- 8.Turpie AG, Chin BS, Lip GY. Venous thromboembolism: pathophysiology, clinical features, and prevention. Bmj. 2002;325:887–90. doi: 10.1136/bmj.325.7369.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79–84. doi: 10.1111/j.1365-2249.2007.03321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mok GC, Menahem S. Large pericardial effusions of inflammatory origin in childhood. Cardiol Young. 2003;13:131–6. doi: 10.1017/s104795110300026x. [DOI] [PubMed] [Google Scholar]
- 11.Ferrans VJ, Rodriguez ER, McAllister HA., Jr Granulomatous inflammation of the heart. Heart Vessels Suppl. 1985;1:262–70. doi: 10.1007/BF02072406. [DOI] [PubMed] [Google Scholar]
- 12.Grumach AS, Jacob CM, Stolf NG, Maksoud JG, Carneiro-Sampaio MM. Constrictive pericarditis as a complication of chronic granulomatous disease of childhood. Rev Hosp Clin Fac Med Sao Paulo. 1987;42:30–2. [PubMed] [Google Scholar]
- 13.Haas M. Histology and immunohistology of IgA nephropathy. J Nephrol. 2005;18:676–80. [PubMed] [Google Scholar]
- 14.Narsipur SS, Shanley PF. IgA nephropathy in a patient with chronic granulomatous disease. J Nephrol. 2002;15:713–5. [PubMed] [Google Scholar]
- 15.Sato M, Kinugasa E, Ideura T, Koshikawa S. Phagocytic activity of polymorphonuclear leucocytes in patients with IgA nephropathy. Clin Nephrol. 1983;19:166–71. [PubMed] [Google Scholar]
- 16.Borchers AT, Selmi C, Cheema G, Keen CL, Shoenfeld Y, Gershwin ME. Juvenile idiopathic arthritis. Autoimmun Rev. 2006;5:279–98. doi: 10.1016/j.autrev.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 17.Cordoba-Guijarro S, Feal C, Dauden E, Fraga J, Garcia-Diez A. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409–11. doi: 10.1046/j.1468-3083.2000.00113.x. [DOI] [PubMed] [Google Scholar]
- 18.Sillevis Smitt JH, Bos JD, Weening RS, Krieg SR. Discoid lupus erythematosus-like skin changes in patients with autosomal recessive chronic granulomatous disease. Arch Dermatol. 1990;126:1656–8. doi: 10.1001/archderm.126.12.1656b. [DOI] [PubMed] [Google Scholar]
- 19.Etzioni A. Immune deficiency and autoimmunity. Autoimmun Rev. 2003;2:364–9. doi: 10.1016/s1568-9972(03)00052-1. [DOI] [PubMed] [Google Scholar]
- 20.Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376–9. doi: 10.1111/j.1525-1470.1986.tb00544.x. [DOI] [PubMed] [Google Scholar]
- 21.Manzi S, Urbach AH, McCune AB, Altman HA, Kaplan SS, Medsger TA, Jr, et al. Systemic lupus erythematosus in a boy with chronic granulomatous disease: case report and review of the literature. Arthritis Rheum. 1991;34:101–5. doi: 10.1002/art.1780340116. [DOI] [PubMed] [Google Scholar]
- 22.Trelinski J, Chojnowski K, Kurenko-Deptuch M, Kasznicki M, Bernatowska E, Robak T. Successful treatment of refractory autoimmune thrombocytopenia with rituximab and cyclosporin A in a patient with chronic granulomatous disease. Ann Hematol. 2005;84:835–6. doi: 10.1007/s00277-005-1094-5. [DOI] [PubMed] [Google Scholar]
- 23.Matsuura R, Kagosaki Y, Tanaka Y, Kashiwa H, Sakano T, Kobayashi Y, et al. A female case of chronic granulomatous disease (CGD) associated with chronic idiopathic thrombocytopenic purpura. Hiroshima J Med Sci. 1980;29:83–6. [PubMed] [Google Scholar]
- 24.Barese CN, Podesta M, Litvak E, Villa M, Rivas EM. Recurrent eosinophilic cystitis in a child with chronic granulomatous disease. J Pediatr Hematol Oncol. 2004;26:209–12. doi: 10.1097/00043426-200403000-00014. [DOI] [PubMed] [Google Scholar]
- 25.Hartl D, Belohradsky BH, Griese M, Nicolai T, Krauss-Etschmann S, Roos D, et al. Celiac disease and pulmonary hemosiderosis in a patient with chronic granulomatous disease. Pediatr Pulmonol. 2004;38:344–8. doi: 10.1002/ppul.20059. [DOI] [PubMed] [Google Scholar]
- 26.Witebsky E. Concept of autoimmune disease. Ann N Y Acad Sci. 1966;135:443–50. doi: 10.1111/j.1749-6632.1966.tb45493.x. [DOI] [PubMed] [Google Scholar]
- 27.Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky’s postulates revisited) Immunol Today. 1993;14:426–30. doi: 10.1016/0167-5699(93)90244-F. [DOI] [PubMed] [Google Scholar]
- 28.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–8. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–9. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 30.Brown JR, Goldblatt D, Buddle J, Morton L, Thrasher AJ. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD) J Leukoc Biol. 2003;73:591–9. doi: 10.1189/jlb.1202599. [DOI] [PubMed] [Google Scholar]
- 31.Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol. 2004;5:818–27. doi: 10.1038/ni1096. [DOI] [PubMed] [Google Scholar]
- 32.Romani L, Fallarino F, De Luca A, Montagnoli C, D’Angelo C, Zelante T, et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–5. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]