

Abstract
Type 1 pilus directs bladder epithelial binding and invasion by uropathogenic Escherichia coli (UPEC) in the initial stage of cystitis, but the bacterial determinants of postinvasion events in the pathogenesis of cystitis are largely undetermined. We show here that the UPEC outer membrane protein A (OmpA), a monomeric, major, integral protein component of the bacterial outer membrane, functions as a critical determinant of intracellular virulence for UPEC, promoting persistent infection within bladder epithelium. Using a murine urinary tract infection (UTI) model, we demonstrate that whereas deletion of the UPEC ompA gene did not disrupt initial epithelial binding and invasion by UPEC, it did preclude completion of the intracellular bacterial community (IBC) pathway, accompanied by diminishing bacterial loads in the bladder. This defect in epithelial persistence of the ompA mutant was enhanced in competitive infections with wild-type UPEC. Microscopic examinations revealed that the ompA mutant formed significantly fewer IBCs, and those that were initiated were unable to progress past the early stages of maturation. These defects could be corrected by complementation of ompA. In addition, expression of ompA during wild-type UTI was sharply increased at time points correlated with IBC development and the arrival of host immune effector cells. Our findings establish OmpA as a key UPEC virulence factor that functions after epithelial invasion to facilitate IBC maturation and chronic bacterial persistence.
The American patient population suffers each year from approximately 11 million urinary tract infections (UTIs), which are associated with an estimated $1.6 billion in clinical and indirect economic costs (13, 15). This substantial morbidity is exacerbated by the high frequency of recurrent infections despite appropriate antimicrobial therapy. Many recurrences are caused by organisms that are genotypically identical to the initial infecting strain, suggesting that in addition to reinoculation from a gastrointestinal source, recurrence might arise after incomplete eradication of the organism from the bladder (8). Uropathogenic Escherichia coli (UPEC) are the primary etiology of UTIs, causing up to 90% of community-acquired infections (45, 46). Persistence of UPEC within the urinary tracts of experimentally infected mice has been associated with their ability to invade superficial epithelial (umbrella) cells of the bladder (34, 36), establishing a niche for host colonization beyond the luminal surface to the intracellular compartment and sheltering the pathogen from host defenses. Acute cystitis is characterized by the development of intracellular bacterial communities (IBCs) in both mice and humans (3, 47). The IBCs represent clonal expansion of invaded bacteria proceeding through a complex maturation cycle (24) that facilitates the subsequent development of quiescent reservoirs or nests of UPEC within Lamp1-positive intracellular vesicles (40). These long-term resident bacteria are sequestered from antibiotic therapy and remain apparently undetected by host immune mechanisms (37).
While type 1 pili and flagella are critical and contributing factors, respectively (16, 18, 53), for the early events of binding and invasion, additional factors that promote the intracellular phenotypes of UPEC are largely unidentified. We recently demonstrated that the periplasmic chaperone SurA is required for both invasion and intracellular maturation of UPEC (27). This chaperone, conserved across numerous gram-negative bacteria, facilitates transit of nascent polypeptides destined for insertion into the outer membrane (OM) as β-barrel porins (11, 31). SurA deletion abrogates production of the type 1 pilus usher FimD, thus precluding expression of type 1 pili and epithelial colonization. However, SurA-dependent proteins also contribute to postinvasion phenotypes of UPEC (19, 25). More recently, deletion of the hfq gene (encoding an RNA chaperone) was shown to impair the ability of UPEC to form IBCs, perhaps through downstream effects on membrane integrity (30). Further, it is logical to surmise that the extracellular domains of OM proteins might mediate pathogenesis by participating in interactions with the host and potentially with other bacteria in a community. Therefore, we sought to specify SurA-dependent OM proteins that contribute to the postinvasion phenotypes of UPEC during cystitis, namely, IBC maturation and intraepithelial persistence.
In the present study, we explored the pathogenic role of outer membrane protein A (OmpA), a SurA-dependent, major protein component of the E. coli OM that assumes a β-barrel conformation with eight transmembrane segments and four extracellular loops (4). Multiple cellular functions have been proposed for OmpA, including maintaining cell structural integrity, serving as a phage and colicin receptor, and mediating F-factor-dependent conjugation (29). Although its role as a porin is debated (5, 7), some data suggest that OmpA may form nonspecific channels for diffusion of solutes. With regard to bacterial virulence, OmpA is necessary for successful infection by E. coli K-1, the leading gram-negative cause of neonatal septicemia and meningitis (51). E. coli K-1 ompA mutants are significantly less serum resistant (42, 52) and show decreased capacity to invade cultured human brain microvascular endothelial cells, which model the blood-brain barrier (41, 43). Other authors have speculated that OmpA represents a pathogen-associated molecular pattern that interacts with antigen-presenting cells (20-22). In UPEC the pathogenic function of OmpA has yet to be elucidated, although it was one of several OM proteins whose expression was augmented during in vitro growth of UPEC in urine (1). We demonstrate here that OmpA provides a pathogenic advantage to UPEC during establishment of its intracellular niche and promotes persistence of UPEC within the bladder following acute cystitis.
MATERIALS AND METHODS
Bacterial strains and media.
E. coli strains were cultured overnight at 37°C in Luria-Bertani (LB) broth (Difco/Becton Dickinson, Franklin Lakes, NJ) under static conditions to promote surface expression of type 1 pili. Where indicated, chloramphenicol was added at 20 μg/ml or ampicillin was added at 100 μg/ml. UPEC strain UTI89 was isolated from a patient with active cystitis. UTI89 ompA::cat was created by linear transformation of UTI89/pKM208 (38) with a fragment amplified from template plasmid pKD3 (12) using the primers 5′-ATGAAAAAGACAGCTATCGCGATTGCAGTGGCACTGGTGTAGGCTGGAGCTGCTTC-3′ (forward) and 5′-TTAAGCCTGCGGCTGAGTTACAACGTCTTTGATGCCCATATGA ATATCCTCCTTAG-3′ (reverse; Integrated DNA Technologies, Coralville, IA). The deletion was verified by direct sequencing, and the absence of OmpA expression by UTI89 ompA::cat was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of the total membranes. Of note, ompA is independently transcribed in UTI89 (9) and other sequenced strains of E. coli, so polar effects were excluded. For complementation experiments, a low-copy-number plasmid (pKW5) encoding ompA under its native promoter was constructed. The ompA gene and upstream region were amplified by high-fidelity PCR using primers 5′-CGTGTCTAGATTTCCTTGCGGAGGCTTGTCTGAAGCGGTTTC-3′ (forward) and 5′-ACCCAAGCTTAACTTAAGCCTGCGGCTGAGTTACAACGTC-3′ (reverse). This fragment was digested with HindIII and XbaI and then ligated into pACYC184 (Cmr Tetr) that had been similarly digested, leaving expression of the marker under the upstream influence of ompA's native promoter. Transformed clones of E. coli Top10 (Invitrogen, Carlsbad, CA) were selected on tetracycline plates and tested by colony PCR, the construct was confirmed by direct sequencing, and expression was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of the total membranes. Prior to mouse inoculations, the complemented ΔompA/pKW5 strain was grown in only 5 μg of tetracycline/ml to keep the plasmid copy number low, since ompA overexpression at high levels was detrimental to growth in vitro. UTI89 ΔfimH, which lacks the gene encoding the type 1 pilus tip adhesin, was kindly provided by K. Wright (53). To facilitate visualization by confocal microscopy, UTI89 and derivatives were transformed with pcomGFP (10).
Electron microscopy and hemagglutination assays.
For immunogold electron microscopy, washed bacteria were adsorbed to Formvar/carbon-coated grids, incubated sequentially with primary rabbit antiserum raised against the adhesin domain of the type 1 pilus adhesin FimH (denoted FimHA) and gold-conjugated antirabbit immunoglobulin G, and washed twice with distilled water before staining with 1% aqueous uranyl acetate. Samples were viewed on a JEOL 1200EX transmission electron microscope (JEOL USA, Peabody, MA). To assess in vitro function of type 1 pili, hemagglutination of guinea pig erythrocytes (Colorado Serum Co., Denver, CO) was assayed as described previously (32).
Ex vivo gentamicin protection assay and murine cystitis.
A well-described model of murine cystitis was used (36). All animal procedures were approved by the Animal Studies Committee at Washington University. Briefly, overnight static LB broth cultures were harvested by centrifugation, washed, resuspended, and diluted to the final inoculum in phosphate-buffered saline (PBS), guided by an optical density at 600 nm. Eight-week-old female C3H/HeN mice (Harlan, Indianapolis, IN) were transurethrally infected with a 50-μl inoculum of ∼107 bacteria. For ex vivo gentamicin protection (invasion) assays, bladders were harvested at the indicated times postinfection, splayed aseptically, and then washed with PBS to recover the luminal bacteria. Bladders processed for titers were rocked gently in PBS containing gentamicin (100 μg/ml) for 1 h at 37°C to kill the remaining extracellular bacteria, homogenized in PBS, and plated to LB agar to recover intracellular bacteria. For time course infections, bladders were homogenized at the indicated times postinfection in sterile PBS with 0.1% Triton X-100, serially diluted, and plated onto LB agar to enumerate the CFU. For competitive infections, equal amounts of UTI89 and UTI89 ompA::cat were mixed immediately prior to inoculation to a final density of ∼107 bacteria in a 50-μl inoculum, and harvested bladder homogenates were plated on LB agar with or without chloramphenicol. For complementation experiments, homogenates were plated to LB agar with or without tetracycline to ensure reporting of data from mice in which the complementing plasmid had been retained. Experiments were repeated at least three times, and aggregate CFU data from these independent experiments were reported.
LacZ staining and fluorescent confocal microscopy.
For lacZ staining, groups of three to four mice were infected with indicated strains; bladders were stretched, fixed for 30 min in 10% neutral buffered formalin, washed with lacZ wash buffer (PBS with 0.01 M MgCl2, 0.01% sodium deoxycholate, and 0.02% Igepal CA-640), incubated in lacZ stain (lacZ wash buffer with 1 mg of X-Gal [5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside]/ml, 5 mM potassium ferrocyanide, and 5 mM potassium ferricyanide) for 16 h, washed three times in PBS, and photographed by stereomicroscopy in whole mount for enumeration of the spots representing IBCs (27). Bladders were then embedded in paraffin and, as needed, sections were deparaffinized and stained with hematoxylin and eosin for light microscopy. For confocal microscopy, freshly harvested bladders from experimental groups of three to four mice were stretched under PBS, fixed with 3% paraformaldehyde in PBS for 1 h, and stained briefly with SYTO 61 red fluorescent nucleic acid stain (Molecular Probes, Carlsbad, CA) to visualize any bacteria that had lost the green fluorescent protein-containing plasmid. Bladders were then washed with PBS, mounted on glass slides with ProLong Gold antifade reagent (Molecular Probes), and viewed by using an LSM510 Meta laser scanning confocal microscope (Carl Zeiss, Inc., Thornwood, NY). Aggregate data from three independent experiments were reported.
RNA isolation and real-time PCR.
Bladders infected with UTI89 as described above were harvested at the indicated time points, homogenized, and stored at −80°C in RNA Protect reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. For the calibrator control sample, a portion of the unused inoculum (109 CFU/ml in sterile PBS) was similarly treated and stored at −80°C. For analysis, stored bladder homogenates were thawed and centrifuged (16,000 × g for 5 min); supernatants were discarded, and the pellets (from five animals per group) were pooled and resuspended in lysis buffer (RLT; Qiagen). Samples were further homogenized with a FastPrep FP120 reciprocal shaking device and a commercially available extraction reagent, lysing matrix B (MP Biomedicals, Solon, OH). For analysis of ompA transcript levels in luminal and intracellular compartments, a gentamicin protection approach was used as described above. An aliquot of the luminal wash plus an aliquot of bladder homogenate after gentamicin treatment were stored at −80°C in RNA Protect reagent until subsequent RNA extraction and analysis.
Total RNA was extracted by silica column purification using an RNeasy minikit (Qiagen) according to the manufacturer's instructions. Contaminating chromosomal DNA was removed by on-column DNase (Qiagen) and, when necessary, a second DNase treatment (Invitrogen). The absence of DNA was confirmed by PCR, and the quality and quantity of RNA were determined by spectrophotometry and agarose gel electrophoresis. First-strand cDNA was synthesized by using Superscript II reverse transcriptase (Invitrogen) and random primers. Real-time PCR was performed in an ABI 7500 Fast thermal cycler using Power SYBR green PCR master mix (both from Applied Biosystems, Foster City, CA) and primers designed to amplify E. coli ompA (forward, 5′-GGGTGTTTCCTACCGTTTCG-3′; reverse 5′-TGGAGCCGGAGCAACTACTG-3′) and the rRNA gene rrsA (forward, 5′-CCAGGGCTACACACGTGCTA-3′; reverse 5′-TCTCGCGAGGTCGCTTCT-3′), chosen as the endogenous control gene because of invariable expression in a variety of conditions and the efficiency of the PCR with the reagents and conditions used in these studies (J. Loughman, unpublished data). Fold changes in message abundance (of triplicate assays) were determined by using the ΔΔCT method (33) by normalizing cycle threshold (CT) values to each gene's CT value obtained from baseline expression in the starting inoculum.
Statistical analysis.
Statistical analysis of numerical data was performed by using the Mann-Whitney U test (two-tailed) or Wilcoxon test within Prism software (GraphPad, La Jolla, CA). A P value of 0.05 was used as the threshold of significance.
RESULTS
OmpA of UPEC is not required for type 1 pilus-dependent binding and invasion of bladder epithelium.
Expression of functional type 1 pili confers upon UPEC the ability to bind mannosylated uroplakin proteins on the luminal surface, permitting invasion of the mammalian bladder epithelium (34, 49). Although SurA is necessary for these type 1 pilus-dependent functions, our objective was to identify SurA-dependent proteins that contribute (independent of type 1 pili) to the postinvasion biology of cystitis. Therefore, we first determined whether deletion of ompA in the cystitis-derived UPEC strain UTI89 affects the presentation or function of type 1 pili. Growth of UTI89 in LB broth and resistance to novobiocin (a measure of membrane integrity [26]) were unchanged by ompA deletion (data not shown). Immunogold electron microscopic images of wild-type and OmpA-deficient UTI89 displayed normal and equivalent numbers of pili, which labeled with anti-FimHA antibody (Fig. 1A and B). The ompA mutant agglutinated guinea pig erythrocytes as effectively as wild-type UTI89 (Fig. 1C), and agglutination by both strains was inhibited by addition of 2% α-d-mannopyranoside (data not shown). To assess type 1 pilus function in mammalian infection, we performed an ex vivo gentamicin protection assay in female C3H/HeN mice. We recovered comparable levels of both luminal and intracellular bacteria from bladders 1 h after infection with ompA mutant or wild-type UTI89 (Fig. 1D). Taken together, these data indicate that lack of OmpA in UPEC does not affect type 1 pilus assembly, presentation, or function and that epithelial binding and invasion are unaffected by UPEC ompA deletion in the murine model of cystitis.
FIG. 1.
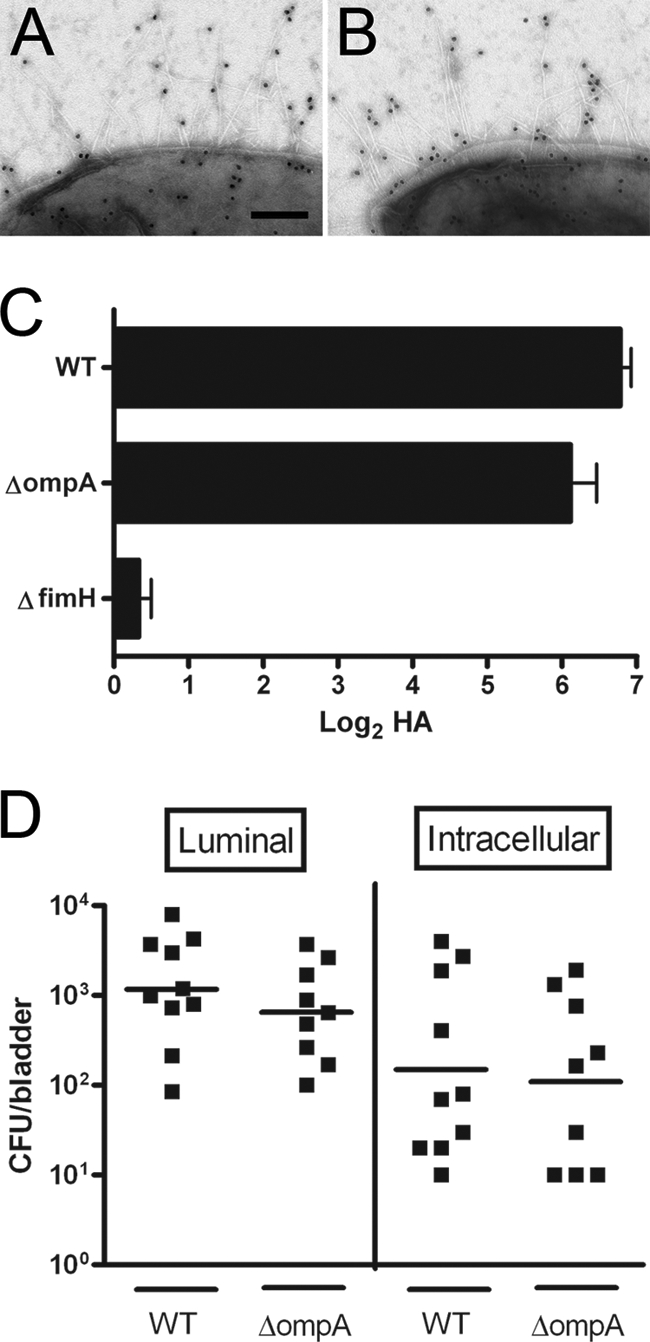
Type 1 pilus expression and function in the UPEC ompA mutant. Immunogold electron microscopy demonstrates equivalent pilus expression in wild-type UTI89 (A) and the ompA mutant (B). (C) The hemagglutination of guinea pig erythrocytes by wild-type (WT) and ompA mutant UTI89 is equivalent; the UTI89 fimH mutant is included as a negative control. In ex vivo gentamicin protection assays, CFU recovered 1 h after infection with either wild type or ompA mutant UTI89 are equivalent in the luminal and intracellular compartments (D). Scale bar for panels A and B, 200 nm.
OmpA promotes acute and persistent UTI.
UPEC attributes that specifically contribute to intracellular pathogenesis, subsequent to initial epithelial colonization, have yet to be demonstrated. To determine whether OmpA has a role in uropathogenesis, bladder bacterial loads of the ompA mutant relative to wild-type UTI89 were evaluated along a time course of murine cystitis. Female C3H/HeN mice were transurethrally inoculated with ∼107 CFU of wild-type or OmpA-deficient UTI89, and bladders were harvested at multiple time points (from 6 h to 2 weeks) for tissue CFU determination. Time points were chosen to reflect distinct stages of IBC development and chronic persistence (2, 24, 40). Up to 24 h postinfection, CFU recovered from bladders infected with wild-type UTI89 or its ompA mutant were similar (Fig. 2). In contrast, there was a trend toward lower CFU in ompA::cat strain-infected bladders at 48 h (an interval associated with late fluxing bacteria and second-round IBC development) and a statistically significant decrease in CFU in ompA::cat strain-infected bladders at 1 and 2 weeks (P < 0.001). At this final time point, concomitant with reservoir formation, many of the ompA::cat strain-infected bladders were found to be sterile (Fig. 2). Compared to wild-type infection, CFU recovered from the kidneys after 1 week were also sharply attenuated by ompA deletion (data not shown). Mice infected with the complemented ΔompA/pKW5 strain demonstrated restoration of wild-type levels of recoverable tetracycline-resistant bladder CFU at these time points (Fig. 2). During competitive infection with both the mutant and wild-type strains, late-stage bladder CFU defects were amplified, with ompA mutant CFU beginning to fall by 24 h and the mutant universally cleared by 2 weeks (P < 0.015; Fig. 3). Recoverable wild-type CFU were unaffected by coinfection with the ompA mutant (compare the wild-type CFU values in Fig. 2 to those in Fig. 3). CFU defects at the observed time points suggest that OmpA is important for completion of the IBC pathway and for subsequent persistence within the bladder.
FIG. 2.
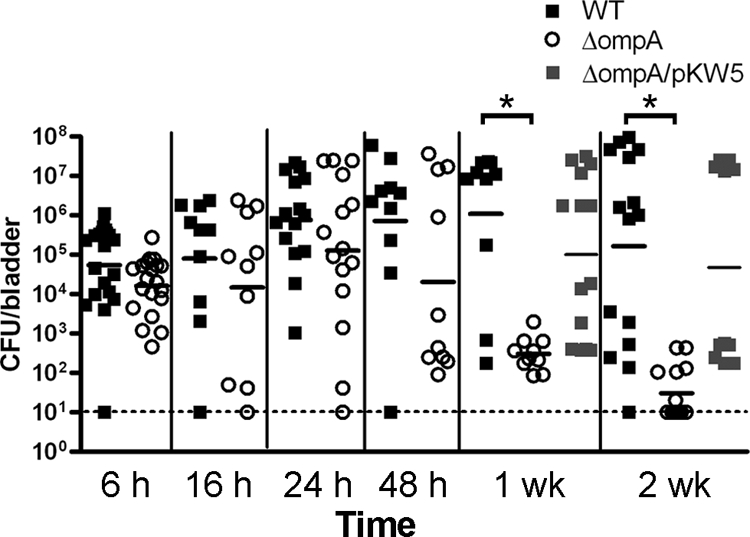
Bacterial loads in the bladders of C3H/HeN mice at the indicated time points after infection with either wild-type UTI89 (▪), the ompA mutant (○), or the complemented ΔompA/pKW5 strain (░⃞), and the dashed line indicates the limit of detection (10 CFU/bladder). Bacterial loads of the ompA mutant begin to fall at 48 h and are significantly lower than the wild type at 1 week and 2 weeks postinfection (*, P < 0.001), and this difference is complemented when ompA is provided in trans.
FIG. 3.

CFU recovered at the indicated time points after competitive coinfection of C3H/HeN mice with both wild-type and ompA mutant UTI89. The competitive fitness of the ompA mutant is sharply attenuated as early as 24 h (*, P = 0.002 to 0.015).
OmpA is necessary for IBC maturation during acute UTI.
Development of IBCs is vital for UPEC's early survival in the face of the bladder's mucosal and innate immune defenses and is required for establishment of chronic infection (3, 24, 26). To examine the morphological correlates of the observed CFU defects of the ompA mutant, we assessed global IBC presentation via LacZ staining of whole-mount bladders, which exploits the fact that UTI89 (like all E. coli strains) expresses β-galactosidase. At 6 h after infection of C3H/HeN mice, bladders infected with UTI89 displayed a mean of 30 IBCs per half-bladder, versus only 8 IBCs per half-bladder after infection with the ompA mutant (P = 0.007; Fig. 4A). Complementation of the mutant with pKW5 restored IBC numbers to 32 per half-bladder (Fig. 4A). Bladder sections taken from mice 6 h after infection with UTI89 or the ompA mutant strain were also viewed by light microscopy. At this early stage, the size and appearance of the few visualized ompA mutant IBCs suggested a modest defect in development compared to wild-type IBCs (Fig. 4B and C).
FIG. 4.
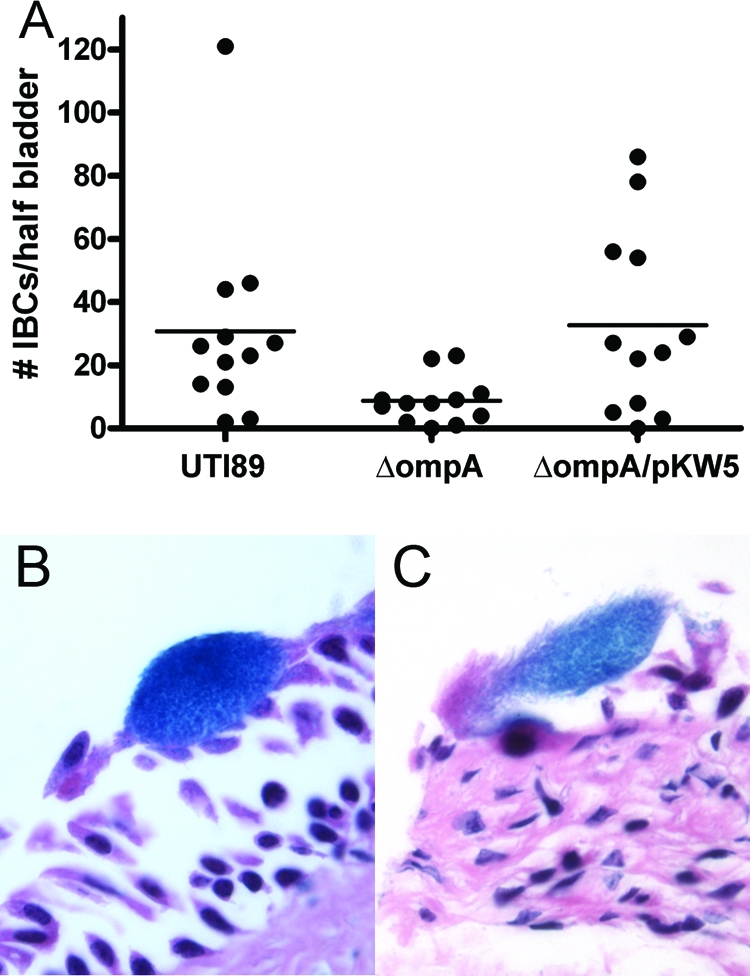
Early IBC formation in C3H/HeN mice. (A) The numbers of IBCs detected at 6 h by LacZ staining are significantly lower after infection with the ompA mutant, compared to wild-type UTI89 (P = 0.007) or the complemented ΔompA/pKW5 strain (P = 0.04). By light microscopy, wild-type early IBCs are readily detected in hematoxylin-and-eosin-stained bladder sections (B); modest intracellular replication was evident in occasional ompA mutant IBCs (C).
To assess the potential of the UPEC ompA mutant to complete IBC maturation and proceed to later stages of the pathogenic cascade of murine cystitis, we examined by fluorescent confocal microscopy the bladders of C3H/HeN mice infected with UTI89 or ompA::cat carrying gfp. Consistent with previous results (24), wild-type UTI89 formed normal early IBCs by 6 h (data not shown), and mature IBCs and many filamentous forms by 16 h (Fig. 5A and B). In animals infected with OmpA-deficient bacteria, we located no IBCs at 6 h by this technique (data not shown). At 16 h rare, small intracellular collections of ΔompA mutant were visualized, but these poorly formed OmpA-deficient IBCs contained fewer bacteria and appeared smaller in dimension than wild-type IBCs (Fig. 5C). Similarly, few examples of stunted filamentous forms were seen after ompA::cat strain infection (Fig. 5D). These defects were restored by complementation with pKW5 (Fig. 5E and F). Although bladders infected with wild-type UTI89 also demonstrated IBCs at 24 h postinfection, mice infected with the mutant lacked persisting IBCs at this time point, also restored by complementation with pKW5 (data not shown). These findings indicate that OmpA is necessary for UPEC to form mature IBCs, expand within their intracellular niche, and develop filaments that initiate subsequent rounds of invasion and IBC formation (24, 27).
FIG. 5.

Confocal microscopic examination of IBC maturation and filamentation. Consistent with previous results, mature IBCs were readily identified 16 h after wild-type UPEC infection (A), whereas a rare, stunted intracellular collection was found at this time point after infection with the ompA mutant (C). Similarly, robust filamentous bacterial populations were evident 16 h after wild-type infection (B), in contrast to the sparse filamentous forms seen after infection with the ompA mutant (D). In mice infected with the complemented ΔompA/pKW5 strain, the formation of mature IBCs (E) and filaments (F) was restored. Scale bars, 10 μm.
Expression of ompA is temporally regulated during murine cystitis.
Loss of OmpA leads to pathogenic defects that are evident at discrete stages of pathogenesis, namely, morphological IBC abnormalities in the acute phase and subsequent defects in persistence. We tested the hypothesis that ompA expression would be regulated in correlation with the various stages of pathogenesis by measuring transcript levels in vivo with reverse transcription-PCR (RT-PCR). After standard infection of C3H/HeN mice with wild-type UTI89, bladders were harvested at time points from 6 h to 2 weeks, and the abundance of ompA transcript was determined and normalized to expression of the endogenous control gene rrsA. The data were expressed as the fold change in transcript abundance compared to that in the initial infecting inoculum. Expression of ompA was upregulated (20- to 35-fold) at 16 to 24 h, returning nearer to baseline at subsequent time points (Fig. 6A). To determine whether ompA expression was elevated specifically by bacteria within the extracellular and/or intracellular compartments of the bladder, RT-PCR was performed on luminal washes and gentamicin-treated bladder homogenates from mice 16 and 48 h after infection with UTI89. Analysis at 16 h demonstrated upregulation of ompA in both fractions (Fig. 6B), with higher expression in the luminal compartment. At 48 h, expression levels remained elevated over the baseline but more comparable between extracellular and intracellular compartments. These data suggest that ompA is upregulated by intracellular bacteria, supporting IBC formation and maturation, but also in extracellular bacteria, which are predominantly exposed to host innate immune pressures.
FIG. 6.
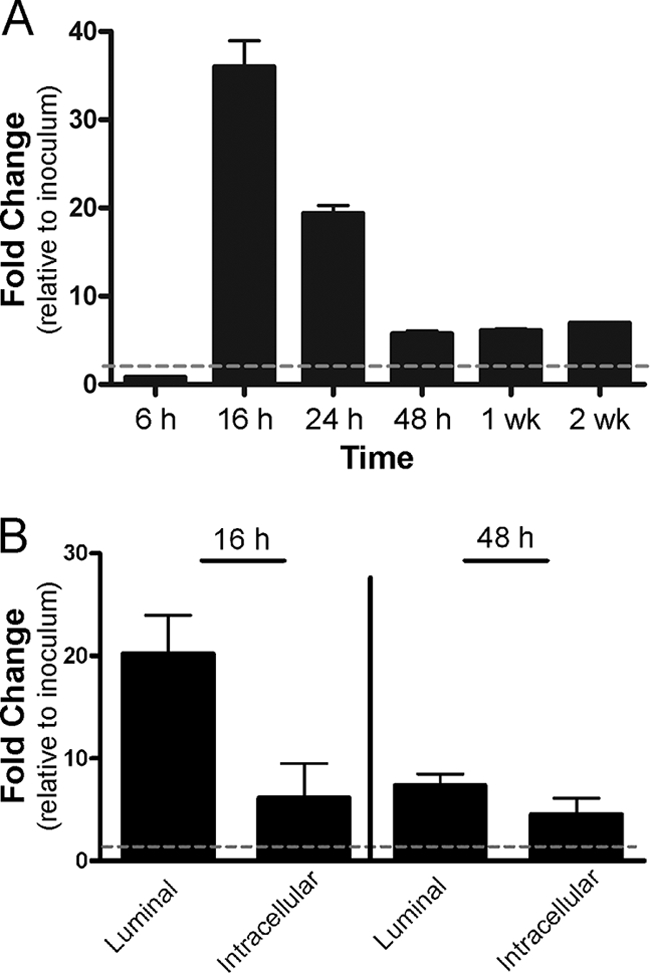
RT-PCR analysis of ompA expression in bladder RNA extracts. (A) RT-PCR performed on whole-bladder extracts demonstrates upregulation of ompA expression at 16 and 24 h, which subsides to still-elevated levels at 48 h to 2 weeks after infection with UTI89. (B) RNA isolated separately from the luminal and gentamicin-protected (intracellular) fractions reveals upregulation of ompA in both compartments at 16 and 48 h. The dashed line in each graph depicts the baseline (inoculum) level of ompA expression, used for comparison.
DISCUSSION
The present study demonstrates the SurA-dependent OmpA as a critical player in the pathogenesis of UPEC, necessary for progression of cystitis beyond the early stage of IBC formation and essential for the intraepithelial persistence that is thought to underlie recurrence of UTI (35, 39, 40). The loss of OmpA in UPEC had no discernible effect on the assembly or function of surface-expressed type 1 pili, as demonstrated by electron microscopy and in vitro hemagglutination. Further, there was no effect of ompA lack on bladder epithelial binding and invasion in vivo, indicating that OmpA also does not augment or facilitate these type 1 pilus-mediated functions. In contrast, OmpA of E. coli K-1 was found to confer binding of cultured human brain microvascular endothelial cells, a model of the blood-brain barrier (44). In these same studies, however, OmpA did not facilitate binding of E. coli K-1 to other endothelial cell types, suggesting specific receptor expression by human brain microvascular endothelial cells (41, 44). We conclude that the UPEC ompA mutant was able to accomplish key early steps in the pathogenesis of cystitis, namely, luminal colonization, epithelial invasion, and initial intracellular replication.
The relative equivalence of observed bladder bacterial loads at early time points after wild-type and ompA mutant infections suggests only a modest effect on luminal and early intracellular replication. However, bacterial loads of the ompA mutant fell sharply at later time points, a difference even more evident during competitive coinfections. Our microscopic investigations demonstrated that the UPEC ompA mutant could indeed form initial intracellular collections but was incompetent for progression past the early stage of IBC maturation; these data indicate that successful epithelial invasion is insufficient to ensure completion of the IBC pathway. An additional finding that supports the importance of OmpA during IBC maturation was that ompA expression was upregulated by intracellular bacteria at this stage. Interestingly, ompA was not differentially regulated (compared to growth in LB broth) in bacteria from sequential urine samples of CBA/J mice inoculated repeatedly with another UPEC strain, CFT073 (isolated from a patient with pyelonephritis) (17). However, in our study, the infecting strain (a cystitis isolate) and method of infection were distinct, and our data specifically measured intracellular as well as extracellular bacterial expression of ompA.
The observed failure of the ompA mutant to complete the IBC maturation cycle could be interpreted in several ways. First, cells containing early ompA mutant IBCs might be more easily or rapidly exfoliated by the host, leading to a lack of visible IBCs at later time points. However, we did not observe increased exfoliation in ompA mutant-infected mice. Also, such a stochastic explanation is unlikely because decreased numbers of ΔompA IBCs are evident microscopically before exfoliation occurs in these mice, and continued defects in intracellular replication and filamentation are observed at later time points. We also speculate that OmpA might be necessary for coalescence of the IBC through interbacterial interactions or might act as a porin for traversal of nutrients (7) or other solutes to support optimal growth in the intracellular compartment, but the specific role(s) of OmpA in this venue have yet to be determined.
Later stages of the IBC pathway are tailored to subvert host immune responses: intracellular bacteria are provided a sanctuary from neutrophils (24), and filamentous bacteria transiting the extracellular space between rounds of IBC formation are resistant to polymorphonuclear leukocyte phagocytosis and killing (26). Establishment of chronic residence within the bladder requires bacteria to survive these extracellular periods of exposure to host immune effectors. On this basis, we speculate that OmpA deficiency may hamper the organism's resistance to the action of neutrophils or other components of innate and/or adaptive immunity. For example, several studies have demonstrated the in vitro capacity of UPEC strains to downregulate the secretion of proinflammatory cytokines by cultured epithelial cells (6, 19, 28). In vitro epithelial cytokine suppression requires SurA (19), implying that one or more integral OM proteins are involved; indeed, OmpA of E. coli K-1 inhibits proinflammatory cytokine production from isolated monocytes (48). Alternatively, OmpA of UPEC might protect the organism from antimicrobial peptides secreted by polymorphonuclear leukocytes, as demonstrated in vitro for OmpA of E. coli K-1 (14). Such a need for OmpA during these extracellular phases of cystitis is also evidenced by upregulation of ompA in luminal bacteria, which are exposed as innate responses are being mustered. Our ongoing studies aim to further specify the mechanism(s) of OmpA interactions with the host. Whatever the mechanism, the effect of ompA deletion is specific, rather than reflecting more generally the lack of a major OM protein, as ompC deletion does not recapitulate the ΔompA phenotypes (S. Chen, unpublished data).
OmpA has not previously been demonstrated to contribute to urovirulence, although it was identified as an antigen expressed during cystitis (17) and during bacterial growth in urine (1). Strong immunoreactivity against an array of OM proteins, including OmpA, was shown when OM extracts from the pyelonephritis strain CFT073 were separated by two-dimensional gel electrophoresis and probed with sera from mice chronically infected with the strain (17). The ability of OmpA to stimulate mammalian adaptive responses also makes it an attractive vaccine candidate (21, 23, 50). Our results support this potential exploitation of OmpA; priming the adaptive immune system with the capacity to respond to this important determinant of later-stage pathogenic phenotypes might offer benefit in preventing the establishment of chronic bacterial reservoirs, thereby precluding subsequent recurrences of UTI.
Acknowledgments
This study was supported by Public Health Service grants DK067894 and DK076556 (to D.A.H.); the Washington University Child Health Research Center (HD01487); and the Edward Mallinckrodt, Jr., Foundation of St. Louis, MO. T.F.N. was supported by a Ford Foundation Diversity Fellowship administered by the National Academies.
We thank J. Loughman for technical assistance and P. Tarr and M. Caparon for critical review of the manuscript.
Editor: V. J. DiRita
Footnotes
Published ahead of print on 21 September 2009.
REFERENCES
- 1.Alteri, C. J., and H. L. Mobley. 2007. Quantitative profile of the uropathogenic Escherichia coli outer membrane proteome during growth in human urine. Infect. Immun. 75:2679-2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, G. G., K. W. Dodson, T. M. Hooton, and S. J. Hultgren. 2004. Intracellular bacterial communities of uropathogenic Escherichia coli in urinary tract pathogenesis. Trends Microbiol. 12:424-430. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, G. G., J. J. Palermo, J. D. Schilling, R. Roth, J. Heuser, and S. J. Hultgren. 2003. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 301:105-107. [DOI] [PubMed] [Google Scholar]
- 4.Arora, A., F. Abildgaard, J. H. Bushweller, and L. K. Tamm. 2001. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat. Struct. Biol. 8:334-338. [DOI] [PubMed] [Google Scholar]
- 5.Arora, A., D. Rinehart, G. Szabo, and L. K. Tamm. 2000. Refolded outer membrane protein A of Escherichia coli forms ion channels with two conductance states in planar lipid bilayers. J. Biol. Chem. 275:1594-1600. [DOI] [PubMed] [Google Scholar]
- 6.Billips, B. K., A. J. Schaeffer, and D. J. Klumpp. 2008. Molecular basis of uropathogenic Escherichia coli evasion of the innate immune response in the bladder. Infect. Immun. 76:3891-3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bond, P. J., J. D. Faraldo-Gomez, and M. S. P. Sansom. 2002. OmpA: a pore or not a pore? Simulation and modeling studies. Biophys. J. 83:763-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brauner, A., B. Kaijser, and I. Kuhn. 1994. Recurrent Escherichia coli bacteraemia: clinical characteristics and bacterial properties. J. Infect. 28:49-57. [DOI] [PubMed] [Google Scholar]
- 9.Chen, S. L., C. S. Hung, J. Xu, C. S. Reigstad, V. Magrini, A. Sabo, D. Blasiar, T. Bieri, R. R. Meyer, P. Ozersky, J. R. Armstrong, R. S. Fulton, J. P. Latreille, J. Spieth, T. M. Hooton, E. R. Mardis, S. J. Hultgren, and J. I. Gordon. 2006. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc. Natl. Acad. Sci. USA 103:5977-5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cormack, B. P., R. H. Valdivia, and S. Falkow. 1996. FACS-optimized mutants of the green fluorescent protein. Gene 173:33-38. [DOI] [PubMed] [Google Scholar]
- 11.Danese, P. N., and T. J. Silhavy. 1998. Targeting and assembly of periplasmic and outer-membrane proteins in Escherichia coli. Annu. Rev. Genet. 32:59-94. [DOI] [PubMed] [Google Scholar]
- 12.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foxman, B. 2002. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am. J. Med. 113:5S-13S. [DOI] [PubMed] [Google Scholar]
- 14.Fu, H., A. A. Belaaouaj, C. Dahlgren, and J. Bylund. 2003. Outer membrane protein A deficient Escherichia coli activates neutrophils to produce superoxide and shows increased susceptibility to antibacterial peptides. Microbes Infect. 5:781-788. [DOI] [PubMed] [Google Scholar]
- 15.Griebling, T. L. 2007. Urinary tract infections in women, p. 587-620. In M. S. Litwin and C. S. Saigal (ed.), Urologic diseases in America. U.S. Government Printing Office, Washington, DC.
- 16.Gunther, I. N., J. A. Snyder, V. Lockatell, I. Blomfield, D. E. Johnson, and H. L. Mobley. 2002. Assessment of virulence of uropathogenic Escherichia coli type 1 fimbrial mutants in which the invertible element is phase-locked on or off. Infect. Immun. 70:3344-3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagan, E. C., and H. L. Mobley. 2007. Uropathogenic Escherichia coli outer membrane antigens expressed during urinary tract infection. Infect. Immun. 75:3941-3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hultgren, S. J., T. N. Porter, A. J. Schaeffer, and J. L. Duncan. 1985. Role of type 1 pili and effects of phase variation on lower urinary tract infections produced by Escherichia coli. Infect. Immun. 50:370-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunstad, D. A., S. S. Justice, C. S. Hung, S. R. Lauer, and S. J. Hultgren. 2005. Suppression of bladder epithelial cytokine responses by uropathogenic Escherichia coli. Infect. Immun. 73:3999-4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeannin, P., B. Bottazzi, M. Sironi, A. Doni, M. Rusnati, M. Presta, V. Maina, G. Magistrelli, J. F. Haeuw, G. Hoeffel, N. Thieblemont, N. Corvaia, C. Garlanda, Y. Delneste, and A. Mantovani. 2005. Complexity and complementarity of outer membrane protein A recognition by cellular and humoral innate immunity receptors. Immunity 22:551-560. [DOI] [PubMed] [Google Scholar]
- 21.Jeannin, P., G. Magistrelli, L. Goetsch, J. F. Haeuw, N. Thieblemont, J. Y. Bonnefoy, and Y. Delneste. 2002. Outer membrane protein A (OmpA): a new pathogen-associated molecular pattern that interacts with antigen presenting cells-impact on vaccine strategies. Vaccine 20(Suppl. 4):A23-A27. [DOI] [PubMed] [Google Scholar]
- 22.Jeannin, P., G. Magistrelli, N. Herbault, L. Goetsch, S. Godefroy, P. Charbonnier, A. Gonzalez, and Y. Delneste. 2003. Outer membrane protein A renders dendritic cells and macrophages responsive to CCL21 and triggers dendritic cell migration to secondary lymphoid organs. Eur. J. Immunol. 33:326-333. [DOI] [PubMed] [Google Scholar]
- 23.Jeannin, P., T. Renno, L. Goetsch, I. Miconnet, J. P. Aubry, Y. Delneste, N. Herbault, T. Baussant, G. Magistrelli, C. Soulas, P. Romero, J. C. Cerottini, and J. Y. Bonnefoy. 2000. OmpA targets dendritic cells, induces their maturation and delivers antigen into the MHC class I presentation pathway. Nat. Immunol. 1:502-509. [DOI] [PubMed] [Google Scholar]
- 24.Justice, S. S., C. Hung, J. A. Theriot, D. A. Fletcher, G. G. Anderson, M. J. Footer, and S. J. Hultgren. 2004. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc. Natl. Acad. Sci. USA 101:1333-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Justice, S. S., D. A. Hunstad, J. R. Harper, A. R. Duguay, J. S. Pinkner, J. Bann, C. Frieden, T. J. Silhavy, and S. J. Hultgren. 2005. Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J. Bacteriol. 187:7680-7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Justice, S. S., D. A. Hunstad, P. C. Seed, and S. J. Hultgren. 2006. Filamentation by Escherichia coli subverts innate defenses during urinary tract infection. Proc. Natl. Acad. Sci. USA 103:19884-19889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Justice, S. S., S. R. Lauer, S. J. Hultgren, and D. A. Hunstad. 2006. Maturation of intracellular Escherichia coli communities requires SurA. Infect. Immun. 74:4793-4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klumpp, D. J., A. C. Weiser, S. Sengupta, S. G. Forrestal, R. A. Batler, and A. J. Schaeffer. 2001. Uropathogenic Escherichia coli potentiates type 1 pilus-induced apoptosis by suppressing NF-κB. Infect. Immun. 69:6689-6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koebnik, R. 1999. Structural and functional roles of the surface-exposed loops of the beta-barrel membrane protein OmpA from Escherichia coli. J. Bacteriol. 181:3688-3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulesus, R. R., K. Diaz-Perez, E. S. Slechta, D. S. Eto, and M. A. Mulvey. 2008. Impact of the RNA chaperone Hfq on the fitness and virulence potential of uropathogenic Escherichia coli. Infect. Immun. 76:3019-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazar, S. W., M. Almiron, A. Tormo, and R. Kolter. 1998. Role of the Escherichia coli SurA protein in stationary-phase survival. J. Bacteriol. 180:5704-5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazar, S. W., and R. Kolter. 1996. SurA assists the folding of Escherichia coli outer membrane proteins. J. Bacteriol. 178:1770-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 34.Martinez, J. J., M. A. Mulvey, J. D. Schilling, J. S. Pinkner, and S. J. Hultgren. 2000. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 19:2803-2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mulvey, M. A. 2002. Adhesion and entry of uropathogenic Escherichia coli. Cell. Microbiol. 4:257-271. [DOI] [PubMed] [Google Scholar]
- 36.Mulvey, M. A., Y. S. Lopez-Boado, C. L. Wilson, R. Roth, W. C. Parks, J. Heuser, and S. J. Hultgren. 1998. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 282:1494-1497. [DOI] [PubMed] [Google Scholar]
- 37.Mulvey, M. A., J. D. Schilling, and S. J. Hultgren. 2001. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 69:4572-4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy, K. C., and K. G. Campellone. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic Escherichia coli. BMC Mol. Biol. 4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mysorekar, I. U., and S. J. Hultgren. 2006. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. USA 103:14170-14175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mysorekar, I. U., M. A. Mulvey, S. J. Hultgren, and J. I. Gordon. 2002. Molecular regulation of urothelial renewal and host defenses during infection with uropathogenic Escherichia coli. J. Biol. Chem. 277:7412-7419. [DOI] [PubMed] [Google Scholar]
- 41.Prasadarao, N. V. 2002. Identification of Escherichia coli outer membrane protein A receptor on human brain microvascular endothelial cells. Infect. Immun. 70:4556-4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prasadarao, N. V., A. M. Blom, B. O. Villoutreix, and L. C. Linsangan. 2002. A novel interaction of outer membrane protein A with C4b binding protein mediates serum resistance of Escherichia coli K1. J. Immunol. 169:6352-6360. [DOI] [PubMed] [Google Scholar]
- 43.Prasadarao, N. V., P. K. Srivastava, R. S. Rudrabhatla, K. S. Kim, S. H. Huang, and S. K. Sukumaran. 2003. Cloning and expression of the Escherichia coli K1 outer membrane protein A receptor, a gp96 homologue. Infect. Immun. 71:1680-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prasadarao, N. V., C. A. Wass, J. N. Weiser, M. F. Stins, S. H. Huang, and K. S. Kim. 1996. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect. Immun. 64:146-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ronald, A. 2003. The etiology of urinary tract infection: traditional and emerging pathogens. Dis. Mon. 49:71-82. [DOI] [PubMed] [Google Scholar]
- 46.Ronald, A., and E. Ludwig. 2001. Urinary tract infections in adults with diabetes. Int. J. Antimicrob. Agents 17:287-292. [DOI] [PubMed] [Google Scholar]
- 47.Rosen, D. A., T. M. Hooton, W. E. Stamm, P. A. Humphrey, and S. J. Hultgren. 2007. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 4:e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Selvaraj, S. K., and N. V. Prasadarao. 2005. Escherichia coli K1 inhibits proinflammatory cytokine induction in monocytes by preventing NF-κB activation. J. Leukoc. Biol. 78:544-554. [DOI] [PubMed] [Google Scholar]
- 49.Sun, T. T., H. Zhao, J. Provet, U. Aebi, and X. R. Wu. 1996. Formation of asymmetric unit membrane during urothelial differentiation. Mol. Biol. Rep. 23:3-11. [DOI] [PubMed] [Google Scholar]
- 50.Torres, A. G., Y. Li, C. B. Tutt, L. Xin, T. Eaves-Pyles, and L. Soong. 2006. Outer membrane protein A of Escherichia coli O157:H7 stimulates dendritic cell activation. Infect. Immun. 74:2676-2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang, Y., and K. S. Kim. 2002. Role of OmpA and IbeB in Escherichia coli K1 invasion of brain microvascular endothelial cells in vitro and in vivo. Pediatr. Res. 51:559-563. [DOI] [PubMed] [Google Scholar]
- 52.Weiser, J. N., and E. C. Gotschlich. 1991. Outer membrane protein A (OmpA) contributes to serum resistance and pathogenicity of Escherichia coli K-1. Infect. Immun. 59:2252-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright, K. J., P. C. Seed, and S. J. Hultgren. 2007. Development of intracellular bacterial communities of uropathogenic Escherichia coli depends on type 1 pili. Cell. Microbiol. 9:2230-2241. [DOI] [PubMed] [Google Scholar]