

Abstract
The largest part of the Earth's microbial biomass is stored in cold environments, which represent almost untapped reservoirs of novel species, processes, and genes. In this study, the first metagenomic survey of the metabolic potential and phylogenetic diversity of a microbial assemblage present in glacial ice is presented. DNA was isolated from glacial ice of the Northern Schneeferner, Germany. Pyrosequencing of this DNA yielded 1,076,539 reads (239.7 Mbp). The phylogenetic composition of the prokaryotic community was assessed by evaluation of a pyrosequencing-derived data set and sequencing of 16S rRNA genes. The Proteobacteria (mainly Betaproteobacteria), Bacteroidetes, and Actinobacteria were the predominant phylogenetic groups. In addition, isolation of psychrophilic microorganisms was performed, and 13 different bacterial isolates were recovered. Analysis of the 16S rRNA gene sequences of the isolates revealed that all were affiliated to the predominant groups. As expected for microorganisms residing in a low-nutrient environment, a high metabolic versatility with respect to degradation of organic substrates was detected by analysis of the pyrosequencing-derived data set. The presence of autotrophic microorganisms was indicated by identification of genes typical for different ways of carbon fixation. In accordance with the results of the phylogenetic studies, in which mainly aerobic and facultative aerobic bacteria were detected, genes typical for central metabolism of aerobes were found. Nevertheless, the capability of growth under anaerobic conditions was indicated by genes involved in dissimilatory nitrate/nitrite reduction. Numerous characteristics for metabolic adaptations associated with a psychrophilic lifestyle, such as formation of cryoprotectants and maintenance of membrane fluidity by the incorporation of unsaturated fatty acids, were detected. Thus, analysis of the glacial metagenome provided insights into the microbial life in frozen habitats on Earth, thereby possibly shedding light onto microbial life in analogous extraterrestrial environments.
More than 75% of the Earth's biosphere is constantly exposed to temperatures below 5°C. Permafrost and ice contain one of the lowest temperature settings of all habitats on Earth and exhibit high stability with respect to environmental conditions. These habitats represent a long-term, chronological archive of microorganisms. It has been postulated that permafrost and glacial ice harbor the oldest prokaryotes on Earth (54). Ionic impurities prevent freezing of veins in ice and thin films in permafrost and permit transport of nutrients to and products from microorganisms (35). Microorganisms living in these environments have evolved unique features of their proteins, membranes, and genetic responses to thermal shifts. Since there is evidence for the presence of ice on Mars and Jupiter's moon Europa, the interest in the investigation of microbial life in frozen habitats has increased. Glacial ice is regarded as an environment, which is equivalent to extraterrestrial cold habitats (32). Recently, high-throughput pyrosequencing technology has been applied for the metagenomic characterization of environmental microbial communities (3, 16). The most important advantages of this cloning-independent approach are the avoidance of cloning bias and bias introduced by application of PCR amplification. Metagenomic analyses of environmental samples have been proposed to be the most accurate quantitative approach for description of microbial communities present in a habitat (52). In addition to the assessment of the taxonomic composition, relative abundances of all genes and metabolic profiles can be determined. To date, a pyrosequencing-based metagenomic analysis of a permanently frozen habitat has not been conducted. In several studies, the prokaryotic diversity of glacial and subglacial habitats in America (9, 44), Asia (10, 55), Antarctica (4, 37), Greenland (32, 42), and New Zealand (19) has been analyzed based on cultivation and analysis of 16S rRNA genes. However, studies of the microbial composition of European glaciers are rare. These glaciers have been mainly investigated with respect to the presence of yeasts (51) and bacterial population sizes (41).
Since the 1980s, glaciers in the European Alps have been receding quickly. According to Haeberli et al. (21), these glaciers lost half of their total volume between 1850 and 1975. Recently, the speed of mass and volume losses of these glaciers accelerated. This is probably a result of the global climate change. It has been estimated that an almost complete deglaciation of the European Alps will happen within this century (21).
In the present study, we have analyzed the phylogenetic composition and metabolic potential of the microbial assemblage present in glacier ice of the Northern Schneeferner, which is the largest and highest glacier of the five glaciers located in the German Alps. The Northern Schneeferner was first described in 1820 and covers an area of 340,000 m2. The present study is the first to assess the taxonomic and metabolic diversity of a glacial microbial assemblage by analysis of a large pyrosequencing-derived data set. To complement this approach, traditional methods for taxonomic assessment, such as PCR amplification of 16S rRNA genes and isolation of microorganisms, were used.
MATERIALS AND METHODS
Sampling, media, and growth conditions.
Sampling of glacier ice from the Northern Schneeferner (Germany; 47°25′N, 10°59′E) in June 2005 and the subsequent DNA isolation have been described previously (43). Briefly, the glacier ice was collected from the surface of the ice core after melting of the snow cover. Sampling of glacial ice was performed by using clean surface-sterilized tools. Ice blocks up to a depth of 0.5 m were collected and transferred into sterile polypropylene bags (Sarstedt AG & Co., Nümbrecht, Germany), which were stored in sterile plastic containers. The first 30 cm of the sampled glacial ice core exposed to the glacier surface was removed and discarded. The samples for DNA extraction were returned frozen to the laboratory. Subsequently, the ice was melted at 4°C and filtered by using a sterile cellulose acetate membrane (pore size, 0.2 μm; Whatman, Dassel, Germany). The melted glacial ice contained (per liter): As, <0.01 mg; Pb, 0.022 mg; Ca, 4.7 mg; Fe, <0.02 mg; K, 1.1 mg; Cu, 0.122 mg; Mg, 0.23 mg; Mn, 0.083 mg; Na, 2.4 mg; Ni, 0.007 mg; and Zn, 0.81 mg. Total genomic DNA from glacier ice samples was extracted from the membranes by using a NucleoSpin tissue kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instructions with slight modifications (43). The concentration of extracted DNA was quantified by using a NanoDrop ND-1000 spectrophotometer (Peqlab Biotechnologie GmbH, Erlangen, Germany). Cell-containing membranes were stored until use at −80°C. Control experiments in which frozen sterile water instead of glacial ice was used and then subjected to the above-mentioned procedures confirmed that we did not introduce any contamination that could be detected by PCR or cultivation.
To isolate microorganisms, 100-μl portions of molten glacier ice were spread directly onto the following agar plates: full strength R2A (38), 0.25 strength R2A, 0.5 strength tryptic soy agar (Oxoid, Cambridge, United Kingdom), and 0.2 strength Luria-Bertani medium. Plates were incubated at 4°C. Isolates that formed colonies with different morphologies were purified by repeated transfer on agar plates. The ability of each isolate to form colonies at 4, 18, 30, and 37°C was determined.
Amplification of 16S rRNA genes and construction of clone libraries.
The PCR mixture (50 μl) for amplification of 16S rRNA genes contained 5 μl of Mg-free polymerase buffer (MBI Fermentas, St. Leon-Rot, Germany), 200 μM concentrations of each of the four deoxynucleoside triphosphates, 1.75 mM MgCl2, 2 μM concentrations of each of the primers, 1 U of Taq DNA polymerase (MBI Fermentas), and 20 ng of isolated DNA as a template. The following thermal cycling scheme was used: initial denaturation at 95°C for 2 min, 25 cycles of denaturation at 95°C for 1.5 min, and annealing for 1 min at a temperature gradient ranging from 55 to 66°C for the archaeal primer pairs (Arch8F/Arch958R, Arch8F/Arch1041R, Arch21F/Arch958R, and Arch21F/Arch1041R) and at 55, 63, and 55°C for the bacterial primer pairs 349F/1114R, 8F/1114R, and 8F/1492R, respectively (for primer sequences, see Table S1 in the supplemental material). Subsequently, extension at 72°C (1 min per 1,000 bp), followed by a final extension period at 72°C for 10 min, was carried out. In the case of isolates, a cell suspension (1 μl) derived from single colonies was directly used as a template for the PCR. Negative controls contained the entire reaction mixture without the template DNA. Finally, all obtained PCR products were analyzed for appropriate size and then purified by using the QIAquick PCR purification kit (Qiagen, Hilden, Germany). PCR products obtained from isolates were sequenced directly, whereas products derived from environmental DNA were cloned into pCR2.1-TOPO as recommended by the manufacturer (Invitrogen, Karlsruhe, Germany). The PCR product-containing vectors were used to transform Escherichia coli TOP10. A total of 419 recombinant plasmids were isolated from randomly selected E. coli clones and sequenced. To analyze the microbial community present in the glacier ice by denaturing gradient gel electrophoresis, 16S rRNA genes were amplified by using the primer pair 341F/907R (see Table S1 in the supplemental material). To primer 341F a GC clamp (5′-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG-3′) was attached at the 5′ terminus. PCRs were performed by using the above-mentioned reaction mixture containing 10 μg of bovine serum albumin.
Sequence analysis of the 16S rRNA gene sequences.
The Göttingen Genomics Laboratory (Göttingen, Germany) determined the 16S rRNA gene sequences. The 16S rRNA gene sequences from clone libraries and isolates were edited by using the gap4 program from the Staden Package (6). All sequences were checked for chimeric artifacts by using Mallard (2) and the CHIMERA_CHECK program (28) of the Ribosomal Database Project (RDP) II database (12). Taxonomic classification was performed by using the RDP sequence match tool (http://rdp.cme.msu.edu/seqmatch/seqmatch_intro.jsp) and the BLAST tool of the National Center for Biotechnology Information (NCBI) database (1). Operational taxonomic units (OTUs) were determined at sequence similarity levels of 99, 97, and 80% by using the furthest-neighbor method of DOTUR (39). To determine the number of observed unique OTUs as a function of the distance between sequences and the number of sequences sampled, rarefaction analysis was performed. In addition to the Shannon-Weaver diversity index, the Chao1 richness estimator, and the abundance-based coverage estimator (ACE) were calculated by using DOTUR (39). Only one sequence per OTU (>99% identity cutoff) was used for construction of phylogenetic trees. Sequences of the nearest neighbors were retrieved from the NCBI and the RDP II databases. The 16S rRNA gene sequences from clone libraries and the most similar neighbors were imported into the most recent SSU Ref SILVA database (www.arb-silva/download/) of the ARB program package (26). Multiple sequence alignments were checked manually and improved by using the ARB editor tool. Phylogenetic trees were created by using the maximum-parsimony algorithm implemented in ARB. The robustness of obtained tree topologies was evaluated by bootstrap analysis with 100 resamplings.
Pyrosequencing and analysis of metagenomic GS-FLX data.
Glacial DNA purified from approximately 8 kg of ice has been used as starting material for pyrosequencing. DNA was sequenced by conducting two full runs (70x75 picotitre plates) on a Roche GS-FLX pyrosequencer (Roche, Mannheim, Germany). The pyrosequencing-derived data set was analyzed with the phylogenetic algorithm CARMA (25). Conserved Pfam domain and protein families were determined and classified into a higher-order taxonomy as described by Krause et al. (25). In addition, the data set was compared to the NCBI-nr, RDP II, KEGG, and COG databases. BLASTX (1) was used to query the NCBI-nr, COG (48), and the KEGG (24) databases. Comparisons of the pyrosequencing-derived data set to the NCBI-nr and COG databases were performed at a cutoff e-value of 10−5. For comparisons to the KEGG database, a cutoff e-value of 10−3 was chosen. These values were also used for analysis of the metagenomes from the Yellowstone National Park Octopus and Mushroom hot springs (NCBI project ID 20953), Waseca County farm soil (AAFX00000000) (50), and a microbial community isolated from an aquatic lake in Antarctica (NCBI project ID 33179), which were used for comparison to the glacial ice metagenome. In order to identify taxonomic marker genes in the pyrosequencing-derived glacial data set, sequences were analyzed by a BLASTN search against the RDP II database. Matches with an e-value of <10−3 and a match length of >200 nucleotides were accepted. For assignation to phylogenetic groups, matches were reanalyzed by using the RDP Classifier (53). Matches with a RDP confidence estimate below 60% were designated “unclassified.”
Nucleotide sequence accession numbers.
The 16S rRNA gene sequences have been deposited in the GenBank database under accession numbers EU978474 to EU978633, EU978636 to EU978652, and EU978654 to EU978854. The sequences derived from pyrosequencing have been deposited in the NCBI Short Read Archive under accession number SRA001163.
RESULTS AND DISCUSSION
Phylogenetic composition of the bacterial community.
Environmental DNA was extracted from the samples of the Northern Schneeferner glacier. For this purpose, the ice samples were melted at 4°C. Subsequently, cells were concentrated by filtration. The cell-containing membrane filters were used as starting material for DNA isolation (43). Approximately 5 μg of DNA per kg of melted glacier ice were recovered. As expected from the low microbial community size of glacier ice (31), the obtained DNA yield is much lower than that of DNA isolations from other habitats such as marine sediments (2.8 to 14.6 μg/g) (27) and soils (1 to 500 μg/g) (14). Further analysis revealed that the isolated DNA was suitable for PCR amplification and pyrosequencing. In a previous study, isolated glacial DNA has been successfully used for construction and screening of metagenomic libraries (43). Pyrosequencing of glacial DNA yielded 1,076,539 reads, with an average read length of 223 bp.
The phylogenetic diversity present in the glacial ice metagenome was assessed by the following three approaches: analysis of a constructed 16S rRNA gene library, identification and classification of 16S rRNA gene sequences in the pyrosequencing-derived data set, and evaluation of this data set based on similarities to conserved proteins families and domains using the CARMA algorithm (25) (Fig. 1).
FIG. 1.

Distribution of phylogenetic groups in glacier ice. Three different approaches were applied: analysis of amplified 16S rRNA gene sequences (A), comparison of pyrosequencing-derived sequences with the RDP II database (B) (cutoffs: match length >200 nucleotides; RDP Classifier [53] confidence estimate >60%), and taxonomic assignment of the pyrosequencing-derived sequences using CARMA (C) (25). Shown are the percentages of the phylogenetically classified sequences. Phylogenetic groups accounting for <1% of the sequences are summarized in the artificial group “Other.”
A total of 419 bacterial 16S rRNA gene sequences were amplified from glacial DNA, sequenced, and analyzed. After removal of potential chimeras (66 sequences) and duplicates (15 sequences), the remaining database contained 338 unique 16S rRNA gene sequences. In most cases, the nearest neighbors were 16S rRNA gene sequences of uncultured organisms, which were derived from cold habitats such as the ice cover of Antarctica (DQ521501), glacial ice (AY315166), or the snow cover at Spitzberg (DQ497241). In addition, many sequences were related to genera such as Polaromonas, Sphingomonas, Cryobacterium, and Stenotrophomonas, which have been found in other permanently cold environments (19, 32, 55) (see Fig. S1 to S4 in the supplemental material). To identify unique phylotypes and to estimate the bacterial richness, DOTUR analysis (39) was performed. This revealed that the 338 16S rRNA genes sequences represented 108 OTUs based on a >99% identity cutoff (Table 1) . Rarefaction curves reached saturation at a distance level of 20% (phylum level) but not at a distance level of 3% (species level) (Fig. 2). In addition, the Shannon-Weaver index of 3.44 at a genetic distance of 3% indicated a high bacterial diversity. These results together with Chao1 and ACE richness estimates (Table 1) indicated that a substantial fraction of the bacterial species diversity was assessed. More important, almost all phyla predicted to be present in glacial ice by the ACE and Chao1 richness estimators were recovered (Table 1).
TABLE 1.
Richness and diversity estimates of the bacterial 16S rRNA gene clone libraries constructed from glacier ice of the Northern Schneeferner
| Genetic distance | Richness or diversity estimatea |
|||
|---|---|---|---|---|
| Richnessb | ACEc | Chao1d | Shannone | |
| 0.01 | 108 | 173 | 153 | 4.09 |
| 0.03 | 72 | 132 | 107 | 3.44 |
| 0.1 | 28 | 38 | 35 | 2.19 |
| 0.2 | 13 | 15 | 14 | 1.66 |
The estimates were calculated by using DOTUR (39).
Richness is expressed as the number of observed unique OTUs.
ACE is a nonparametric richness estimator based on the distribution of abundant (>10) and rare (≤10) OTUs.
Chao1 is a nonparametric richness estimator based on distribution of singletons and doubletons.
Shannon, Shannon-Weaver index of diversity. A higher number indicates more diversity.
FIG. 2.

Rarefaction curves indicating the observed number of OTUs within the 16S rRNA gene libraries derived from glacial ice. The curves were calculated with DOTUR (39). OTUs are shown at the 1, 3, and 20% genetic distance level. Error bars represent the 95% confidence interval.
The analyzed bacterial sequences were affiliated to the following 11 phylogenetic groups: Proteobacteria (190 sequences, 55 OTUs), Bacteroidetes (101 sequences, 31 OTUs), Actinobacteria (33 sequences, 11 OTUs), Cyanobacteria (4 sequences, 2 OTUs), Gemmatimonadetes (3 sequences, 2 OTUs), Chloroflexi (1 sequence, 1 OTU), Verrucomicrobia (1 sequence, 1 OTU), Acidobacteria (1 sequence, 1 OTU), Deinococcus-Thermus (2 sequences, 2 OTUs), candidate division TM7 (1 sequence, 1 OTU), and unclassified Bacteria (1 sequence, 1 OTU) (Fig. 1). Phylogenetic trees were constructed using one 16S rRNA gene sequence per OTU (see Fig. S1 to S4 in the supplemental material). The predominant phylogenetic groups were Proteobacteria and Bacteroidetes. The results were supported by denaturing gradient gel electrophoresis analysis of the bacterial community, in which only representatives of these phyla were detected (see Fig. S5 and Table S2 in the supplemental material).
In order to validate the results of the clone library the pyrosequencing-derived data set was searched for 16S rRNA fragments. In this way, 1,116 16S rRNA gene fragments >200 bp were identified, of which 468 were affiliated to distinct phylogenetic groups (Fig. 1 and Data Set S1 in the supplemental material). Overall, classification of the thereby recovered sequences confirmed the results of the 16S rRNA gene clone library.
The above-reported approaches are based on analysis of a phylogenetic marker gene, whereas CARMA is based on similarities of the unassembled pyrosequencing-derived reads to conserved Pfam domains and protein families. According to Krause et al. (25), conserved gene fragments as short as 27 amino acids can be precisely classified with an average specificity of 97% (superkingdom level) to 93% (order level) by the latter approach. However, the accuracy depends on the grade of representation of taxonomic groups in the Pfam database. In the present study, a taxonomic classification was achieved at the superkingdom and phylum level for 14 and 11% of the reads, respectively. These values for classified sequences are consistent with those of a synthetic metagenome (25) but are higher than those of metagenomes from other habitats (3, 17, 20).
Most of the classified reads were assigned to Bacteria (83% of the sequences) (Fig. 3). The bacterial composition determined by CARMA was highly similar to that of the 16S rRNA-based approach with one exception (Fig. 1). In addition to the 11 phylogenetic groups detected by analysis of the 16S rRNA gene clone library the presence of at least 11 additional phyla was indicated, but these phyla were represented by <1% of all classified sequences (Fig. 1).
FIG. 3.
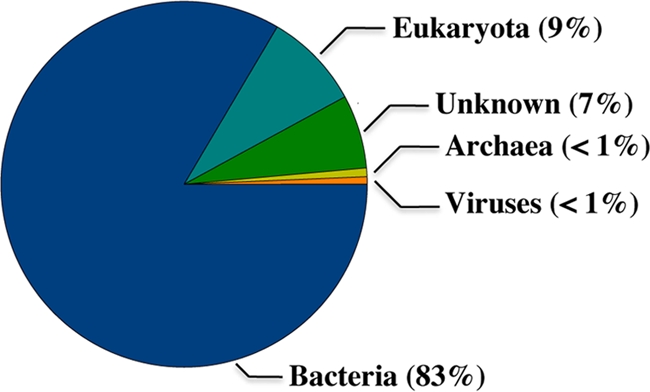
Distribution of phylogenetic groups in glacier ice determined by using the CARMA algorithm. A total of 14% of the pyrosequencing-derived data set (150,478 sequences) were phylogenetically classified at the superkingdom level. Shown are the percentages of all classified sequences. The group “Unknown” represents sequences, which match Pfam database entries that cannot be assigned to a taxonomic group.
The dominant phylogenetic group established by all approaches was the phylum Proteobacteria (56 to 65% of all classified sequences). Most proteobacterial sequences were assigned to the class Betaproteobacteria (36 to 42% of all classified sequences). This corresponded to other studies of glacier ice (19), subglacial habitats (9), and mountain snow (40). Within this class, the family Comamonadaceae includes many genera which have been found in polar and alpine environments. This indicated that these organisms are important members of glacial bacterial communities (19, 44). The second most abundant phylogenetic group was the phylum Bacteroidetes (10 to 30% of all classified sequences). In addition, a large part of the sequences grouped into the gram-positive bacteria (mainly Actinobacteria). It has been shown that Actinobacteria have a higher ability to survive in old permafrost than low GC gram-positive and gram-negative bacteria (54). This is probably caused by the ability of Actinobacteria to develop resting forms with low metabolic activity. All other bacterial phyla were represented by <4% of the classified sequences. Thus, the Proteobacteria, Bacteroidetes, and Actinobacteria represented the predominant phyla in glacier ice from the Northern Schneeferner. These groups were also abundant in ice derived from several other glaciers (32, 54, 51) and other permanently cold habitats, such as subglacial meltwater (9, 19, 44), snow (40), or Antarctic lakes (33). These results support the perception that related phylotypes exist in geographically diverse cold environments because of similar strategies for survival and remaining active at low temperatures (36). However, in most of the previous studies, the composition of bacterial communities present in glacier ice has been analyzed by culture-dependent methods.
Isolation of bacteria from glacial ice.
We also cultivated and classified bacteria from glacial ice of the Northern Schneeferner. For this purpose, melted ice was used to inoculate low-nutrient agar media. The first colonies appeared after 5 days of incubation at 4°C. Over a time period of 12 weeks, approximately 90 colonies with different morphologies were selected and the organisms were purified by repeated transfer (four to five times) to agar plates. The analysis of the 16S rRNA gene sequences of the purified organisms revealed the presence of 13 different isolates, designated glbI1 to glbI13 (see Table S3 in the supplemental material). Analysis of the 16S rRNA gene sequences showed that the isolated organisms belonged to the three most abundant phyla established by the above-described approaches. Seven of the bacterial isolates were affiliated to the Proteobacteria and three each to the Bacteroidetes and Actinobacteria (see Table S3 in the supplemental material). All of the isolates exhibited 16S rRNA gene sequence identities of 98 and 99% to their nearest described relatives, except isolate glbI4. This isolate displayed a sequence similarity of 95% to Sphingoterrabacterium pocheensis (AB267718) and putatively represented a new species. In addition, most of the 16S rRNA gene sequences of the isolates showed high similarities (97 to 100% identity) to those derived from the 16S rRNA gene clone libraries and the pyrosequencing data set (data not shown). All isolates were able to grow at temperatures of 4 and 18°C. Six isolates showed growth at 30°C, but no isolate was able to grow at 37°C. This implied that all of the isolates are either psychrophilic or psychrotolerant species. In addition, the majority of the isolates formed pigmented colonies (see Table S3 in the supplemental material). Pigmentation has been reported to be necessary for adaptation to low temperatures and resistance to environmental stress (34, 55).
Presence of archaea in glacial ice.
It has been shown that archaea dominate in young, seasonal Antarctic and Arctic sea ice and the cold interior and deep waters of the ocean (15, 23). However, archaeal 16S rRNA genes were not detected in published studies of glacial or subglacial ice. For example, Sheridan et al. (42), Christner et al. (10, 11), and Skidmore et al. (44) used archaeal primers for analyses of glacial or subglacial ice, but either no or no unambiguous PCR product was detected. It has been suggested that the lack of archaea in ancient glacial ice is due to out-competition by bacteria or to a high susceptibility of archaea to death and decay in ice (33). In the present study, all attempts to amplify archaeal 16S rRNA gene sequences were also unsuccessful. In contrast to the results of the 16S rRNA gene analyses, the presence of a small number of archaea in glacial ice was indicated by CARMA analysis, but <1% of the reads were assigned to archaea (Fig. 3). In addition, functional evaluation of the pyrosequencing-derived data set revealed a few sequences that were related to ether lipid metabolism (see Data Set S2 in the supplemental material), which is a typical feature of archaea. Nevertheless, the prokaryotic glacial ice community is dominated by bacteria.
General analysis of the metabolic potential encoded by the glacial ice metagenome.
To date, little is known about the dominant metabolic functions in permanently frozen ecosystems. We established the functional diversity of the Northern Schneeferner by comparisons of all pyrosequencing-derived sequences (1,076,539 sequences) to the KEGG and COG databases. A total of 29% of all sequences were assigned to 4,876 unique KO (KEGG Orthology) groups at a cutoff e-value of 10−3. In addition, 33% of all sequences showed matches to the COG database, which correspond to 3,709 unique COGs at a cutoff e-value of 10−5.
The functional category metabolism in the COG and KEGG databases was represented by 45 and 57% of all assigned sequences, respectively. Genetic information processing (KEGG) and cellular processes and signaling (COG) yielded 22 and 20% of all matches, respectively. The category environmental information processing (KEGG) and information storage and processing (COG) included 16 and 19% of all classified sequences, respectively (see Fig. S6 in the supplemental material). Within the KEGG categories, matches were separated into different subcategories and several sequences occupied more than one subcategory (Table 2 and see Data Set S2 in the supplemental material). Most of the glacial sequences in the subcategory carbohydrate metabolism shared homologies to known genes involved in pyruvate (8,523 sequences), butanoate (7,975 sequences), propanoate (7,990 sequences), starch and sucrose (4,218 sequences), and glyoxylate and dicarboxylate (4,041 sequences) metabolism. In addition, sequences were homologous to genes responsible for glycolysis/gluconeogenesis (6,450 sequences), the citrate cycle (5,677 sequences), and the pentose-phosphate pathway (4,077 sequences) (Table 2). The fraction of genes in the category methane metabolism was higher in the glacial metagenome (0.63%) than in the metagenomes derived from farm soil (0.42%), Octopus/Mushroom hot springs of the Yellowstone National Park (0.12%), and the microbial community of an aquatic lake in Antarctica (0.34%). The glacial sequences were mainly related to catalases/peroxidases, formate dehydrogenases, CO dehydrogenases, and glycine hydroxymethyltransferase. The latter enzyme is involved in assimilation of C1 substrates via the serine pathway. The metabolism of secondary metabolites was mostly associated with limonene and pinene degradation (2,924 sequences) (see Data Set S2 in the supplemental material). In addition, a large fraction of sequences (4.3%) showed similarities to genes involved in degradation and metabolism of xenobiotics (see Data Set S2 in the supplemental material). The abundance of these genes in the glacial ice metagenome is higher than that of metagenomes with different temperature settings such as the metagenomes derived from farm soil (3.37%) and the Yellowstone National Park hot springs (1.43%). In summary, the above-mentioned results indicated a high metabolic versatility with respect to the use of different carbon sources.
TABLE 2.
Number of sequences showing homologies to genes associated with KEGG pathways in the categories “carbohydrate metabolism” and “energy metabolism”a
| KEGG class ID | KEGG category [ko no.] | No. of matches (% relative abundance) |
|---|---|---|
| 1110 | Carbohydrate metabolism | 41,723 (12.33) |
| 10 | Glycolysis/gluconeogenesis [PATH:ko00010] | 6,450 (1.52) |
| 20 | Citrate cycle (tricarboxylic acid cycle) [PATH:ko00020] | 5,677 (1.33) |
| 30 | Pentose phosphate pathway [PATH:ko00030] | 4,077 (0.96) |
| 31 | Inositol metabolism [PATH:ko00031] | 518 (0.12) |
| 40 | Pentose and glucuronate interconversions [PATH:ko00040] | 1,323 (0.31) |
| 51 | Fructose and mannose metabolism [PATH:ko00051] | 3,340 (0.79) |
| 52 | Galactose metabolism [PATH:ko00052] | 1,761 (0.41) |
| 53 | Ascorbate and aldarate metabolism [PATH:ko00053] | 1,181 (0.28) |
| 500 | Starch and sucrose metabolism [PATH:ko00500] | 4,218 (0.99) |
| 520 | Nucleotide sugar metabolism [PATH:ko00520] | 1,982 (0.47) |
| 530 | Aminosugar metabolism [PATH:ko00530] | 2,949 (0.69) |
| 562 | Inositol phosphate metabolism [PATH:ko00562] | 785 (0.18) |
| 620 | Pyruvate metabolism [PATH:ko00620] | 8,523 (2.00) |
| 630 | Glyoxylate and dicarboxylate metabolism [PATH:ko00630] | 4,041 (0.95) |
| 640 | Propanoate metabolism [PATH:ko00640] | 7,990 (1.99) |
| 650 | Butanoate metabolism [PATH:ko00650] | 7,975 (1.88) |
| 660 | C5-branched dibasic acid metabolism [PATH:ko00660] | 1,072 (0.25) |
| 1120 | Energy metabolism | 28,859 (8.53) |
| 190 | Oxidative phosphorylation [PATH:ko00190] | 8,954 (2.10) |
| 191 | Pyruvate/oxoglutarate oxidoreductases | 1,068 (0.25) |
| 192 | ATPases | 1,976 (0.46) |
| 195 | Photosynthesis [PATH:ko00195] | 1,511 (0.36) |
| 196 | Photosynthesis-antenna proteins [PATH:ko00196] | 92 (0.02) |
| 680 | Methane metabolism [PATH:ko00680] | 2,694 (0.63) |
| 710 | Carbon fixation [PATH:ko00710] | 3,676 (0.86) |
| 720 | Reductive carboxylate cycle (CO2 fixation) [PATH:ko00720] | 4,676 (1.10) |
| 910 | Nitrogen metabolism [PATH:ko00910] | 5,539 (1.30) |
| 920 | Sulfur metabolism [PATH:ko00920] | 1,866 (0.44) |
The values were determined by comparison of sequences derived from pyrosequencing of the glacial DNA to the KEGG database (24) at a cutoff e-value of 10−5. Data Set S2 in the supplemental material shows a detailed list of all matches to KEGG pathways and associated KO and COG groups.
The majority of the glacial sequences in the category energy metabolism were related to genes that participate in oxidative phosphorylation (8,954 sequences). Most sequences related to photosynthesis (1,511 sequences) were associated with ATPase-like genes, and only a few sequences showed similarities to genes encoding components of photosystems and proteorhodopsin-like proteins (Table 2). This is in accordance with the low number of photosynthetic organisms detected in glacial ice. The low abundance of genes involved in photosynthesis and photosynthetic organisms might be due to the fact that the ice core is covered by snow most of the year. The abundance of genes involved in nitrogen metabolism (1.3%) in the glacial metagenome was in the range of the values determined for the other metagenomes (lake in Antarctica, 1.53%; farm soil, 1.12%; hot springs, 1.35%). This included assimilatory and respiratory nitrate/nitrite reductase-like genes as well as glutamate/glutamine synthase-like genes. The presence of these genes in the glacial ice metagenome implied that assimilation and respiration of inorganic nitrogen sources is important in glacial ice. Most sequences associated with sulfur metabolism (1,866 sequences) showed similarities to cysteine synthases, sulfate adenylyltransferases, and sulfite reductases, which participate in sulfur assimilation (Table 2). Hodson et al. (22) reported the presence of inorganic and organic forms of nitrogen and phosphorus for two glaciers in Arctic Svalbard. These authors proposed the occurrence of NH4+ assimilation and nitrification on the glacier surface and denitrification and sulfate reduction in subglacial environments. In the present study, no evidence for nitrification or respiratory sulfate reduction was detected, but dissimilatory and assimilatory nitrate/nitrite reduction was found. In addition, genes involved in assimilatory sulfate reduction were detected, indicating the presence of inorganic sulfur sources in glacier ice as suggested by Bottrell and Tranter (7). Based on the central metabolic pathways, it can be concluded that the prokaryotes present in the upper layer of the Northern Schneeferner glacier are predominantly aerobic or facultative aerobic and nonphototrophic bacteria. In addition, strictly anaerobic bacteria such as acetogenic, methanogenic, or sulfate/sulfur-reducing bacteria were not detected by phylogenetic classification of the assemblage. The presence of genes homologous to denitrifying enzymes revealed the alternative use of nitrate/nitrite as electron acceptor in the absence of oxygen. This is in accordance with the vast majority of Proteobacteria detected in the ice of the Northern Schneeferner. Most denitrifying bacteria belong to the Proteobacteria, which are known to be metabolically versatile facultative aerobic bacteria.
Functions related to a psychrophilic lifestyle and to living at low organic carbon concentrations.
Glacial ice is a low-nutrient environment. Stibal et al. (47) proposed that most of the organic matter present in glacial ice originates from allochthonous airborne material and atmospheric deposition on the glacier surface. An additional source of nutrients is glacial flour, which is generated by glacial erosion and abrasion of solid material (47). Numerous genes related to the degradation of xenobiotics, different biopolymers, and other carbon sources, indicated a high degradative capacity of the microorganisms present in the glacial ice. This might be a result of survival in a habitat with low concentrations of many different organic carbon sources. The importance of an autotrophic lifestyle in glacial ice was indicated by the presence of a significant number of genes involved in carbon fixation via the Calvin cycle, the reductive carboxylate cycle and the serine pathway (Table 2).
To maintain the fluidity of the membranes the incorporation of unsaturated fatty acids into membranes is an important characteristic of bacteria living at low temperatures (30). Correspondingly, 1,075 sequences (0.25%) of the pyrosequencing-derived data set were similar to genes associated with the biosynthesis of unsaturated fatty acids (see Data Set S2 in the supplemental material). In contrast, in metagenomes derived from environments for thermophilic microorganisms such as the Octopus and Mushroom hot springs of the Yellowstone National Park the abundance of these genes is lower (0.15%). In addition, a large number of desaturases that are essential for the conversion of saturated to unsaturated fatty acids were found in the glacial ice metagenome (Table 3). The synthesis of carotenoids (599 sequences) was indicated (see Data Set S2 in the supplemental material). These compounds have been also reported to be important pigments of cell membranes of psychrophilic bacteria, since they modulate the membrane fluidity and stability at a low temperature and high osmotic pressure (15, 18). This is consistent with the above-reported pigmentation of the organisms isolated from the Northern Schneeferner. Maintenance of protein folding is indicated by the presence of genes encoding peptidyl-prolyl cis-trans isomerases, which are typical for a cold-adapted lifestyle (13).
TABLE 3.
Enzymes, amino acids, and compounds associated with a psychrophilic lifestyle and their functions
| Keyword | No. of sequence annotations containing keyworda | Selected function (reference) |
|---|---|---|
| Desaturase | 1,012 | Catalyzes synthesis of unsaturated fatty acids (5) |
| Peptidyl-prolyl cis-trans isomerase | 1,021 | Maintains protein-folding rates at low temperatures (13) |
| Glycine | 2,644 | Osmoprotectant (49) |
| Betaine | 471 | Osmoprotectant and cryoprotectant (30) |
| Sarcosine | 233 | Intermediate in the metabolism of choline to glycine (30) |
| Choline | 827 | Precursor of glycine betaine (30) |
| Glutamate | 5,782 | Osmoprotectant (29) |
| Dioxygenase | 2,706 | Antioxidative enzyme (29) |
| Superoxide dismutase | 257 | Antioxidative enzyme (30) |
NCBI-nr database matches of the pyrosequencing-derived sequence annotations (cutoff e-value 10−5) containing the keyword.
Since the solubility of gasses increases rapidly at low temperatures, the ability to respond to reactive oxygen species is an essential function for organisms living at low temperatures. It has been shown that exposure to low temperature is associated with enhancement of oxidative stress (8, 45). To prevent oxidative harm, genes similar to antioxidative enzymes such as catalases/peroxidases and superoxide dismutases were present (Table 3).
The avoidance of cell damage by formation of ice crystals is also a prerequisite for living at subzero temperatures. Correspondingly, many genes involved in the synthesis of well-known cryo- and osmoprotectants, such as glycine, betaine, choline, sarcosine, and glutamate, were detected (Table 3). Strikingly, the KEGG category environmental information processing contained 4,043 sequences exhibiting similarities to genes associated with the type II secretion system but only 580 and 671 sequences for type III and IV secretion systems, respectively (see Data Set S2 in the supplemental material). Recently, it has been shown that type II secretion systems possess important functions with respect to survival in environmental niches. It has been demonstrated that type II exoproteins promote growth at low temperatures. (46).
Conclusions.
The diversity and metabolic capacity of a microbial assemblage derived from a permanently frozen habitat has been elucidated for the first time by evaluation of a large pyrosequencing-derived data set in addition to traditional approaches. Analyses of the taxonomic composition and the metabolic potential revealed that the glacial ice metagenome of the Northern Schneeferner is dominated by aerobic and facultative aerobic bacteria (mainly Proteobacteria). A wide metabolic diversity was established, including a large degradative capacity and the ability to assimilate inorganic and organic nitrogen and sulfur sources. Genes for growth under anaerobic conditions by utilizing nitrate/nitrite as an electron acceptor were detected. In addition, several enzymes and compounds essential for a psychrophilic lifestyle and adaptation to living in a low-nutrient environment were present. The reported results will contribute to our understanding of microbial activity and strategies for cell survival and growth in subzero temperature ecosystems. This might also contribute to unravel prerequisites for life in frozen extraterrestrial habitats.
Supplementary Material
Acknowledgments
We thank Gerhard Gottschalk for critical reading of the manuscript.
This study was supported by a grant from the German Bundesministerium für Bildung und Forschung.
Footnotes
Published ahead of print on 2 October 2009.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 2.Ashelford, K. E., N. A. Chuzhanova, J. C. Fry, A. J. Jones, and A. J. Weightman. 2006. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl. Environ. Microbiol. 72:5734-5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biddle, J. F., S. Fitz-Gibbon, S. C. Schuster, J. E. Brenchley, and C. H. House. 2008. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. USA 105:10583-10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bidle, K. D., S. Lee, D. R. Marchant, and P. G. Falkowski. 2007. Fossil genes and microbes in the oldest ice on Earth. Proc. Natl. Acad. Sci. USA 104:13455-13460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolter, M. 2004. Ecophysiology of psychrophilic and psychrotolerant microorganisms. Cell Mol. Biol. 50:563-573. [PubMed] [Google Scholar]
- 6.Bonfield, J. K., K. Smith, and R. Staden. 1995. A new DNA sequence assembly program. Nucleic Acids Res. 23:4992-4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bottrell, S., and M. Tranter. 2002. Sulphide oxidation under partially anoxic conditions at the bed of Haut Glacier d'Arolla, Switzerland. Hydrolog. Process 16:2363-3468. [Google Scholar]
- 8.Chattopadhyay, M. K. 2006. Mechanism of bacterial adaptation to low temperature. J. Biosci. 31:157-165. [DOI] [PubMed] [Google Scholar]
- 9.Cheng, S. M., and J. M. Foght. 2007. Cultivation-independent and -dependent characterization of Bacteria resident beneath John Evans Glacier. FEMS Microbiol. Ecol. 59:318-330. [DOI] [PubMed] [Google Scholar]
- 10.Christner, B. C., E. Mosley-Thompson, L. G. Thompson, and J. N. Reeve. 2003. Bacterial recovery from ancient glacial ice. Environ. Microbiol. 5:433-436. [DOI] [PubMed] [Google Scholar]
- 11.Christner, B. C., E. Mosley-Thompson, L. G. Thompson, and J. N. Reeve. 2001. Isolation of bacteria and 16S rDNAs from Lake Vostok accretion ice. Environ. Microbiol. 3:570-577. [DOI] [PubMed] [Google Scholar]
- 12.Cole, J. R., B. Chai, T. L. Marsh, R. J. Farris, Q. Wang, S. A. Kulam, S. Chandra, D. M. McGarrell, T. M. Schmidt, G. M. Garrity, and J. M. Tiedje. 2003. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 31:442-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Amico, S., T. Collins, J. C. Marx, G. Feller, and C. Gerday. 2006. Psychrophilic microorganisms: challenges for life. EMBO Rep. 7:385-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daniel, R. 2005. The metagenomics of soil. Nat. Rev. Microbiol. 3:470-478. [DOI] [PubMed] [Google Scholar]
- 15.Deming, J. W. 2002. Psychrophiles and polar regions. Curr. Opin. Microbiol. 5:301-309. [DOI] [PubMed] [Google Scholar]
- 16.Dinsdale, E. A., R. A. Edwards, D. Hall, F. Angly, M. Breitbart, J. M. Brulc, M. Furlan, C. Desnues, M. Haynes. L. Li, L. McDaniel, M. A. Moran, K. E. Nelson, C. Nilsson, R. Olson, J. Paul, B. R. Brito, Y. Ruan, B. K. Swan, R. Stevens, D. L. Valentine, R. V. Thurber, L. Wegley, B. A. White, and F. Rohwer. 2008. Functional metagenomic profiling of nine biomes. Nature 452:629-632. [DOI] [PubMed] [Google Scholar]
- 17.Edwards, R. A., B. Rodriguez-Brito, L. Wegley, M. Haynes, M. Breitbart, D. M. Peterson, M. O. Saar, S. Alexander, E. C. Alexander, Jr., and F. Rohwer. 2006. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feller, G., and C. Gerday. 2003. Psychrophilic enzymes: hot topics in cold adaptation. Nat. Rev. Microbiol. 1:200-208. [DOI] [PubMed] [Google Scholar]
- 19.Foght, J., J. Aislabie, S. Turner, C. E. Brown, J. Ryburn, D. J. Saul, and W. Lawson. 2004. Culturable bacteria in subglacial sediments and ice from two Southern Hemisphere glaciers. Microb. Ecol. 47:329-340. [DOI] [PubMed] [Google Scholar]
- 20.Gill, S. R., M. Pop, R. T. DeBoy, P. B. Eckburg, P. J. Turnbaugh, B. S. Samuel, J. I. Gordon, D. A. Relman, C. M. Fraser-Liggett, and K. E. Nelson. 2006. Metagenomic analysis of the human distal gut microbiome. Science 312:1355-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haeberli, W., M. Hoelzle, F. Paul, and M. Zemp. 2007. Integrated monitoring of mountain glaciers as key indicators of global climate change: the European Alps. Ann. Glaciol. 46:150-160. [Google Scholar]
- 22.Hodson, A. J., P. N. Mumford, J. Kohler, and P. M. Wynn. 2005. The high arctic glacial ecosystem: new insights from nutrient budgets. Biogeochemistry 72:233-256. [Google Scholar]
- 23.Junge, K., H. Eicken, and J. W. Deming. 2004. Bacterial activity at −2 to −20°C in Arctic wintertime sea ice. Appl. Environ. Microbiol. 70:550-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa, M., M. Araki, S. Goto, M. Hattori, M. Hirakawa, M. Itoh, T. Katayama, S. Kawashima, S. Okuda, T. Tokimatsu, and Y. Yamanishi. 2008. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36:D480-D484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krause, L., N. N. Diaz, A. Goesmann, S. Kelley, T. W. Nattkemper, F. Rohwer, R. A. Edwards, and J. Stoye. 2008. Phylogenetic classification of short environmental DNA fragments. Nucleic Acids Res. 36:2230-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ludwig, W., O. Strunk, R. Westram, L. Richter, H. Meier, A. Yadhukumar, A. Buchner, T. Lai, S. Steppi. G. Jobb, W. Förster, I. Brettske, S. Gerber, A. W. Ginhart, O. Gross, S. Grumann, S. Hermann, R. Jost, A. König, T. Liss, R. Lüβmann, M. May, B. Nonhoff, B. Reichel, R. Strehlow, A. Stamatakis, N. Stuckmann, A. Vilbig, M. Lenke, T. Ludwig, A. Bode, and K.-H. Schleifer. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luna, G. M., A. Dell'Anno, and R. Danovaro. 2006. DNA extraction procedure: a critical issue for bacterial diversity assessment in marine sediments. Environ. Microbiol. 8:308-320. [DOI] [PubMed] [Google Scholar]
- 28.Maidak, B. L., J. R. Cole, T. G. Lilburn, C. T. Parker, P. R. Saxman, R. J. Farris, G. M. Garrity, G. J. Olsen, T. M. Schmidt, and J. M. Tiedje. 2001. The RDP-II (Ribosomal Database Project). Nucleic Acids Res. 29:173-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Medigue, C., E. Krin, G. Pascal, V. Barbe, A. Bernsel, P. N. Bertin, F. Cheung, S. Cruveiller, S. D'Amico, A. Duilio, G. Fang, G. Feller, C. Ho, S. Mangenot, G. Marino, J. Nilsson, E. Parrilli, E. P. C. Rocha, Z. Rouy, A. Sekowska, M. L. Tutino, D. Vallenet, G. von Heijne, and A. Danchin. 2005. Coping with cold: the genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res. 15:1325-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Methe, B. A., K. E. Nelson, J. W. Deming, B. Momen, E. Melamud, X. Zhang, J. Moult, R. Madupu, W. C. Nelson, R. J. Dodson, L. M. Brinkac, S. C. Daugherty, A. S. Durkin, R. T. DeBoy, J. F. Kolonay, S. A. Sullivan, L. Zhou, T. M. Davidsen, M. Wu, A. L. Huston, M. Lewis, B. Weaver, J. F. Weidman, H. Khouri, T. R. Utterback, T. V. Feldblyum, and C. M. Fraser. 2005. The psychrophilic lifestyle as revealed by the genome sequence of Colwellia psychrerythraea 34H through genomic and proteomic analyses. Proc. Natl. Acad. Sci. USA 102:10913-10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miteva, V. I. 2008. Bacteria in snow and glacier ice, p. 31-50. In R. Margesin, F. Schinner, J.-C. Marx, and C. Gerday (ed.), Psychrophiles: from biodiversity to biotechnology. Springer-Verlag, Heidelberg, Germany.
- 32.Miteva, V. I., P. P. Sheridan, and J. E. Brenchley. 2004. Phylogenetic and physiological diversity of microorganisms isolated from a deep Greenland glacier ice core. Appl. Environ. Microbiol. 70:202-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosier, A. C., A. E. Murray, and C. H. Fritsen. 2007. Microbiota within the perennial ice cover of Lake Vida, Antarctica. FEMS Microbiol. Ecol. 59:274-288. [DOI] [PubMed] [Google Scholar]
- 34.Ponder, M. A., S. J. Gilmour, P. W. Bergholz, C. A. Mindock, R. Hollingsworth, M. F. Thomashow, and J. M. Tiedje. 2005. Characterization of potential stress responses in ancient Siberian permafrost psychroactive bacteria. FEMS Microbiol. Ecol. 53:103-115. [DOI] [PubMed] [Google Scholar]
- 35.Price, P. B., and T. Sowers. 2004. Temperature dependence of metabolic rates for microbial growth, maintenance, and survival. Proc. Natl. Acad. Sci. USA 101:4631-4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Priscu, J. C., and B. C. Christner. 2004. Earth's icy biosphere, p. 130-145. In A. T. Bull (ed.), Microbial diversity and bioprospecting. ASM Press, Washington, DC.
- 37.Priscu, J. C., E. F. Adams, W. B. Lyons, M. A. Voytek, D. W. Mogk, R. L. Brown, C. P. McKay, C. D. Takacs, K. A. Welch, C. F. Wolf, J. D. Kirshtein, and R. Avci. 1999. Geomicrobiology of subglacial ice above Lake Vostok, Antarctica. Science 286:2141-2144. [DOI] [PubMed] [Google Scholar]
- 38.Reasoner, D. J., and F. E. Geldreich. 1985. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 49:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schloss, P. D., and J. Handelsman. 2005. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 71:1501-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Segawa, T., K. Miyamoto, K. Ushida, K. Agata, N. Okada, and S. Kohshima. 2005. Seasonal change in bacterial flora and biomass in mountain snow from the Tateyama Mountains, Japan, analyzed by 16S rRNA gene sequencing and real-time PCR. Appl. Environ. Microbiol. 71:123-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharp, M., J. Parkes, B. Cragg, l. J. Fairchild, H. Lamb, and M. Tranter. 1999. Widespread bacterial populations at glacier beds and their relationship to rock weathering and carbon cycling. Geology 27:107-110. [Google Scholar]
- 42.Sheridan, P. P., V. I. Miteva, and J. E. Brenchley. 2003. Phylogenetic analysis of anaerobic psychrophilic enrichment cultures obtained from a Greenland glacier ice core. Appl. Environ. Microbiol. 69:2153-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simon, C., J. Herath, S. Rockstroh, and R. Daniel. 2009. Rapid identification of genes encoding DNA polymerases by function-based screening of metagenomic libraries derived from glacial ice. Appl. Environ. Microbiol. 75:2964-2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skidmore, M., S. P. Anderson, M. Sharp, J. Foght, and B. D. Lanoil. 2005. Comparison of microbial community compositions of two subglacial environments reveals a possible role for microbes in chemical weathering processes. Appl. Environ. Microbiol. 71:6986-6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smirnova, G. V., O. N. Zakirova, and O. N. Oktiabr'skii. 2001. Role of the antioxidant system in response of Escherichia coli bacteria to cold stress. Mikrobiologiia 70:55-60. [PubMed] [Google Scholar]
- 46.Soderberg, M. A., O. Rossier, and N. P. Cianciotto. 2004. The type II protein secretion system of Legionella pneumophila promotes growth at low temperatures. J. Bacteriol. 186:3712-3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stibal, M., M. Tranter, L. G. Benning, and J. Rehak. 2008. Microbial primary production on an Arctic glacier is insignificant in comparison with allochthonous organic carbon input. Environ. Microbiol. 10:2172-2178. [DOI] [PubMed] [Google Scholar]
- 48.Tatusov, R. L., D. A. Natale, I. V. Garkavtsev, T. A. Tatusova, U. T. Shankavaram, B. S. Rao, B. Kiryutin, M. Y. Galperin, N. D. Fedorova, and E. V. Koonin. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29:22-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas, D. N., and G. S. Dieckmann. 2002. Antarctic Sea ice: a habitat for extremophiles. Science 295:641-644. [DOI] [PubMed] [Google Scholar]
- 50.Tringe, S. G., C. von Mering, A. Kobayashi, A. A. Salamov, K. Chen, H. W. Chang, M. Podar, J. M. Short, E. J. Mathur, J. C. Detter, P. Bork, P. Hugenholtz, and E. M. Rubin. 2005. Comparative metagenomics of microbial communities. Science 308:554-557. [DOI] [PubMed] [Google Scholar]
- 51.Turchetti, B., J. P. Buzzini, M. Goretti, E. Branda, G. Diolaiuti, C. D'Agata, C. Smiraglia, and A. Vaughan-Martini. 2008. Psychrophilic yeasts in glacial environments of Alpine glaciers. FEMS Microbiol. Ecol. 63:73-83. [DOI] [PubMed] [Google Scholar]
- 52.von Mering, C., P. Hugenholtz, S. G. Tringe, T. Doerks, L. J. Jensen, N. Ward, and P. Bork. 2007. Quantitative phylogenetic assessment of microbial communities in diverse environments. Science 315:1126-1130. [DOI] [PubMed] [Google Scholar]
- 53.Wang, Q., G. M. Garrity, J. M. Tiedje, and J. R. Cole. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261-5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willerslev, E., A. J. Hansen, and H. N. Poinar. 2004. Isolation of nucleic acids and cultures from fossil ice and permafrost. TRENDS Ecol. Evol. 19:141-147. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, X. F., T. D. Yao, L. D. Tian, S. J. Xu, and L. Z. An. 2008. Phylogenetic and physiological diversity of bacteria isolated from Puruogangri ice core. Microb. Ecol. 55:476-488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.