

Carbohydrate domains mediate the stability, folding, and biological activity of glycoproteins.1 Quality investigations into the impact of specific glycosylation patterns are complicated by formation of horrific mixtures of difficultly separable glycoforms. A major program underway in our laboratory seeks to address the challenge of obtaining single glycoforms of biologics through the de novo chemical synthesis of homogeneous glycopeptides and glycoproteins of established therapeutic value.2
Accordingly, we became interested in monomeric immunoglobulin G (IgG) purified from intravenous immunoglobulin (IVIG). This therapeutically valuable anti-inflammatory agent is commonly employed in the treatment of autoimmune disorders, such as rheumatoid arthritis and immune thrombocytopenia. The biological activity of IVIG derives from its Fc fragment, which presents a biantennary tridecasaccharide, attached through an N-linkage to the Asn297 residue. Recently, Ravetch and coworkers discovered the critical role of the terminal 2,6-linked sialic acids of the glycan in mediating the anti-inflammatory activity of IVIG.3 Although the IVIG glycan is biosynthesized as a heterogeneous mixture, containing both 2,3- and 2,6-sialylated carbohydrate domains, only the Fc fragment possessing the 2,6-sialylated glycoform demonstrates appreciable anti-inflammatory activity in arthritic mice. Moreover, an IVIG Fc fragment possessing exclusively 2,6-sialic acid linkages was shown to be 10-fold more active in suppressing inflammation in mice than was an Fc fragment isolated from IVIG (containing both 2,6- and 2,3- linkages). These results suggest that an Fc peptide fragment presenting a homogeneous 2,6-sialylated glycan domain (cf. 1) could be more potent and effective than the heterogeneous mixture currently employed in clinical settings.
We describe herein the chemical synthesis of homogeneous tridecasaccharide, 1, possessing terminal 2,6-sialic acid linkages. We further describe the aspartylation of 1 with a peptide domain, thereby demonstrating the ability to convert it into a polyglycopolypeptide (see 28). Accordingly, glycopeptidic constructs corresponding to the carbohydrate domain of immunoglobulin G can now be synthesized and screened.
The power of prioritized strategic bond disconnection as a means of guiding synthetic analysis, formalized by the Corey school,4 is well appreciated by students of chemical synthesis of complex target systems. Less well appreciated is a type of pattern recognition analysis which can be very helpful in devising total synthesis programs directed toward biologic level oligosaccharides.5 System 1 constitutes a significant challenge to chemical synthesis. Thus, the 13-mer contains an often troublesome (L)-α fucosyl ring (ring 5) branching from the “reducing end” GlcNAc of the terminal chitobiose (rings 1 and 2). It also presents the complex ß-mannose linkage joining a ß-mannose (ring 3) to the chitobiose core system. The ß-mannoside (ring 3), in turn, is linked in a biantennary fashion from its C3 and C6 hydroxyls to two α-linked mannosides (see rings 6 and 7). The C2 hydroxyls of these mannosides are linked in a ß-fashion to two lactosamines, joined in 2,6 linkages to sialic acids (see rings 8-13). The ß-linked mannose (ring 3) is also joined at its equatorial C4 oxygen, in a ß-linkage to a GlcNAc residue (see ring 4).
Based on earlier literature in this general area,6 it seemed likely that the most demanding phase of the synthesis would involve the attachment of the two trisaccharide ensembles – rings 8, 9, and 10 as well as rings 11, 12, and 13 – to their respective axial hydroxyl acceptor sites in ring 6 and ring 7. Looming particularly difficult was the prospect of introducing additional appendages at the interior C2 axial hydroxyl of ring 7. Indeed, it was our hope for conciseness to conduct simultaneous azaglycosylations at both of these two acceptor sites to introduce ß-glucosamine donor residues (see rings 8 and 11). These rings would in turn be joined at their C4 equatorial hydroxyl groups, via ß-linkages, to C6 sialylated galactosyl donors. Ring 7 emanates from the particularly hindered C3 hydroxyl site of ring 3 nestled between the glucosamine moiety at C4, the axial hydroxyl at C2, and the complex core (rings 1 and 2) substructure. In fact, previous attempts to use the more exposed C6 hydroxyl of ring 3 as a viable acceptor site met with only the slightest of success. No glycosylation at all had been achieved at the much more hindered secondary hydroxyl acceptor site at C3 of ring 3. Indeed, the key breakthrough involved the use of a phenylsulfamido function on the future ring 4 (see asterisk, compound 4) to allow for introduction of rings 5, 6, and 7 (vide infra).
In Scheme 1, we recapitulate, by focusing on key building blocks, the strategic analysis, which eventually achieved success. Thus, as shown, we envisioned reaching tetrasaccharide 4 through coupling of trisaccharide 5 with sulfonamide donor 6.7 As noted, the presence of the sulfonamide functionality on diol 4 would prove key to the efficient installation of the “wing” mannose and fucose units, furnishing core heptasaccharide 3.8 Elongation of the mannose units with a ß-GlcNAc donor would yield the nonasaccharide, 2, which would finally be subjected to diglycosylation with disaccharide 10, to afford the target compound, 1. Happily, in this synthetic analysis, we were able to revisit the chemistry developed in connection with our longstanding glycal assembly program.9
Scheme 1.

Synthetic Strategy Toward Tridecasaccharide 1.
Our synthesis of key intermediate 2 commenced with the known monosaccharides 1110 and 12 (Scheme 2). Glycosylation of 12 with donor 11, through exposure to NIS/TMSOTf11 under sonication conditions, proceeded rapidly to afford the disaccharide in 82% yield.12 Subsequent saponification furnished acceptor 13. The latter was then glycosylated with donor 14,13 to give trisaccharide 15 with good selectivity (β/α = 7:1). The β-isomer was isolated in 65% yield. Following removal of the benzylidene acetal14 and selective benzoylation of the resulting 4,6-diol, compound 5 was in hand. MeOTf-promoted15 glycosylation with glucosamine building block 6 afforded a 92% yield of 16 (β/α = 2.7:1).
Scheme 2.

Synthesis of Intermediate 2.
Sequential removal of the benzoate and PMB protecting groups of tetrasaccharide 16 afforded the 3,6-diol, 4, in 77% yield over two steps. Two-fold glycosylation of 4 was accomplished with an excess of donor 7 and NIS/TMSOTf, to provide hexasaccharide 17 in 90% yield. The TBDPS group was cleaved upon exposure to HF/Py (88% yield). To the resultant free hydroxyl group was appended fluoride donor 8,16 under Cp2Zr(OTf)2 mediation,17 to afford compound 19 in 93% yield and excellent selectivity (α/β = 12:1). Following removal of the levulinoyl protecting group, intermediate 3 was in hand. BF3·OEt2-promoted diglycosylation of 3 was achieved through the use of excess donor 9.18 Finally, removal of the chloroacetate groups on the glucosamine units provided the target nonasaccharide 2 in 52% yield over two steps.
Having completed the synthesis of compound 2, we now turned to the assembly of the disaccharide donor, 10 (Scheme 3). In the event, coupling of phosphite 2119 and galactal 2220 proceeded smoothly to afford disaccharide 23 with good selectivity (α/β = 11:1). The predominant α isomer was isolated in 74% yield. Oxidation of 23 with PhI(OAc)2,21 followed by acetylation furnished disaccharide 24 in one pot.22 Finally, deprotection of the anomeric acetate was accomplished through exposure to hydrazine acetate, to provide the hemiacetal, which was further reacted with DAST to afford the target fluoride donor 10.
Scheme 3.
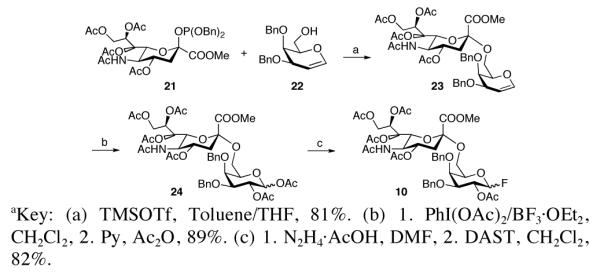
Synthesis of Disaccharide Donor 10.
With the two component pieces in hand, we now turned our attention to the completion of the tridecasaccharide synthesis. As outlined in Scheme 4, BF3·OEt2-mediated coupling of excess amounts of donor 10 with 2 provided mainly tridecasaccharide 25 (54% yield), along with a small amount of mono-coupled product. The global deprotection of 25 was accomplished in five steps, as shown. Thus, the methyl ester and acetyl groups were removed using excess NaOMe. Next, the phthalimide groups were cleaved, and the resultant free amines were acetylated. We were pleased to find that the 25 benzyl groups and the sulfonamide group could be efficiently cleaved upon exposure to sodium in liquid ammonia.23 Finally, selective acetylation was accomplished through treatment with acetic anhydride in saturated NaHCO3 to afford 1.
Scheme 4.

a Synthesis of Tridecasaccharide 1 and Glycopeptide 28
Finally, we examined the merger of tridecasaccharide 1 with a peptide domain. Thus, exposure of 1 to Kochetkov conditions24 provided 26 (Scheme 5). Aspartylation of 26 with 27 provided the target glycopeptide 28, albeit in modest yield.
In summary, we have synthesized homogeneous 2,6-sialylated tridecasaccharide of the Fc sector of IVIG, and succesfully coupled the carbohydrate with a peptide domain to provide a glycopeptide adduct (28). The merger of 1 with appropriate Fc-derived peptide fragments, and the results of biological evaluations of the glycopeptides, will be reported in due course.
Supplementary Material
Acknowledgement
Support was provided by the NIH (CA28824 to SJD). We thank Dr. George Sukenick, Hui Fang, and Sylvi Rusli of SKI’s NMR core facility for mass spectral and NMR analysis; Rebecca Wilson for editorial assistance; and Dana Ryan for assistance with the preparation of the manuscript.
Footnotes
Supporting Information Available: Experimental procedures, copies of spectral data, and characterization (PDF). This material is available free of charge via the Internet at http://pubs.acs.org
References
- (1)(a).Connor SEO, Imperiali B. Chem. Biol. 1996;3:803. doi: 10.1016/s1074-5521(96)90064-2. [DOI] [PubMed] [Google Scholar]; (b) Kobata A. Acc. Chem. Res. 1993;26:319. [Google Scholar]; (b) Feizi T. Nature. 1985;314:53. doi: 10.1038/314053a0. [DOI] [PubMed] [Google Scholar]
- (2)(a).For a review of glycoprotein synthesis, see: Davis BJ. Chem. Rev. 2002;102:579. doi: 10.1021/cr0004310.Though unrelated to our current effort, dramatic advances have been made in the field of enzymatic glycoprotein synthesis:Gamblin DP, Scanlan EM, Davis BJ. Chem. Rev. 2009;109:131. doi: 10.1021/cr078291i.For recent glycoprotein synthetic efforts from our laboratory, see:Kan C, Trzupek JD, Wu B, Wan Q, Chen G, Tan Z, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5438. doi: 10.1021/ja808707w.Yuan Y, Chen J, Wan Q, Tan Z, Chen G, Kan C, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5432. doi: 10.1021/ja808705v.Tan Z, Shang S, Halkina T, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5424. doi: 10.1021/ja808704m.Nagorny P, Fasching B, Li X, Chen G, Aussedat B, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5792. doi: 10.1021/ja809554x.
- (3)(a).Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Science. 2008;320:373. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kaneko V, Nimmerjahn F, Ravetch JV. Science. 2006;313:670. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- (4).Corey EJ, Cheung X-M. The Logic of Chemical Synthesis. Wiley-Interscience; New York: 1995. [Google Scholar]
- (5).Wilson RM, Danishefsky SJ. J. Org. Chem. 2007;72:4293. doi: 10.1021/jo070871s. [DOI] [PubMed] [Google Scholar]
- (6)(a).Yamazaki F, Nukada T, Ito Y, Sato S, Ogawa T. Tetrahedron Lett. 1989;33:4417.Yamazaki F, Sato S, Nukada T, Ito Y, Ogawa T. Carbohydrate Research. 1990;201:31. doi: 10.1016/0008-6215(90)84223-h.Paulsen H, Heume M, Nurnberger H. Carbohydrate Research. 1990;200:127. doi: 10.1016/0008-6215(90)84187-y.Weiler S, Schmidt RR. Tetrahedron Lett. 1998;39:2299.Unverzagt C. Angew. Chem. 1997;109:2078.Eller S, Schuberth R, Gundel G, Seifert J, Unverzagt C. Angew. Chem. Int. Ed. 2007;46:4173. doi: 10.1002/anie.200604788.Paulsen H. Angew. Chem. Int. Ed. 1990;29:823.For syntheses of selected complex bisected glycans, see also:Jonke S, Liu KG, Schmidt RR. Chem. Eur. J. 2006;12:1274. doi: 10.1002/chem.200500707.Matsuo I, Wada M, Manabe S, Yamaguchi Y, Otake K, Kato K, Ito Y. J. Am. Chem. Soc. 2003;125:3402. doi: 10.1021/ja021288q.
- (7).Griffith DA, Danishefsky SJ. J. Am. Chem. Soc. 1990;112:5811. [Google Scholar]
- (8).Our initial efforts to elongate the heptasaccharide in the presence of an NPhth functionality were unsuccessful, presumably due to the steric bulk of the phthalimide group. The sulfonamide-protected heptasaccharide was much more amenable to elongation.
- (9).Danishefsky SJ, Bilodeau MT. Angew. Chem. Int. Ed. 1996;35:1380. [Google Scholar]
- (10).Geeta S, Ole H. Journal of Carbohydrate Chemistry. 1991;10(5):927. [Google Scholar]
- (11).Mootoo DR, Konradson P, Udodong U, Fraser-Reid B. J. Am. Chem. Soc. 1988;110:5583. [Google Scholar]
- (12).Deng S, Gangadharmath U, Chang C. J. Org. Chem. 2006;71:5179. doi: 10.1021/jo060374w. [DOI] [PubMed] [Google Scholar]
- (13).Crich D, Sun S. Tetrahedron. 1998;54:8321. [Google Scholar]
- (14).Nicolaou KC, Veale CA, Hwang CK, Hutchinson J, Prasad CVC, Ogilvie WW. Angew. Chem. Int. Ed. 1991;30:299. [Google Scholar]
- (15).Seeberger PH, Beebe X, Sukenick GD, Pochapsky S, Danishefsky SJ. Angew. Chem. Int. Ed. 1997;36:491. [Google Scholar]
- (16).Wu B, Tan Z, Chen G, Chen J, Hua Z, Wan Q, Ranganathan K, Danishefsky SJ. Tetrahedron Lett. 2006;47:8009. doi: 10.1016/j.tetlet.2006.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Allen JR, Allen JG, Zhang X, Williams LJ, Zatorski A, Ragupathi G, Livingston PO, Danishefsky SJ. Chem. Eur. J. 2000;6:1366. doi: 10.1002/(sici)1521-3765(20000417)6:8<1366::aid-chem1366>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- (18).Unverzagt C. Chem. Eur. J. 2003;9:1369. doi: 10.1002/chem.200390156. [DOI] [PubMed] [Google Scholar]
- (19).Bhattacharya SK, Danishefsky SJ. J. Org. Chem. 2000;65:144. doi: 10.1021/jo9912496. [DOI] [PubMed] [Google Scholar]
- (20).Chen XT, Sames D, Danishefsky SJ. J. Am. Chem. Soc. 1998;120:7760. [Google Scholar]
- (21)(a).Shi L, Kim Y-J, Gin DY. J. Am. Chem. Soc. 2001;123:6939. doi: 10.1021/ja015991a. [DOI] [PubMed] [Google Scholar]; (b) Plante OJ, Palmacci ER, Andrade RB, Seeberger PH. J. Am. Chem. Soc. 2001;123:9545. doi: 10.1021/ja016227r. [DOI] [PubMed] [Google Scholar]; (19) Hanashima S, Castagner B, Esposito D, Nokami T, Seeberger PH. Org. Lett. 2000;2:3881. doi: 10.1021/ol0704946. [DOI] [PubMed] [Google Scholar]
- (22).Hanashima S, Castagner B, Esposito D, Nokami T, Seeberger PH. Org. Lett. 2007;9:1777. doi: 10.1021/ol0704946. [DOI] [PubMed] [Google Scholar]
- (23)(a).Iserloh U, Dudkin V, Wang Z, Danishefsky SJ. Tetrahedron Lett. 2002;43:7027. [Google Scholar]; (b) Wang Z-G, Warren JD, Dukin VY, Zhang X, Iserloh V, Visser M, Eckhardt M, Seeberger PH, Danishefsky SJ. Tetrahedron. 2006;62:4954. [Google Scholar]
- (24).Cohen-Anisfeld ST, Lansbury PT. J. Am. Chem. Soc. 1993;115:10531. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.