

Abstract
Polymyxins are often the only option to treat acquired multidrug-resistant Pseudomonas aeruginosa. Polymyxin susceptibility in P. aeruginosa PAO1 is associated with the lipopolysaccharide structure that is determined by arnBCADTEF and modulated by phoPQ and pmrAB. We examined five clonally unrelated clinical isolates of polymyxin B-resistant P. aeruginosa to investigate the molecular basis of polymyxin resistance. All isolates grew with 4 μg/ml polymyxin B (MIC, 8 μg/ml), whereas P. aeruginosa PAO1 grew with 0.25 μg/ml polymyxin B (MIC, 0.5 μg/ml). The resistant isolates were converted to susceptible ones (the MICs fell from 8 to 0.5 μg/ml) following the introduction of phoPQ (four isolates) and pmrAB (one isolate), which had been cloned from strain PAO1. DNA sequence analysis revealed that a single-nucleotide substitution in three isolates replaced a single amino acid of PhoQ, the deletion of 17 nucleotides in one isolate truncated the protein of PhoQ, and two nucleotide substitutions in one isolate replaced two amino acids of PmrB. The involvement of these amino acid substitutions or the truncated protein of PhoQ and PmrB in polymyxin B resistance was confirmed using strain PAO1 lacking phoPQ or pmrAB that was transformed by phoPQ or pmrAB containing the amino acid substitutions or the truncated protein. The resistant clinical isolates were sensitized by the inactivation of arnBCADTEF (the MICs fell from 8 to 0.5 μg/ml). These results suggest that polymyxin B resistance among clinical isolates of P. aeruginosa is associated with alterations in two-component regulatory systems of phoPQ or pmrAB.
Pseudomonas aeruginosa is a nosocomial gram-negative opportunistic pathogen that causes a variety of infections (e.g., urinary tract, respiratory, skin, soft tissue, etc.) (3, 12, 18, 19). P. aeruginosa accounts for 11 to 14% of all nosocomial infections and is a major problem for people hospitalized with cancer, cystic fibrosis, or burns (3). Treatment usually involves the use of one or more antibiotics, such as β-lactams, aminoglycosides, or quinolones. Combination therapy usually is recommended for P. aeruginosa infections, as it decreases the risk of antibiotic resistance and enhances the eradication rate. Despite the use of combination therapy, there are numerous reports of the emergence of multidrug-resistant P. aeruginosa. Polymyxins (polymyxin B and colistin) often have been the last resort to treat such isolates (1, 3, 27). However, polymyxin B resistance in multidrug-resistant clinical isolates has been reported (4, 6, 11, 25).
The mode of action and the resistance mechanism to polymyxin B has been studied extensively using the reference P. aeruginosa strain PAO1. Polymyxin B is a polycationic lipopeptide antibiotic that interacts with a negatively charged lipid A moiety of the lipopolysaccharide (LPS) of gram-negative bacteria and leads to cell lysis and death (26). Resistance to polymyxin B is caused by the inhibition of the interactions between the antibiotic and the lipid A moiety of the LPS, and the inhibition is based on modifications of lipid A so that it is less negatively charged. LPS modification occurs by the addition of 4-amino-4-deoxy-l-arabinose to lipid A under limiting nutrient conditions, such as 20 μM magnesium or calcium, and is directed by an arnBCADTEF operon modulated by two-component regulatory systems of phoPQ and pmrAB (9, 13, 14, 20). In normal growth conditions, the two-component regulatory systems strictly repress arnBCADTEF, resulting in a phenotype of intrinsic susceptibility to polymyxins (8, 16, 17). Therefore, it is postulated that any interruption of the regulatory systems derepresses arnBCADTEF with resulting resistance to polymyxins. Indeed, in vitro-acquired polymyxin B-resistant P. aeruginosa PAK carried a single-amino-acid substitution in PmrB (20).
Although polymyxin-resistant clinical isolates of P. aeruginosa have been reported increasingly worldwide (5, 6, 10, 11, 25), the genetic basis for the resistance in these clinical isolates is unclear. This study aimed to examine the molecular details of polymyxin B resistance among clinical isolates of P. aeruginosa. We characterized five polymyxin B-resistant clinical isolates of P. aeruginosa and found that four isolates carried a single-amino-acid substitution or protein truncation in PhoQ, and one isolate carried dual amino acid substitutions in PmrB. The involvement of the amino acid substitutions or protein truncation in polymyxin B resistance was confirmed by introducing phoPQ or pmrAB containing the amino acid substitutions or protein truncation into P. aeruginosa PAO1 lacking phoPQ or pmrAB.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Previously identified polymyxin B-resistant clinical isolates of P. aeruginosa were obtained from State University of New York (SUNY) Downstate Medical Center (Brooklyn, NY) (11), and the reference P. aeruginosa strain PAO1 also was used. Escherichia coli DH5α was used for general cloning experiments. P. aeruginosa and E. coli DH5α were grown routinely in Luria-Bertani (LB) medium. When needed, ampicillin (100 μg/ml), chloramphenicol (10 μg/ml), gentamicin (15 μg/ml), or tetracycline (10 μg/ml) was added to the LB medium for E. coli; carbenicillin (100 μg/ml), gentamicin (100 μg/ml), or tetracycline (80 μg/ml) also was added to the LB medium for P. aeruginosa. Divalent cation-adjusted Mueller-Hinton (MH) (Oxoid, Ogdensburg, New York) broth (pH 7.0) was used for antibiotic susceptibility testing. All antibiotics and other chemicals used in this study were purchased from Sigma (St. Louis, MO).
Antibiotic susceptibility testing.
Antibiotic susceptibility was determined as MICs using the broth dilution method as guided by the CLSI (2). The following polymyxin B susceptibility breakpoints were used: resistance, MIC ≥ 8 μg/ml; susceptible, MIC ≤ 2 μg/ml.
Genotype analysis.
Genotype analysis was performed by PCR-based random amplified polymorphic DNA (RAPD) fingerprint using arbitrary primers of 5′-ACGGCCGACC-3′ (primer 1) and 5′-GCTGGGCCGA-3′ (primer 2) as described previously (15). RAPD products were separated by electrophoresis in 1.5% agarose gels with Tris-acetate-EDTA running buffer at 9 V/cm for 3 h. Molecular size standards were included on the agarose gels (1-kb ladder; Invitrogen).
Cloning of full-length pmrAB and phoPQ.
The full length of pmrAB was cloned from a cosmid clone that was generated from P. aeruginosa PAO1 and obtained from the Pseudomonas Genetic Stock Center (www.pseudomonas.med.ecu.edu/). The cosmid clone pMO0010107 was digested by HindIII and SacI to isolate a 9.2-kb DNA fragment containing pmrAB, which was inserted into the same restriction enzyme sites of a P. aeruginosa/E. coli shuttle vector, pUCP18 (22), and was named pAU126. Since a cosmid clone containing full-length phoPQ was not available from the Pseudomonas Genetic Stock Center, full-length oprH-phoPQ was amplified from P. aeruginosa PAO1 using the PCR primer pair 5′-CAGGCAGATCACGAGAAACA-3′ and 5′-CAGCCGAACAGACTTCAGCG-3′ (3,060 bp from positions 1276710 to 1279770; http://www.pseudomonas.com/). The PCR fragment was inserted into the shuttle vector pUCP18 and was named pAU124.
Full-length oprH-phoPQ (3,060 bp) from clinical isolates (NY214, NY215, NY217, and NY219) was cloned by PCR methods using the same PCR primer pair as that used above and were named pAU146, pAU147, pAU148, and pAU149, respectively. A portion of pmrB from the clinical isolate NY220 also was cloned by PCR methods. A PCR primer pair (5′-GCGGTGGAGGCGGTACCGCTGG-3′ and 5′-GGGAATTCTCAGATATGTGACCGCCCGC-3′) was used to amplify the PCR fragment (803 bp from positions 5365390 to 5366193; http://www.pseudomonas.com/) that comprised the putative histidine box motif (20) and the C terminus with KpnI/SacI restriction enzyme sites at both ends of the PCR fragment. The PCR fragment digested by KpnI/SacI then was replaced by the same restriction fragment from the pmrAB clone (pAU126). The new pmrAB clone containing the histidine box motif and C terminus from NY220 was named pAU154. All cloned PCR amplicons were subjected to confirmatory sequencing (SeqWright DNA Sequencing Services, Houston, TX) to rule out the presence of unwanted mutations introduced by PCR.
Gene replacement.
A gentamicin (Gm) resistance cassette from pGMΩ1 (21) was inserted into BsmI/NruI to delete 1,870 bp from oprH-phoPQ (pAU124). The Gm cassette also was inserted into PstI to delete 1,466 bp from pmrAB (pAU126). The DNA fragments carrying a deletion of oprH-phoPQ and pmrAB were inserted into the conjugative plasmid pRTP1 (23), and the resulting plasmids were used to delete the genes of P. aeruginosa PAO1 by biparental conjugation as described previously (7). A portion of the gene arnB also was amplified using a PCR primer pair of 5′-CGGCCCCCTGGCGGAGCGAT-3′ and 5′-AGGTCGGCCAGGTTGTATTT-3′ (1,077 bp from positions 3979487 to 3980564; http://www.pseudomonas.com/). The PCR fragment was cloned into pBluescript SK (Stratagene, La Jolla, CA), and a tetracycline (Tc) resistance cassette from Tn5 (21) was inserted into the HincII site of the PCR fragment. The PCR fragment inactivated by the Tc cassette was used to knock out the arnB gene as mentioned above. The gene knockouts were confirmed for authenticity by PCR methods as described previously (24).
Nucleotide sequence accession numbers.
The nucleotide sequences of phoQ from polymyxin B-resistant clinical isolates of P. aeruginosa (NY214, NY215, NY217, and NY219) and pmrB from a polymyxin B-resistant clinical isolate of P. aeruginosa (NY220) have been submitted to the GenBank nucleotide sequence databases under the accession numbers GQ266138, GQ266139, GQ266140, GQ266141, and GQ266142, respectively.
RESULTS AND DISCUSSION
Antibiotic susceptibility of clinical isolates.
The antibiotic susceptibility of the five clinical isolates of P. aeruginosa was determined as MICs of polymyxin B, β-lactams (azlocillin, carbenicillin, ceftriaxone, and ticarcillin), fluoroquinolones (ciprofloxacin and nalidixic acid), and aminoglycosides (gentamicin and tobramycin). The polymyxin B MICs for all of the isolates were 8 μg/ml. Also, the MICs of all other antibiotics except fluoroquinolones were higher (at least fourfold) than those for the reference strain P. aeruginosa PAO1. The fluoroquinolone MICs for the two isolates NY217 and NY219 were similar to those for the reference strain PAO1 (Table 1). Genotypic relatedness among the five clinical isolates also was examined by RAPD genotype fingerprinting patterns. Two arbitrary PCR primers showed quite a distinct genotype pattern for each isolate, including strain PAO1 (Fig. 1). This result suggests that the isolates are of different lineages.
TABLE 1.
Antibiotic susceptibility of P. aeruginosa clinical isolates
| Antibiotic | MIC (μg/ml) for P. aeruginosa straina: |
|||||
|---|---|---|---|---|---|---|
| PAO1 | NY214 | NY215 | NY217 | NY219 | NY220 | |
| Azlocillin | 2 | 8 | 8 | 16 | 8 | 16 |
| Carbenicillin | 64 | 256 | 256 | >256 | 256 | >256 |
| Ceftriaxone | 4 | 16 | 32 | 16 | 32 | 32 |
| Ticarcillin | 8 | 32 | 32 | 32 | 32 | 32 |
| Ciprofloxacin | 0.5 | 8 | 4 | 1 | 0.5 | 8 |
| Nalidixic acid | 64 | >256 | >256 | 64 | 64 | >256 |
| Gentamicin | 0.25 | 4 | 32 | 4 | 4 | 4 |
| Tobramycin | 0.25 | 2 | 2 | 1 | 2 | 8 |
| Polymyxin B | 0.5 | 8 | 8 | 8 | 8 | 8 |
MIC measurements were repeated three times with identical results.
FIG. 1.
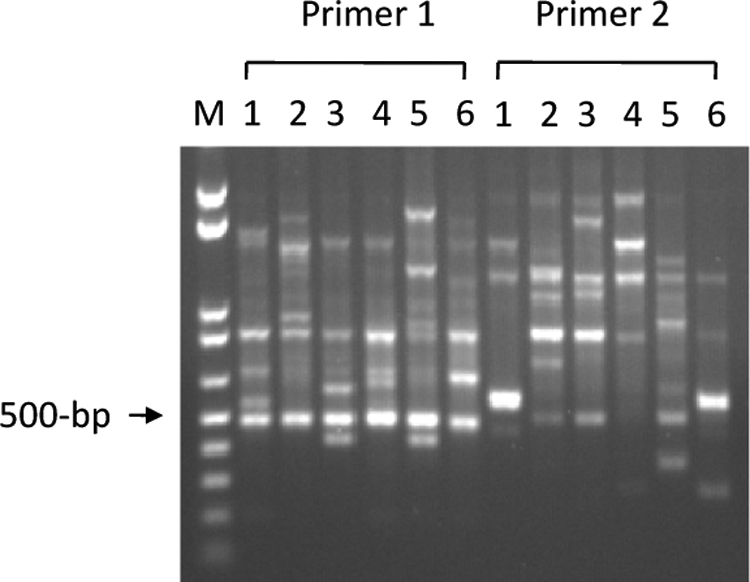
Genotype fingerprinting analysis of clinical isolates of P. aeruginosa. RAPD fingerprinting genotype analysis was performed using arbitrary PCR primers 1 and 2 as described in Materials and Methods. Lane 1 shows reference strain PAO1, and lanes 2 to 6 are clinical isolates of NY214, NY215, NY217, NY219, and NY220, respectively. Lane M is a 0.1- to 1-kb ladder (Invitrogen).
Genetic complementation of polymyxin B resistance by phoPQ and pmrAB.
It has been reported that polymyxin B susceptibility is associated with the LPS structure that is directed by arnBCADTEF and modulated by two-component regulatory systems of pmrAB and phoPQ (13, 14, 16). To examine the role of the two-component regulatory systems in polymyxin B resistance, full-length phoPQ (pAU124) and pmrAB (pAU126) cloned from the reference strain PAO1 were introduced into each of the five clinical isolates, and the polymyxin B MICs for the clones were compared to those for the parental isolates. Polymyxin B MICs for four isolates (NY214, NY215, NY217, and NY219) harboring pAU124 were reduced to 0.5 or 1 μg/ml. However, the MICs for the same isolates harboring pAU126 were unchanged. The polymyxin B MIC for isolate NY220 harboring pAU126 was reduced to 1 or 2 μg/ml, whereas the MIC for the same isolate harboring pAU124 was unchanged. The MICs for the five clinical isolates were unchanged by the cloning vector (pUCP18) (Table 2). These results suggest that polymyxin B resistance from the four clinical isolates (NY214, NY215, NY217, and NY219), which is sensitized by an intact phoPQ, is associated with a phoPQ mutation, and that polymyxin B resistance from the isolate NY220, which is sensitized by an intact pmrAB, is associated with a pmrAB mutation.
TABLE 2.
Complementation analysis of polymyxin B-resistant clinical isolates of P. aeruginosa
| Clinical isolate | Polymyxin B MIC (μg/ml) for strain harboring: |
||
|---|---|---|---|
| pAU124a (phoPQ) | pAU126 (pmrAB) | pUCP18 (cloning vector) | |
| NY214 | 1 | 4 (8)b | 8 |
| NY215 | 0.5 | 8 | 8 |
| NY217 | 1 | 8 | 8 |
| NY219 | 1 | 8 | 8 |
| NY220 | 8 | 1 (2) | 8 |
Full-length phoPQ and pmrAB were cloned from P. aeruginosa PAO1 as described in Materials and Methods.
Numbers in parentheses denote repeated MIC measurements performed more than three times.
DNA sequence analysis of phoQ and pmrB from the polymyxin B-resistant clinical isolates.
In vitro-acquired polymyxin B-resistant P. aeruginosa PAK showed nucleotide sequence substitutions that caused amino acid substitutions in a histidine sensor kinase of PmrB (20). To examine whether any amino acid substitution in the histidine sensor kinases of PhoQ and PmrB was associated with polymyxin B resistance, the genes of the resistant isolates were compared to those of the strain PAO1. Results revealed that a single-nucleotide substitution in phoQ substituted a single amino acid for isolates NY214 (V to G), NY217 (H to R), and NY219 (V to G) or a nucleotide deletion (17-bp deletion) in phoQ that shifted a reading frame at the 152nd amino acid and the truncated protein for isolate NY215. Nucleotide substitutions in pmrB substituted two amino acids (A to T and Y to H) for the sequence of isolate NY220 (Table 3 ). Nucleotide sequences of full-length phoQ from isolate NY220 and a putative histidine box motif of pmrB (542 bp from positions 5365240 to 5365782) from the isolates NY214, NY2215, NY217, and NY219 also were determined. Results showed that none of the DNA sequences resulted in amino acid substitutions for PhoQ and PmrB compared to the sequence of the strain PAO1 (Table 3).
TABLE 3.
DNA sequence analysis of phoQ and pmrB from polymyxin B-resistant clinical isolates of P. aeruginosa
| Isolate | Amino acid alteration for: |
|
|---|---|---|
| PhoQa | PmrBb | |
| NY214 | Nucleotide substitution (T at position 1779141 to G) resulting in amino acid substitution V260G | No change |
| NY215 | 17 nucleotides deletion from position 1278818 generated a frameshift with a truncated protein at the 152nd amino acid | No change |
| NY217 | Nucleotide substitution (A at position 1279029 to G) resulting in amino acid substitution H223R | No change |
| NY219 | Nucleotide substitution (T at position 1779141 to G) resulting in amino acid substitution | No change |
| NY220 | No change | Nucleotide substitution (G at position 5365488 to A) resulting in amino acid substitution A247T; nucleotide substitution (T at position 5365792 to C) resulting in amino acid substitution Y345H |
The sequence of full-length phoQ (1,347 bp; 448 amino acids) was compared to that of P. aeruginosa PAO1 (www.pseudomonas.com).
A putative histidine box (H-box; 550 bp) as designated by Moskowitz et al. (20) was analyzed for NY214, NY215, NY217, and NY219; the full-length sequence of pmrB (1,434 bp; 477 amino acids) was analyzed for NY220.
We found that four of the polymyxin B-resistant clinical isolates carried alterations in PhoQ, and the other one carried alterations in PmrB. Although the majority of the alterations were amino acid substitutions, one isolate showed a truncated protein for PhoQ. The insertion inactivation of PhoQ in P. aeruginosa PAO1 showed the increased expression of arnBCADTEF (8), suggesting that the clinical isolate (NY215) carrying truncated PhoQ has the increased expression of arnBCADTEF. In addition to the amino acid substitutions in PhoQ or PmrB, the complete inactivation of PhoQ also is one of genetic alterations responsible for polymyxin B resistance.
Expression of phoQ and pmrB from the polymyxin B-resistant clinical isolates.
Since the genes phoQ and pmrB are controlled by their upstream genes (oprH and pmrA) (13, 16), full-length oprH-phoPQ from NY214, NY215, NY217, and NY219 and full-length pmrAB from NY220 were cloned. Mutant strains of PAO1(ΔoprH-phoPQ) or PAO1(ΔpmrAB) were constructed as described in Materials and Methods. The mutant PAO1 strains were transformed by each clone of oprH-phoPQ or pmrAB from the resistant clinical isolates including PAO1, and the transformants were used to determine polymyxin B susceptibility. MICs for the two mutant strains (ΔoprH-phoPQ and ΔpmrAB) and the same mutant strains harboring each clone of oprH-phoPQ or pmrAB from PAO1 were unchanged compared to those for the parental strain PAO1, suggesting that lacking the two-component regulatory systems has no effect on polymyxin B susceptibility. However, the mutant strain (ΔoprH-phoPQ) harboring oprH-phoPQ from the four resistant clinical isolates (NY214, NY215, NY217, and NY219) restored their polymyxin B resistance (MICs from 0.5 to 4 μg/ml), and the mutant strain (ΔpmrAB) harboring pmrAB from the resistant clinical isolate NY220 also restored its polymyxin B resistance (MICs from 0.25 to 4 μg/ml). The MICs for the mutant strains harboring the cloning vector (pUCP18) were unchanged (Table 4).
TABLE 4.
Polymyxin B susceptibility in mutant P. aeruginosa strains
| P. aeruginosa strain | Polymyxin B MICa (μg/ml) |
|---|---|
| PAO1 | 0.5 |
| PAO1(ΔoprΗ-phoPQ::Gm) | 0.5 |
| PAO1(ΔoprΗ-phoPQ::Gm)/pAU124 (oprH-phoPQ | |
| from PAO1) | 0.25 |
| PAO1(ΔoprΗ-phoPQ::Gm)/pAU146 (oprH-phoPQ | |
| from NY214) | 4 |
| PAO1(ΔoprΗ-phoPQ::Gm)/pAU147 (oprH-phoPQ | |
| from NY215) | 4 |
| PAO1(ΔoprΗ-phoPQ::Gm)/pAU148 (oprH-phoPQ | |
| from NY217) | 4 |
| PAO1(ΔoprΗ-phoPQ::Gm)/pAU149 (oprH-phoPQ | |
| from NY219) | 4 |
| PAO1(ΔoprΗ-phoPQ::Gm)/pUCP18 (vector) | 0.25 |
| PAO1(ΔpmrAB::Tc) | 0.25 |
| PAO1(ΔpmrAB::Tc)/pAU126 (pmrAB from PAO1) | ≤0.25 |
| PAO1(ΔpmrAB::Tc)/pAU154 (pmrAB from NY220) | 4 |
| PAO1(ΔpmrAB::Tc)/pUCP18 | 0.25 |
| PAO1(arnB::Tc) | 0.5 |
| NY214(arnB::Tc) | 0.5 |
| NY215(arnB::Tc) | 0.5 |
| NY217(arnB::Tc) | 0.5 |
| NY219(arnB::Tc) | NDb |
| NY220(arnB::Tc) | 0.5 |
MIC measurements were repeated three times with identical results.
ND, not determined.
Involvement of arnBCADTEF in polymyxin B resistance of clinical isolates.
To clarify the involvement of LPS modifications in the polymyxin B resistance of the clinical isolates, arnB was inactivated by inserting a Tc resistance cassette. All clinical isolates with the inactivation of arnB became susceptible to polymyxin B at the same level as that of susceptible P. aeruginosa PAO1 (MICs fell from 8 to 0.5 μg/ml) (Table 4). These results suggest that the polymyxin B resistance of the clinical isolates is fully explained by the LPS modifications directed by arnBCADTEF.
Overall, we found alterations in signal sensor kinases of PhoQ or PmrB from polymyxin B-resistant clinical isolates and confirmed that these alterations were associated with resistance to polymyxin B. These findings suggest that polymyxin B resistance in clinical isolates of P. aeruginosa usually is caused by alterations in either PhoQ or PmrB, which may have become evident in response to the increased use of polymyxins in recent years.
Acknowledgments
We are grateful to David Landman and John Quale for providing polymyxin B-resistant clinical isolates of P. aeruginosa.
Footnotes
Published ahead of print on 14 September 2009.
REFERENCES
- 1.Arnold, T. M., G. N. Forrest, and K. J. Messmer. 2007. Polymyxin antibiotics for gram-negative infections. Am. J. Health Syst. Pharm. 64:819-826. [DOI] [PubMed] [Google Scholar]
- 2.CLSI. 2007. Performance standards for antimicrobial susceptibility testing: 17th informational supplement M100-S17. Clinical and Laboratory Standards Institute, Wayne, PA.
- 3.Driscoll, J. A., S. L. Brody, and M. H. Kollef. 2007. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67:351-368. [DOI] [PubMed] [Google Scholar]
- 4.Falagas, M. E., and I. A. Bliziotis. 2007. Pandrug-resistant gram-negative bacteria: the dawn of the post-antibiotic era? Int. J. Antimicrob. Agents 29:630-636. [DOI] [PubMed] [Google Scholar]
- 5.Falagas, M. E., P. K. Koletsi, and I. A. Bliziotis. 2006. The diversity of definitions of multidrug-resistant (MDR) and pandrug-resistant (PDR) Acinetobacter baumannii and Pseudomonas aeruginosa. J. Med. Microbiol. 55:1619-1629. [DOI] [PubMed] [Google Scholar]
- 6.Falagas, M. E., P. I. Rafailidis, D. K. Matthaiou, S. Virtzili, D. Nikita, and A. Michalopoulos. 2008. Pandrug-resistant Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii infections: characteristics and outcome in a series of 28 patients. Int. J. Antimicrob. Agents 32:450-454. [DOI] [PubMed] [Google Scholar]
- 7.Gambello, M. J., and B. H. Iglewski. 1991. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J. Bacteriol. 173:3000-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gooderham, W. J., S. L. Gellatly, F. Sanschagrin, J. B. McPhee, M. Bains, C. Cosseau, R. C. Levesque, and R. E. Hancock. 2009. The sensor kinase PhoQ mediates virulence in Pseudomonas aeruginosa. Microbiology 155:699-711. [DOI] [PubMed] [Google Scholar]
- 9.Gooderham, W. J., and R. E. Hancock. 2009. Regulation of virulence and antibiotic resistance by two-component regulatory systems in Pseudomonas aeruginosa. FEMS Microbiol. Rev. 33:279-294. [DOI] [PubMed] [Google Scholar]
- 10.Kuo, L. C., C. J. Yu, L. N. Lee, J. L. Wang, H. C. Wang, P. R. Hsueh, and P. C. Yang. 2003. Clinical features of pandrug-resistant Acinetobacter baumannii bacteremia at a university hospital in Taiwan. J. Formos. Med. Assoc. 102:601-606. [PubMed] [Google Scholar]
- 11.Landman, D., S. Bratu, M. Alam, and J. Quale. 2005. Citywide emergence of Pseudomonas aeruginosa strains with reduced susceptibility to polymyxin B. J. Antimicrob. Chemother. 55:954-957. [DOI] [PubMed] [Google Scholar]
- 12.Lodise, T. P., C. D. Miller, J. Graves, J. P. Furuno, J. C. McGregor, B. Lomaestro, E. Graffunder, and L. A. McNutt. 2007. Clinical prediction tool to identify patients with Pseudomonas aeruginosa respiratory tract infections at greatest risk for multidrug resistance. Antimicrob. Agents Chemother. 51:417-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macfarlane, E. L., A. Kwasnicka, and R. E. Hancock. 2000. Role of Pseudomonas aeruginosa PhoP-phoQ in resistance to antimicrobial cationic peptides and aminoglycosides. Microbiology 146:2543-2554. [DOI] [PubMed] [Google Scholar]
- 14.Macfarlane, E. L., A. Kwasnicka, M. M. Ochs, and R. E. Hancock. 1999. PhoP-PhoQ homologues in Pseudomonas aeruginosa regulate expression of the outer-membrane protein OprH and polymyxin B resistance. Mol. Microbiol. 34:305-316. [DOI] [PubMed] [Google Scholar]
- 15.Mahenthiralingam, E., M. E. Campbell, J. Foster, J. S. Lam, and D. P. Speert. 1996. Random amplified polymorphic DNA typing of Pseudomonas aeruginosa isolates recovered from patients with cystic fibrosis. J. Clin. Microbiol. 34:1129-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McPhee, J. B., M. Bains, G. Winsor, S. Lewenza, A. Kwasnicka, M. D. Brazas, F. S. Brinkman, and R. E. Hancock. 2006. Contribution of the PhoP-PhoQ and PmrA-PmrB two-component regulatory systems to Mg2+-induced gene regulation in Pseudomonas aeruginosa. J. Bacteriol. 188:3995-4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McPhee, J. B., S. Lewenza, and R. E. Hancock. 2003. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 50:205-217. [DOI] [PubMed] [Google Scholar]
- 18.McVay, C. S., M. Velasquez, and J. A. Fralick. 2007. Phage therapy of Pseudomonas aeruginosa infection in a mouse burn wound model. Antimicrob. Agents Chemother. 51:1934-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mittal, R., R. K. Khandwaha, V. Gupta, P. K. Mittal, and K. Harjai. 2006. Phenotypic characters of urinary isolates of Pseudomonas aeruginosa and their association with mouse renal colonization. Indian J. Med. Res. 123:67-72. [PubMed] [Google Scholar]
- 20.Moskowitz, S. M., R. K. Ernst, and S. I. Miller. 2004. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J. Bacteriol. 186:575-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park, S. M., C. D. Lu, and A. T. Abdelal. 1997. Cloning and characterization of argR, a gene that participates in regulation of arginine biosynthesis and catabolism in Pseudomonas aeruginosa PAO1. J. Bacteriol. 179:5300-5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schweizer, H. P. 1991. Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene 97:109-121. [DOI] [PubMed] [Google Scholar]
- 23.Stibitz, S., W. Black, and S. Falkow. 1986. The construction of a cloning vector designed for gene replacement in Bordetella pertussis. Gene 50:133-140. [DOI] [PubMed] [Google Scholar]
- 24.Trieber, C. A., and D. E. Taylor. 2002. Mutations in the 16S rRNA genes of Helicobacter pylori mediate resistance to tetracycline. J. Bacteriol. 184:2131-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang, C. Y., J. S. Jerng, K. Y. Chen, L. N. Lee, C. J. Yu, P. R. Hsueh, and P. C. Yang. 2006. Pandrug-resistant Pseudomonas aeruginosa among hospitalised patients: clinical features, risk-factors and outcomes. Clin. Microbiol. Infect. 12:63-68. [DOI] [PubMed] [Google Scholar]
- 26.Yeaman, M. R., and N. Y. Yount. 2003. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 55:27-55. [DOI] [PubMed] [Google Scholar]
- 27.Zavascki, A. P., L. Z. Goldani, J. Li, and R. L. Nation. 2007. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J. Antimicrob. Chemother. 60:1206-1215. [DOI] [PubMed] [Google Scholar]