

Abstract
Epstein–Barr virus (EBV) is the causative agent of infectious mononucleosis and a risk factor for developing a variety of lymphomas and carcinomas. EBV nuclear antigen 1 (EBNA1) is the only viral protein found in all EBV-related malignancies. It plays a key role in establishing and maintaining the altered state of cells transformed with EBV. EBNA1 is required for a variety of functions, including gene regulation, replication and maintenance of the viral genome, but the regulation of EBNA1's functions is poorly understood. We demonstrate that phosphorylation affects the functions of EBNA1. By using electron-transfer dissociation tandem mass spectrometry, ten specific phosphorylated EBNA1 residues were identified. A mutant derivative preventing the phosphorylation of all ten phosphosites retained the unusually long half-life and the ability to translocate into the nucleus of wild-type EBNA1. This phosphorylation-deficient mutant, however, had a significantly reduced ability to activate transcription and to maintain EBV's plasmids in cells.
INTRODUCTION
Epstein–Barr virus (EBV) is a double-stranded DNA tumour virus in the subfamily Gammaherpesvirinae (Munz, 2004). This lymphotropic virus latently infects >95 % of the human population worldwide. It is the causative agent of infectious mononucleosis and a risk factor for a variety of malignancies, including Burkitt's lymphoma, Hodgkin's disease and nasopharyngeal carcinoma (Crawford, 2001; Young & Rickinson, 2004). EBV induces and maintains proliferation of infected B cells and, in doing so, predisposes the cell to malignant transformation.
EBV nuclear antigen 1 (EBNA1) is the only viral protein found in all EBV-related cancers (Leight & Sugden, 2000). The N terminus of EBNA1 consists of a glycine–glycine–alanine-rich (GGA) region that inhibits both proteasome-mediated degradation of the EBNA1 protein and translation of EBNA1 mRNA (Fahraeus, 2005; Levitskaya et al., 1997; Yin et al., 2003). An EBNA1 derivative lacking this region still maintains the ability to support replication and transcription in cell culture (Lee et al., 1999; Yates & Camiolo, 1988; Yates et al., 1985). EBNA1 contains two glycine–arginine-rich linking regions (GR-1 and GR-2), which allow EBNA1 dimers bound to DNA to associate with other DNA-bound EBNA1 dimers and thereby loop intervening DNA sequences in cis or link two DNA molecules in trans (Frappier & O'Donnell, 1991a; Goldsmith et al., 1993; Mackey et al., 1995; Su et al., 1991). Within GR-1 resides unique region 1 (UR1), aa 65–89, which comprises a transcription-activation domain (Kennedy & Sugden, 2003; Wu et al., 2002). The C terminus of EBNA1 contains a dimerization domain and a DNA-binding domain.
EBNA1 is necessary for latent replication of the EBV genome through site-specific binding to the origin of plasmid replication (oriP) and is required for maintenance of the oriP plasmid (Leight & Sugden, 2000). It is also involved in regulating latent gene expression (Gahn & Sugden, 1995; Sugden & Warren, 1989; Yates et al., 1984). Although it has a variety of functions, EBNA1 has no known enzymic activity. It is hypothesized that EBNA1 carries out its various functions through protein–protein interactions. For example, EBNA1 binds to components of the origin-recognition complex (ORC) (Dhar et al., 2001; Ritzi et al., 2003), suggesting that this interaction is important for EBNA1's replication function. Also, the localization of EBNA1 may be determined through protein–protein interactions. Importin-α (Kim et al., 1997) and karyopherin-α1 (Ito et al., 2000), known nuclear transport factors, interact with EBNA1.
These interactions and others may be influenced by EBNA1's post-translational modifications. EBNA1 is phosphorylated when expressed in human and insect cells (Frappier & O'Donnell, 1991b; Hearing & Levine, 1985; Polvino-Bodnar et al., 1988), but the details of this modification, including the sites of phosphorylation and the biological importance of this modification, are not well understood. Detectable levels of phosphorylated EBNA1 are found following purification of EBNA1 from lymphoblastoid cell lines (Hearing & Levine, 1985). EBNA1 expressed in Sf-9 insect cells after baculovirus expression contains detectable levels of phosphoserine (Frappier & O'Donnell, 1991b), potentially at residues found between aa 325 and 376 (Shire et al., 2006).
To determine the specific sites of EBNA1's phosphorylation, we employed electron-transfer dissociation tandem mass spectrometry (ETD-MS/MS) (Coon et al., 2004; Good et al., 2007; Swaney et al., 2007; Syka et al., 2004). This new MS-based method for fragmenting peptides along the peptide backbone retains post-translational modifications, frequently allowing both sequence assignment and determination of the exact site of modification. By using newly developed murine monoclonal antibodies (mAbs) against EBNA1 (Duellman & Burgess, 2006, 2009) to immunoprecipitate EBNA1 from a Burkitt's lymphoma cell line in combination with multi-dimensional chromatography and ETD-MS/MS, we identified ten sites of phosphorylation on EBNA1. Analysis of an EBNA1 derivative unable to be phosphorylated at these ten sites indicated that modification of these sites in wild-type EBNA1 probably regulates its functions. This EBNA1 derivative was reduced in activation of transcription and in its ability to maintain EBV-derived plasmids.
METHODS
Cell culture.
BJAB is an EBV-negative Burkitt's lymphoma cell line (Menezes et al., 1975). BJAB8 has been described previously (Wang et al., 2006); it stably expresses EBNA1 and hygromycin B phosphotransferase. The EBNA1 derivative expressed in this clone originates from p1553 (Aiyar & Sugden, 1998) and lacks the majority of the GGA-repeat region. Cells were grown in RPMI 1640 medium (Invitrogen) supplemented with 10 % fetal bovine serum (FBS), 200 U penicillin ml−1, 200 μg streptomycin ml−1 and 400 μg hygromycin ml−1 (Calbiochem). BJAB clones expressing EBNA1P10A were grown in RPMI 1640 medium supplemented with 10 % FBS, 200 U penicillin ml−1, 200 μg streptomycin ml−1 and 1 μg puromycin ml−1. Cells were harvested by centrifugation (1500 r.p.m. for 5 min), washed with PBS and centrifugation was repeated. Cell pellets were stored at −80 °C. Human embryonic kidney 293T cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10 % FBS, 200 U penicillin ml−1 and 200 μg streptomycin ml−1. All cells were grown at 37 °C in a humidified 5 % CO2 atmosphere.
Lysate preparation.
Cell pellets from 2×108 cells were resuspended in lysis buffer [50 mM Tris/HCl (pH 7.9), 2 mM MgCl2, 50 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 10 % glycerol, 0.5 % Nonidet P-40] plus EDTA-free protease inhibitor cocktail (Roche), phosphatase inhibitor cocktail set 1 (Calbiochem) and benzonase nuclease (Novagen) at concentrations according to the manufacturers' instructions. Cells were incubated on ice for 10 min and microsonicated (three bursts at 20 % amplitude for 10 s on ice). Lysate was clarified by centrifugation for 5 min at 14 000 r.p.m.
Immunoprecipitation assay.
Purified mAb was conjugated to cyanogen bromide-activated Sepharose 4B at a concentration of 2.5 mg mAb (ml swollen gel)−1 as described previously (Thompson et al., 1992). Resin composed of a cocktail of three mAbs (1EB6, 1EB12 and 3EB13) was applied to a SigmaPrep spin column (Sigma-Aldrich) and equilibrated twice with lysis buffer plus protease and phosphatase inhibitors with two column volumes (CV) of buffer. Cell lysate (2×108 cells in 500 μl buffer) was added to 50 μl resin and incubated with shaking at 23 °C for 30 min. Protein-bound resin was collected by centrifugation (1000 r.p.m. for 30 s). The resin was washed with two buffer conditions: twice with 2 CV lysis buffer plus protease and phosphatase inhibitors and twice with 2 CV Tris/HCl (pH 7.9), 0.1 mM EDTA (TE buffer) and 0.25 M NaCl plus protease and phosphatase inhibitors.
Denaturation and digestion.
EBNA1 was denatured and reduced on resin by incubation in 4 M GuSCN, 0.2 M Tris/HCl (pH 7.9), 5 mM Tris (2-carboxyethyl)phosphine (TCEP) for 2 h at 37 °C. The proteins were alkylated by addition of 10 mM iodoacetamide for 15 min in the dark at 23 °C. The sample was diluted to 0.5 M GuSCN and maintained at 0.2 M Tris/HCl (pH 7.9). LysC protease (250 ng; Princeton Separations) was incubated with the sample (approx. 400 ng target protein in a 325 μl reaction) for 18 h at 37 °C. The complete LysC digest was followed by an incomplete trypsin digest with 500 ng trypsin (Pierce) for 4 h at 37 °C. The sample was collected by centrifugation at 1000 r.p.m. for 1 min and the digestion was stopped by addition of acetic acid to pH 3. The sample was desalted by using a 100 mg C18 Sep-Pak cartridge (Waters), lyophilized to dryness and stored at −20 °C.
Conversion to methyl esters.
Lyophilized peptides were converted to their corresponding methyl esters by the addition of 2 M methanoic acid, which was prepared by the drop-wise addition of 160 μl acetyl chloride to 1 ml anhydrous methanol (both from Alltech). The reaction was mixed for 5 min and 300 μl of the resulting solution was added to the dried peptides. The sample was allowed to react for 2 h at 23 °C and then lyophilized to dryness.
Immobilized metal-affinity chromatography (IMAC).
The sample was resuspended in 70 μl reconstitution buffer (1 : 1 : 1 methanol/acetonitrile/water in 0.01 % acetic acid) followed by iron-immobilized IMAC as described elsewhere (Ndassa et al., 2006). Briefly, a 100 μm inner diameter (ID) column (Polymicro Technologies Inc.) packed with Poros MC-20 particles (Applied Biosystems) was connected via inline microfilters (Upchurch) to a 100 μm ID fused silica capillary. The column was conditioned in four steps of rinsing with: (i) 50 mM EDTA at a flow rate of 20 μl min−1 for 5 min; (ii) purified water at a flow rate of 20 μl min−1 for 5 min; (iii) 100 mM FeCl3 at a flow rate of 20 μl min−1 for 5 min; (iv) reconstitution buffer at a flow rate of 20 μl min−1 for 5 min, and then the flow was restricted to a rate of 1–2 μl min−1. The sample was then loaded onto the IMAC column at a flow rate of 1–2 μl min−1. The column was rinsed with reconstitution buffer for 2 min at a flow rate of 1–2 μl min−1, then for 5 min at 20 μl min−1, and lastly with 0.01 % acetic acid for 3 min at a flow rate of 20 μl min−1. The sample was eluted directly onto a pre-column (50 μm ID) packed with C18 particles to a length of 5 cm and attached to the outlet of the IMAC column (Alltech) with 50 mM sodium phosphate buffer at pH 8.0 for 1 h at a flow rate of 0.5 μl min−1.
Nanoflow HPLC (nHPLC).
The pre-column was connected directly to an analytical column (50 μm ID) with an integrated tip packed with C18 particles to a length of 7 cm (Martin et al., 2000). The phosphopeptide mixture was separated chromatographically via a 0–50 % buffer B linear gradient in 50 min, where buffer A was 100 mM acetic acid and buffer B was 70 % acetonitrile, 100 mM acetic acid.
MS.
Samples were analysed on an ETD-enabled linear ion trap–orbitrap hybrid mass spectrometer (McAlister et al., 2007, 2008; Williams et al., 2007). All MS1 scans were performed in the orbitrap followed by MS2 interrogation via ETD of the five most abundant precursors in the linear ion trap. For all experiments, an MS/MS automatic gain control target of 10 000 ions was used, precursors were excluded dynamically for 30 s and only peptides with assigned charge states ≥2 were selected for MS/MS interrogation.
MS data analysis.
Spectra generated from the experiments were searched against the EBNA1 protein sequence from p1553 by using the Open Mass Spectrometry Search Algorithm (omssa) (Geer et al., 2004). For all searches, parameters were set to consider a static modification of +57 Da on cysteine residues (carbamidomethylation) and a differential modification of +16 Da on methionine and cysteine residues, di-iodination of tyrosine residues, and phosphorylation on threonine, tyrosine and serine residues. All of the phosphorylation sites found were validated manually by spectral interrogation.
Electrophoresis and Western blotting.
Proteins were separated by electrophoresis using 4–12 % Bis–Tris NuPAGE polyacrylamide gels (Invitrogen). Western blots to detect EBNA1 from lymphoma cell lines used the secondary antibody goat anti-mouse IgG, human serum-adsorbed, conjugated to horseradish peroxidase (KPL) and enhanced chemiluminescent (ECL) substrate (Pierce) for detection. Prestained molecular mass markers (MultiMark; Invitrogen) were included on all gels. MagicMark molecular mass markers (Invitrogen) were included on the gels for ECL detection. c-Myc was detected by using c-Myc (N-262) antibody sc-764 (Santa Cruz Biotechnology).
PCR mutagenesis.
All ten of the determined EBNA1 phosphosites were mutated to alanine to generate EBNA1P10A. The EBNA1 sequence from p1553 was ligated into the pT7Blue-2 blunt cloning vector (Novagen). PCR mutagenesis primers were designed to contain the alanine mutations, as listed in Supplementary Table S1 (available in JGV Online). PCRs using Vent polymerase (New England Biolabs) were carried out as follows: one cycle at 95 °C for 5 min; 20 cycles of 95 °C for 30 s, annealing-temperature gradients between 65 and 80 °C as determined for each primer set for 1 min, and 72 °C for 5 min; and one cycle of 72 °C for 10 min. PCR products were digested with DpnI for 2 h at 37 °C. PCR samples that showed new DNA products of the correct size were transformed into Escherichia coli DH5α cells. DNA was isolated and sequenced at the University of Wisconsin Biotechnology Center DNA sequencing facility.
Generation of a stable cell line expressing EBNA1P10A.
EBNA1P10A was PCR-amplified with primers that added an N-terminal NotI site (5′-GAGCGGCCGCATGTCTGACGAGGGGCCAGGT) and a C-terminal XhoI site and two stop codons (5′-TACTCGAGTCATCACTCCTGCCCTTCCTCACCC). This fragment was ligated into the parent retroviral vector p3048, pCMMP-MCS-ires-puro (Lee & Sugden, 2008). Constructs were amplified in DH5α cells and confirmed by DNA sequencing. EBNA1P10A vector was transfected into 293T cells along with other DNAs necessary for retroviral expression (p2842, providing VSV-G; p2843, providing Gag/Pol; p1238, providing NF-κB). For each 100 mm plate of cells, 10 μg DNA was added to 500 μl Opti-MEM (Invitrogen) and combined with 40 μg polyethyleneimine in 500 μl Opti-MEM. After incubation at 23 °C for 25 min, the solution was added to 4 ml DMEM supplemented with 200 U penicillin ml−1 and 200 μg streptomycin ml−1, without FBS, and added to cells at 37 °C for 2 h. Then, 5 ml DMEM supplemented with 200 U penicillin ml−1, 200 μg streptomycin ml−1 and 20 % FBS was added to the cells and they were allowed to grow. After 24 h, medium was changed to DMEM, 10 % FBS, 200 U penicillin ml−1, 200 μg streptomycin ml−1and 50 mM HEPES (Invitrogen). In order to infect BJAB cells with the retrovirus, 4×106 BJAB cells were co-cultivated with the transfected 293T cells for 24 h, then removed. After 48 h, BJAB cells were plated by limiting dilution under selection with 1 μg puromycin ml−1 in order to obtain clonal populations of cells. Two clones, 40 and 44, were selected for further analysis.
Expression-level determination.
A 2-fold serial dilution of whole-cell lysate from BJAB8 and the P10A clones was performed. BJAB8 cell numbers started at 2×106 cells ml−1, while the P10A clone dilutions started at 5×105 cells ml−1. The dilution was analysed by quantitative Western blotting (Bergendahl et al., 2003), probed with anti-EBNA1 mAb 1EB12 labelled fluorescently with Alexa Fluor 647 (Invitrogen).
Protein stability.
Protein synthesis was blocked with 100 μg cycloheximide (CHX) ml−1, whereas control cells were treated with DMSO. Aliquots of treated cells were taken at time points (0, 4, 8, 24 and 48 h) and live cells were separated from dead cells by using a Ficoll-Paque gradient (GE Healthcare). Whole-cell lysate was prepared from an equal number of live cells and analysed for EBNA1 or EBNA1P10A expression by Western blotting, probed with mAb 1EB12.
Cytoplasmic and nuclear fractionation.
NE-PER nuclear and cytoplasmic extraction reagents (Pierce) were used to isolate cytoplasmic and nuclear proteins. Sample preparation was done according to the manufacturer's instructions. Samples were analysed by Western blotting. Nuclear preparations were confirmed with anti-Oct1 mouse mAb (sc-8024) and cytoplasmic samples were confirmed with anti-Hsp90 rabbit polyclonal antibody (sc-7947) (both from Santa Cruz Biotechnology).
Luciferase reporter assay.
The dual-luciferase reporter assay system (Promega) was used to determine the transcription-activation function of EBNA1 and EBNA1P10A. The appropriate stable cell line was electroporated with 200 ng FR-tk-Luc p985, described previously (Middleton & Sugden, 1992), 100 ng Renilla luciferase transfection control p2517, 1 μg ZsGreen expression vector p3031 and 8 μg pcDNA3 vector (Invitrogen). Electroporation was carried out with a custom-built electroporator set at 1500 V and R-adjust max. Forty-eight hours post-transfection, cells were collected by centrifugation at 1500 r.p.m. for 5 min, washed with PBS and resuspended in lysis buffer at a concentration of 5×104 cells μl−1. A total of 20 μl lysate was analysed for luciferase activity, and relative light units (RLU) were normalized for transfection efficiency by comparison to the Renilla luciferase signal. EBNA1's transcriptional activity from BJAB8 cells was set to 100 % and activation of the EBNA1P10A clones is shown as the percentage of wild-type activity.
Long-term replication assay.
In order to measure the ability of EBNA1 and EBNA1P10A to replicate and maintain an oriP plasmid, a colony-formation assay was performed as described previously (Lindner et al., 2008; Wang et al., 2006). Cells of BJAB, BJAB8, clone P10A40 and clone P10A44 were transfected by electroporation (as described above) with 5 μg p994, an oriP vector carrying neomycin resistance, and 1 μg p3031, a ZsGreen-expression vector. Forty-eight hours post-transfection, the percentage of ZsGreen-positive (transfected) cells was determined. The cell population was diluted serially and plated in 96-well plates at ten, three and one green cell(s) per well in RPMI 1640 medium supplemented with 10 % FBS, 200 U penicillin ml−1, 200 μg streptomycin ml−1 and 2.5 mg G418 ml−1. The number of G418-resistant colonies was counted after 3 weeks and colony-formation efficiency was determined. Colonies from each cell type were expanded after 4 weeks and analysed by Western blotting to ensure the expression of EBNA1 and EBNA1P10A. The long-term replication analysis also included two small-scale preliminary experiments that resulted in the same replication-efficiency trend.
RESULTS
Isolation of EBNA1 peptides
The GGA region of EBNA1 (aa 90–325) prevented optimal fragmentation of the EBNA1 protein needed to identify phosphorylated serines at the C-terminal end of the domain. A derivative of EBNA1 that lacks the majority of the GGA region was used for phosphorylation analysis to avoid this problem. This derivative was expressed stably in an EBV-negative Burkitt's lymphoma cell line, BJAB, and will be referred to as EBNA1, as it maintains all of EBNA1's functions in cell culture (Aiyar & Sugden, 1998). Because it is wild-type in function, any modifications required for its known functions would be maintained in this derivative. The expression level of this protein is comparable to that of EBNA1 from EBV in the 721 lymphoblastoid cell line (see Supplementary Fig. S1, available in JGV Online).
To immunoprecipitate EBNA1 from cells, a cocktail of mAbs was used that consisted of antibodies reactive to multiple epitopes throughout the EBNA1 protein [see Supplementary Fig. S2(a), available in JGV Online]. EBNA1 was enriched in the eluate, as shown in the Western blot in Supplementary Fig. S2(b) (available in JGV Online).
EBNA1 has an unusually stable C-terminal protein domain that prevents complete digestion of an approximately 20 kDa fragment. The stability maps to the DNA-binding domain of the protein (Freire et al., 2008; Shah et al., 1992). Work by Freire et al. (2008) shows that extreme denaturation conditions, 4 M GuSCN for 2 h, are required to denature the protein fully. These conditions were necessary to allow complete digestion of EBNA1 (data not shown). After denaturation, the denaturant was diluted and LysC protease was added for complete digestion, followed by an incomplete trypsin digest.
Identification of EBNA1 phosphosites
Phosphorylated peptides were enriched by IMAC and then subjected to nHPLC-ETD-MS/MS for sequencing. These experiments resulted in the mapping of ten phosphorylation sites in EBNA1 (Table 1; Fig. 1a). Both serine and threonine residues were found to be phosphorylated. The peptides recovered did not yield 100 % coverage of the protein (Fig. 1a); 17 of 27 Ser, four of 15 Thr and none of five Tyr residues were isolated. The location of these phosphorylated amino acids is shown in Fig. 1(b).
Table 1.
EBNA1 phosphosites
Peptides recovered for each phosphosite are shown along with the corresponding E value. Those residues conserved in each related virus are indicated; the phosphorylation state of these residues in cynomolgus-EBV and HVP is unknown.
| Phosphosite* | Peptide† | E value‡ | Conserved in: | |
|---|---|---|---|---|
| HVP | Cynomolgus-EBV | |||
| Thr8 | SDEGPGpTGPGNGLGEKGDTSGPEGSGGSGPQR | 4E−11 | No | No |
| Thr20 | GDpTSGPEGSGGSGPQRR | 9E−13 | Yes | No |
| Ser21 | GDTpSGPEGSGGSGPQR | 3E−10 | No | No |
| Ser60 | GGGRPGAPGGpSGSGPRHR | 5E−15 | Yes | Yes |
| Ser62 | GGGRPGAPGGSGpSGPRHR | 8E−13 | Yes | Yes |
| Ser78 | RPpSCIGCK | 2E−03 | Yes | Yes |
| Ser334 | ARGGpSRER | 2E−03 | No | Yes |
| Ser365 | GTHGGTGAGAGAGGAGAGGGGRGRGGpSGGRGR | 2E−299 | No | No |
| Ser383 and Ser393 | RPRpSPSSQSSSSGpSPPRRPPPGR | 5E−12 | Yes | Yes |
*Phosphosite amino acids are numbered according to the B95-8 strain of EBV.
†Phosphosites are indicated in bold type.
‡Expectation scores assigned by omssa. An E value <0.05 is statistically significant. With current search parameters, spectra assigned scores higher than 1E−04 should be subjected to manual interpretation. All of these spectra were validated manually.
Fig. 1.

Identification of EBNA1 phosphosites. (a) Representative spectrum of a phosphorylated EBNA1 peptide. The location of this residue within the protein is shown in bold and underlined. C ions correspond to N-terminal fragments of the peptide recovered, whilst Z ions correspond to C-terminal fragments. The total peptides that were recovered in all of the ETD-MS/MS runs are underlined. The +1, +2 and +3 m/z peaks correspond to the precursor molecule that has undergone electron transfer, but has not dissociated. (b) Schematic of the EBNA1 protein showing the location of the phosphosites within the domain structure. The NLS is located between aa 379 and 386.
Comparison of these residues with those of EBNA1 homologues in the closely related viruses EBV-related herpesvirus of cynomolgus monkey (cynomolgus-EBV; strain Si-IIA) (Ohara et al., 2000) and herpesvirus papio (HVP) (Yates et al., 1996) shows that a subset of these residues are conserved (Table 1). Serines within linking region 1 (GR-1) (Ser60, Ser62 and Ser78) are conserved among all family members, as well as Ser383 [within the nuclear-localization sequence (NLS)] and Ser393. HVP additionally shares Thr20 and cynomolgus-EBV shares Ser334. Residues Thr8, Ser21 and Ser365 are not conserved in either virus. This conservation suggests that the serines within GR-1 and the NLS may be of particular importance for EBNA1's functions. The phosphorylation state of these amino acids in cynomolgus-EBV and HVP EBNA1 homologues is unknown.
To determine whether the phosphorylation of any of these ten residues affects EBNA1's functions in cells, we made a mutant that prevents the phosphorylation of all ten of these sites by substituting them with alanine in the EBNA1P10A derivative.
Properties of the EBNA1P10A derivative
The levels of expression of EBNA1 and EBNA1P10A were determined by quantitative Western blotting (Bergendahl et al., 2003) using a fluorescently labelled primary antibody to detect the derivative in two clones of BJAB cells expressing it constitutively (see Supplementary Fig. S3, available in JGV Online). The derivative is expressed at a higher level than wild-type EBNA1.
Phosphosite Ser383 is located within the EBNA1 NLS, suggesting that prevention of phosphorylation at this residue may change the trafficking of EBNA1 into or out of the nucleus. The localization of EBNA1P10A, however, did not change significantly, as determined by nuclear and cytoplasmic fractionation (see Supplementary Fig. S4, available in JGV Online). For both the wild type and the phosphorylation mutant, EBNA1 was distributed evenly between the cytoplasm and the nucleus.
EBNA1 has a half-life ≥30 h (Tellam et al., 2004). A CHX assay was performed to determine whether phosphorylation of EBNA1 influenced the stability of the protein. CHX was added to the cells and aliquots were taken at various time points up to 48 h. Live cells were separated from non-viable cells with a Ficoll gradient and Western blotting was performed to establish the level of the EBNA1 protein and its derivative in treated cells. There was no difference detected in the stability of EBNA1P10A compared with EBNA1 (Fig. 2).
Fig. 2.
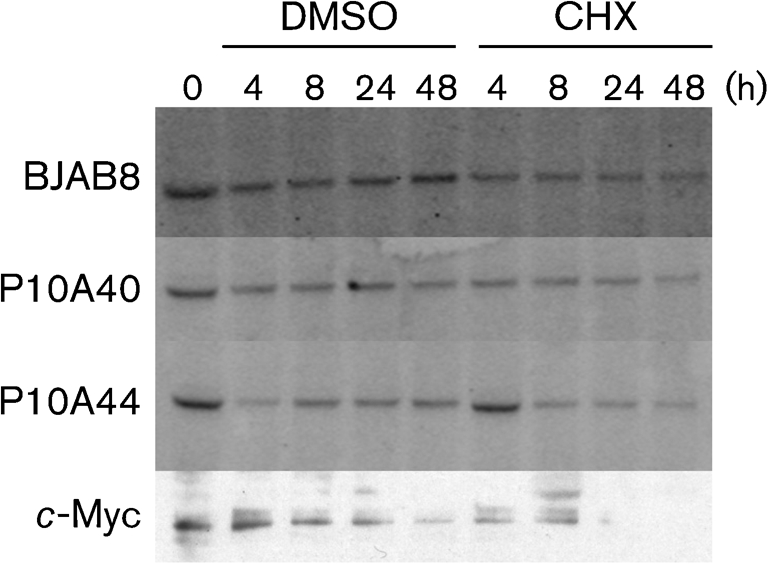
Stability of the EBNA1 and EBNA1P10A proteins. Cycloheximide (CHX) or DMSO was added to cells and aliquots were taken at various time points. The quantitative Western blot, probed with anti-EBNA1 mAb 1EB12 conjugated to Alexa Fluor 647, shows the persistence of both EBNA1 and EBNA1P10A proteins up to 48 h. A representative c-Myc Western blot is included to show that CHX did block translation. This experiment was repeated multiple times with similar results. There was 2-fold more lysate loaded for the BJAB8 sample to account for the difference in expression level.
EBNA1 transcription
To determine whether the phosphorylation-defective EBNA1P10A mutant can activate transcription to the same level as wild-type EBNA1, a luciferase reporter assay was performed. An EBNA1-responsive enhancer element, the family of repeats encoded in oriP, was placed upstream of a thymidine kinase promoter and the luciferase-encoding region (Kennedy & Sugden, 2003). Wild-type EBNA1 activated transcription more efficiently than EBNA1P10A; EBNA1P10A clone 40 supported 83 ± 8.3% of wild-type EBNA1's transcriptional activity (P=0.03; Wilcoxon rank sum test), whilst clone 44 had only 52 ± 7.9% of this activity (P=0.01, Wilcoxon rank sum test).
EBNA1 replication and segregation function
To determine the ability of EBNA1P10A to maintain an oriP plasmid, a long-term replication assay was performed. The maintenance of an oriP plasmid includes EBNA1's ability to both support replication and segregate the plasmid. For replication and maintenance, oriP plasmids introduced into mammalian cells must first undergo establishment (Leight & Sugden, 2001), a cellular process that is being deciphered (Wang & Sugden, 2008). Within the first 10–15 generations, plasmids are lost from the cell population at approximately 15–20 % per generation. After this time, remaining plasmids are lost at only 3–4 % per cell generation and are termed ‘established’.
Cells were transfected with both a plasmid carrying oriP and neomycin resistance and a ZsGreen-expression plasmid. After culturing the cells for 48 h, the number of ZsGreen-positive (transfected) cells was determined and a known number of transfected cells were plated in a 96-well plate under selection with 2.5 mg G418 ml−1. Three weeks later, after establishment, colonies were counted. The efficiency of colony formation measured in BJAB8 cells was set at 1 and the efficiency of the EBNA1P10A clones to support colonies was normalized to it (Table 2). Both clones of BJAB cells expressing EBNA1P10A formed colonies inefficiently. Clone P10A40 had only 51 % of wild-type EBNA1's efficiency, whilst clone P10A44 had 18 % of this efficiency.
Table 2.
Colony-formation efficiencies of EBNA1 and EBNA1P10A
The efficiency of colony formation measured in BJAB8 cells was set at 1 and the efficiency of the EBNA1P10A clones to support colonies was normalized to it.
| Cell line | Normalized colony-formation efficiency |
|---|---|
| BJAB8 (EBNA1) | 1.00 |
| BJAB | <0.008 |
| EBNA1P10A (clone 40) | 0.51 |
| EBNA1P10A (clone 44) | 0.18 |
DISCUSSION
Ten phosphosites were identified on EBV EBNA1. After identification of these ten phosphosites, we made one mutant that prevented the phosphorylation of all sites to assess the contributions that their phosphorylation could make to EBNA1's functions. EBNA1P10A supported transcription between 83 % (clone 40) and 52 % (clone 44) as effectively as the wild type. The EBNA1P10A derivatives supported replication of an EBV-derived plasmid in cells between 51 % (clone 40) and 18 % (clone 44) as efficiently as did wild-type EBNA1. The phosphorylation-deficient derivative retained the unusually long half-life and the ability to translocate into the nucleus of wild-type EBNA1. The difference between the two clones probably represents clonal variation due to additional mutations that arose or the insertion site of the EBNA1 constructs.
EBNA1 probably carries out the majority of its functions through protein–protein interactions, as it has no known independent enzymic activity. These phosphorylated residues may influence the ability of EBNA1 to interact with certain binding partners and thereby stimulate or repress EBNA1's functions. The phosphate groups may be involved directly in the protein–protein interface or may impose a conformational change allowing interaction. Peptidyl–prolyl cis/trans isomerase Pin1 specifically isomerizes pSer/Thr–Pro-containing proteins, thereby inducing a significant conformational change that can have profound effects on function, protein–protein interactions, location and/or stability (reviewed by Lu et al., 2002). EBNA1 phosphosites Ser383 and Ser393 are followed by a proline residue. It is also possible that phosphorylation-dependent modifications occur that are necessary for EBNA1's functions and/or interaction with binding partners. For example, phosphorylation can influence the sumoylation (Hietakangas et al., 2006; Roscic et al., 2006) or ubiquitination (Lin et al., 2006; Yu et al., 2006) of the same protein.
The migration of EBNA1P10A in SDS-PAGE is comparable to that of wild-type EBNA1 (see Supplementary Fig. S3, available in JGV Online). This result suggests that a small amount of EBNA1 protein, undetectable by Western blotting, contains a majority of the phosphorylated residues, because a highly phosphorylated derivative would be expected to migrate slower during gel electrophoresis. It is unlikely that a highly phosphorylated isoform of EBNA1 is required for its prolonged function; however, one or two of these phosphorylated residues could exist for a long time and not be seen by gel electrophoresis, or a highly phosphorylated isoform may be required for a transient interaction followed by removal of all or some of the modifications.
Previous work indicates that a combination of multiple modified residues will be required for EBNA1's functions. For example, deletion of aa 1–40 (which includes phosphosites Thr8, Thr20 and Ser21) had no discernible effect on replication or transcription (Mackey & Sugden, 1999). Phosphosite Ser383, located within the NLS, was mutated to alanine, aspartic acid or phosphoserine, and resulted in only slightly reduced nuclear import and had no effect on transcription (Kitamura et al., 2006). Shire et al. (2006) found that mutation of multiple serines within aa 325–376 was needed to have an effect on segregation, supporting the idea that a specific combination of phosphorylated residues is required for EBNA1's functions.
During the cell cycle, EBNA1 has different functional requirements from recruiting replication machinery to regulating gene expression. Each of these functions probably requires a unique set of binding partners. Phosphorylation is a common means of influencing protein–protein interactions. EBNA1's phosphorylation may be required to recruit subunits of the ORC efficiently to the origin of plasmid replication, or an unknown binding partner necessary for transcriptional activation to promoters bound by EBNA1. For example, bovine papillomavirus (BPV) E2 protein is known to be phosphorylated and that phosphorylation has an effect on its function (McBride et al., 1989). E2's replication function is diminished when phosphosite Ser301 is phosphorylated (McBride & Howley, 1991) and this effect on replication may be due to phosphorylation abrogating the interaction of E2 with E1 (Lusky & Fontane, 1991). Although this example in BPV does not mirror directly the effect that phosphorylation has on EBNA1's functions, it illustrates another virus that has evolved to rely on post-translational modifications, specifically phosphorylation, to influence viral protein function.
EBNA1's transcription and replication functions also rely on this post-translational modification, as a derivative preventing the phosphorylation of these sites is significantly deficient in both functions. A transcription-defective mutant (ΔUR1) acts as a dominant-negative mutant to EBNA1 (Kennedy et al., 2003), demonstrating that the transcription function provided by EBNA1 is essential for EBV. Additionally, EBNA1 is required for the replication and maintenance of the EBV genome and preventing these functions would cause the loss of EBV from latently infected cells. Preventing the phosphorylation of EBNA1, and thereby significantly reducing its ability to function, may provide a novel therapeutic target for EBV-related malignancies.
Supplementary Material
Acknowledgments
We thank Danielle Swaney for assistance with MS. We thank Dr Bill Sugden greatly for significant discussions and help with protocols and reagents. The Beckman Foundation, Eli Lilly and the National Institutes of Health (1R01GM080148 to J. J. C.) provided financial support for this work.
Footnotes
A supplementary table showing primers used for the generation of EBNA1P10A and four supplementary figures are available with the online version of this paper.
References
- Aiyar, A. & Sugden, B. (1998). Fusions between Epstein–Barr viral nuclear antigen-1 of Epstein–Barr virus and the large T-antigen of simian virus 40 replicate their cognate origins. J Biol Chem 273, 33073–33081. [DOI] [PubMed] [Google Scholar]
- Bergendahl, V., Glaser, B. T. & Burgess, R. R. (2003). A fast Western blot procedure improved for quantitative analysis by direct fluorescence labeling of primary antibodies. J Immunol Methods 277, 117–125. [DOI] [PubMed] [Google Scholar]
- Coon, J. J., Syka, J. E. P., Schwartz, J. C., Shabanowitz, J. & Hunt, D. F. (2004). Anion dependence in the partitioning between proton and electron transfer in ion/ion reactions. Int J Mass Spectrom 236, 33–42. [Google Scholar]
- Crawford, D. H. (2001). Biology and disease associations of Epstein–Barr virus. Philos Trans R Soc Lond B Biol Sci 356, 461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar, S. K., Yoshida, K., Machida, Y., Khaira, P., Chaudhuri, B., Wohlschlegel, J. A., Leffak, M., Yates, J. & Dutta, A. (2001). Replication from oriP of Epstein–Barr virus requires human ORC and is inhibited by geminin. Cell 106, 287–296. [DOI] [PubMed] [Google Scholar]
- Duellman, S. J. & Burgess, R. R. (2006). Overproduction in Escherichia coli and purification of Epstein–Barr virus EBNA-1. Protein Expr Purif 47, 434–440. [DOI] [PubMed] [Google Scholar]
- Duellman, S. J. & Burgess, R. R. (2009). Antigen-binding properties of monoclonal antibodies reactive with EBNA1 and use in immunoaffinity chromatography. . PLoS One 4, e4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahraeus, R. (2005). Do peptides control their own birth and death? Nat Rev Mol Cell Biol 6, 263–267. [DOI] [PubMed] [Google Scholar]
- Frappier, L. & O'Donnell, M. (1991a). Epstein–Barr nuclear antigen 1 mediates a DNA loop within the latent replication origin of Epstein–Barr virus. Proc Natl Acad Sci U S A 88, 10875–10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappier, L. & O'Donnell, M. (1991b). Overproduction, purification, and characterization of EBNA1, the origin binding protein of Epstein–Barr virus. J Biol Chem 266, 7819–7826. [PubMed] [Google Scholar]
- Freire, E., Oddo, C., Frappier, L. & de Prat-Gay, G. (2008). Kinetically driven refolding of the hyperstable EBNA1 origin DNA-binding dimeric β-barrel domain into amyloid-like spherical oligomers. Proteins 70, 450–461. [DOI] [PubMed] [Google Scholar]
- Gahn, T. A. & Sugden, B. (1995). An EBNA-1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstein–Barr virus LMP gene. J Virol 69, 2633–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer, L. Y., Markey, S. P., Kowalak, J. A., Wagner, L., Xu, M., Maynard, D. M., Yang, X., Shi, W. & Bryant, S. H. (2004). Open mass spectrometry search algorithm. J Proteome Res 3, 958–964. [DOI] [PubMed] [Google Scholar]
- Goldsmith, K., Bendell, L. & Frappier, L. (1993). Identification of EBNA1 amino acid sequences required for the interaction of the functional elements of the Epstein–Barr virus latent origin of DNA replication. J Virol 67, 3418–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good, D. M., Wirtala, M., McAlister, G. C. & Coon, J. J. (2007). Performance characteristics of electron transfer dissociation mass spectrometry. Mol Cell Proteomics 6, 1942–1951. [DOI] [PubMed] [Google Scholar]
- Hearing, J. C. & Levine, A. J. (1985). The Epstein–Barr virus nuclear antigen (BamHI K antigen) is a single-stranded DNA binding phosphoprotein. Virology 145, 105–116. [DOI] [PubMed] [Google Scholar]
- Hietakangas, V., Anckar, J., Blomster, H. A., Fujimoto, M., Palvimo, J. J., Nakai, A. & Sistonen, L. (2006). PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci U S A 103, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, S., Ikeda, M., Kato, N., Matsumoto, A., Ishikawa, Y., Kumakubo, S. & Yanagi, K. (2000). Epstein–Barr virus nuclear antigen-1 binds to nuclear transporter karyopherin α1/NPI-1 in addition to karyopherin α2/Rch1. Virology 266, 110–119. [DOI] [PubMed] [Google Scholar]
- Kennedy, G. & Sugden, B. (2003). EBNA-1, a bifunctional transcriptional activator. Mol Cell Biol 23, 6901–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, G., Komano, J. & Sugden, B. (2003). Epstein–Barr virus provides a survival factor to Burkitt's lymphomas. Proc Natl Acad Sci U S A 100, 14269–14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, A. L., Maher, M., Hayman, J. B., Ozer, J., Zerby, D., Yates, J. L. & Lieberman, P. M. (1997). An imperfect correlation between DNA replication activity of Epstein–Barr virus nuclear antigen 1 (EBNA1) and binding to the nuclear import receptor, Rch1/importin α. Virology 239, 340–351. [DOI] [PubMed] [Google Scholar]
- Kitamura, R., Sekimoto, T., Ito, S., Harada, S., Yamagata, H., Masai, H., Yoneda, Y. & Yanagi, K. (2006). Nuclear import of Epstein–Barr virus nuclear antigen 1 mediated by NPI-1 (importin α5) is up- and down-regulated by phosphorylation of the nuclear localization signal for which Lys379 and Arg380 are essential. J Virol 80, 1979–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D. Y. & Sugden, B. (2008). The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 111, 2280–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. A., Diamond, M. E. & Yates, J. L. (1999). Genetic evidence that EBNA-1 is needed for efficient, stable latent infection by Epstein–Barr virus. J Virol 73, 2974–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leight, E. R. & Sugden, B. (2000). EBNA-1: a protein pivotal to latent infection by Epstein–Barr virus. Rev Med Virol 10, 83–100. [DOI] [PubMed] [Google Scholar]
- Leight, E. R. & Sugden, B. (2001). Establishment of an oriP replicon is dependent upon an infrequent, epigenetic event. Mol Cell Biol 21, 4149–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitskaya, J., Sharipo, A., Leonchiks, A., Ciechanover, A. & Masucci, M. G. (1997). Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein–Barr virus nuclear antigen 1. Proc Natl Acad Sci U S A 94, 12616–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, D. I., Barbash, O., Kumar, K. G., Weber, J. D., Harper, J. W., Klein-Szanto, A. J., Rustgi, A., Fuchs, S. Y. & Diehl, J. A. (2006). Phosphorylation-dependent ubiquitination of cyclin D1 by the SCFFBX4-αB crystallin complex. Mol Cell 24, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner, S. E., Zeller, K., Schepers, A. & Sugden, B. (2008). The affinity of EBNA1 for its origin of DNA synthesis is a determinant of the origin's replicative efficiency. J Virol 82, 5693–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, K. P., Liou, Y. C. & Zhou, X. Z. (2002). Pinning down proline-directed phosphorylation signaling. Trends Cell Biol 12, 164–172. [DOI] [PubMed] [Google Scholar]
- Lusky, M. & Fontane, E. (1991). Formation of the complex of bovine papillomavirus E1 and E2 proteins is modulated by E2 phosphorylation and depends upon sequences within the carboxyl terminus of E1. Proc Natl Acad Sci U S A 88, 6363–6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey, D. & Sugden, B. (1999). The linking regions of EBNA1 are essential for its support of replication and transcription. Mol Cell Biol 19, 3349–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey, D., Middleton, T. & Sugden, B. (1995). Multiple regions within EBNA1 can link DNAs. J Virol 69, 6199–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, S. E., Shabanowitz, J., Hunt, D. F. & Marto, J. A. (2000). Subfemtomole MS and MS/MS peptide sequence analysis using nano-HPLC micro-ESI Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem 72, 4266–4274. [DOI] [PubMed] [Google Scholar]
- McAlister, G. C., Phanstiel, D., Good, D. M., Berggren, W. T. & Coon, J. J. (2007). Implementation of electron-transfer dissociation on a hybrid linear ion trap-orbitrap mass spectrometer. Anal Chem 79, 3525–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister, G. C., Berggren, W. T., Griep-Raming, J., Horning, S., Makarov, A., Phanstiel, D., Stafford, G., Swaney, D. L., Syka, J. E. & other authors (2008). A proteomics grade electron transfer dissociation-enabled hybrid linear ion trap-orbitrap mass spectrometer. J Proteome Res 7, 3127–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride, A. A. & Howley, P. M. (1991). Bovine papillomavirus with a mutation in the E2 serine 301 phosphorylation site replicates at a high copy number. J Virol 65, 6528–6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride, A. A., Bolen, J. B. & Howley, P. M. (1989). Phosphorylation sites of the E2 transcriptional regulatory proteins of bovine papillomavirus type 1. J Virol 63, 5076–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes, J., Leibold, W., Klein, G. & Clements, G. (1975). Establishment and characterization of an Epstein–Barr virus (EBC)-negative lymphoblastoid B cell line (BJA-B) from an exceptional, EBV-genome-negative African Burkitt's lymphoma. Biomedicine 22, 276–284. [PubMed] [Google Scholar]
- Middleton, T. & Sugden, B. (1992). EBNA1 can link the enhancer element to the initiator element of the Epstein–Barr virus plasmid origin of DNA replication. J Virol 66, 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz, C. (2004). Epstein–Barr virus nuclear antigen 1: from immunologically invisible to a promising T cell target. J Exp Med 199, 1301–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndassa, Y. M., Orsi, C., Marto, J. A., Chen, S. & Ross, M. M. (2006). Improved immobilized metal affinity chromatography for large-scale phosphoproteomics applications. J Proteome Res 5, 2789–2799. [DOI] [PubMed] [Google Scholar]
- Ohara, N., Hayashi, K., Teramoto, N., Oka, T., Fujimoto, K., Yoshikawa, Y., Castanos-Velez, E., Biberfeld, P. & Akagi, T. (2000). Sequence analysis and variation of EBNA-1 in Epstein–Barr virus-related herpesvirus of cynomolgus monkey. Intervirology 43, 102–106. [DOI] [PubMed] [Google Scholar]
- Polvino-Bodnar, M., Kiso, J. & Schaffer, P. A. (1988). Mutational analysis of Epstein–Barr virus nuclear antigen 1 (EBNA 1). Nucleic Acids Res 16, 3415–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzi, M., Tillack, K., Gerhardt, J., Ott, E., Humme, S., Kremmer, E., Hammerschmidt, W. & Schepers, A. (2003). Complex protein–DNA dynamics at the latent origin of DNA replication of Epstein–Barr virus. J Cell Sci 116, 3971–3984. [DOI] [PubMed] [Google Scholar]
- Roscic, A., Moller, A., Calzado, M. A., Renner, F., Wimmer, V. C., Gresko, E., Ludi, K. S. & Schmitz, M. L. (2006). Phosphorylation-dependent control of Pc2 SUMO E3 ligase activity by its substrate protein HIPK2. Mol Cell 24, 77–89. [DOI] [PubMed] [Google Scholar]
- Shah, W. A., Ambinder, R. F., Hayward, G. S. & Hayward, S. D. (1992). Binding of EBNA-1 to DNA creates a protease-resistant domain that encompasses the DNA recognition and dimerization functions. J Virol 66, 3355–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shire, K., Kapoor, P., Jiang, K., Hing, M. N., Sivachandran, N., Nguyen, T. & Frappier, L. (2006). Regulation of the EBNA1 Epstein–Barr virus protein by serine phosphorylation and arginine methylation. J Virol 80, 5261–5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, W., Middleton, T., Sugden, B. & Echols, H. (1991). DNA looping between the origin of replication of Epstein–Barr virus and its enhancer site: stabilization of an origin complex with Epstein–Barr nuclear antigen 1. Proc Natl Acad Sci U S A 88, 10870–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden, B. & Warren, N. (1989). A promoter of Epstein–Barr virus that can function during latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA replication during latent infection. J Virol 63, 2644–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney, D. L., McAlister, G. C., Wirtala, M., Schwartz, J. C., Syka, J. E. & Coon, J. J. (2007). Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal Chem 79, 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syka, J. E., Coon, J. J., Schroeder, M. J., Shabanowitz, J. & Hunt, D. F. (2004). Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellam, J., Connolly, G., Green, K. J., Miles, J. J., Moss, D. J., Burrows, S. R. & Khanna, R. (2004). Endogenous presentation of CD8+ T cell epitopes from Epstein–Barr virus-encoded nuclear antigen 1. J Exp Med 199, 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, N. E., Hager, D. A. & Burgess, R. R. (1992). Isolation and characterization of a polyol-responsive monoclonal antibody useful for gentle purification of Escherichia coli RNA polymerase. Biochemistry 31, 7003–7008. [DOI] [PubMed] [Google Scholar]
- Wang, C. Y. & Sugden, B. (2008). Identifying a property of origins of DNA synthesis required to support plasmids stably in human cells. Proc Natl Acad Sci U S A 105, 9639–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., Lindner, S. E., Leight, E. R. & Sugden, B. (2006). Essential elements of a licensed, mammalian plasmid origin of DNA synthesis. Mol Cell Biol 26, 1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, D. K., Jr, McAlister, G. C., Good, D. M., Coon, J. J. & Muddiman, D. C. (2007). Dual electrospray ion source for electron-transfer dissociation on a hybrid linear ion trap-orbitrap mass spectrometer. Anal Chem 79, 7916–7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, H., Kapoor, P. & Frappier, L. (2002). Separation of the DNA replication, segregation, and transcriptional activation functions of Epstein–Barr nuclear antigen 1. J Virol 76, 2480–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates, J. L. & Camiolo, S. M. (1988). Dissection of DNA replication and enhancer activation functions of Epstein–Barr virus nuclear antigen 1. Cancer Cells 6, 197–205. [Google Scholar]
- Yates, J., Warren, N., Reisman, D. & Sugden, B. (1984). A cis-acting element from the Epstein–Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci U S A 81, 3806–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates, J. L., Warren, N. & Sugden, B. (1985). Stable replication of plasmids derived from Epstein–Barr virus in various mammalian cells. Nature 313, 812–815. [DOI] [PubMed] [Google Scholar]
- Yates, J. L., Camiolo, S. M., Ali, S. & Ying, A. (1996). Comparison of the EBNA1 proteins of Epstein–Barr virus and herpesvirus papio in sequence and function. Virology 222, 1–13. [DOI] [PubMed] [Google Scholar]
- Yin, Y., Manoury, B. & Fahraeus, R. (2003). Self-inhibition of synthesis and antigen presentation by Epstein–Barr virus-encoded EBNA1. Science 301, 1371–1374. [DOI] [PubMed] [Google Scholar]
- Young, L. S. & Rickinson, A. B. (2004). Epstein–Barr virus: 40 years on. Nat Rev Cancer 4, 757–768. [DOI] [PubMed] [Google Scholar]
- Yu, X., Fu, S., Lai, M., Baer, R. & Chen, J. (2006). BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev 20, 1721–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.