

Abstract
VEGF is the key player in tumor angiogenesis. In the current study, the impact of VEGF expression on the response of tumors to the VEGFR2 associated tyrosine kinase inhibitor vandetanib was evaluated.
Materials and Methods
Human colon carcinoma (HT29) and murine squamous carcinoma (SCCVII) clonal cell lines expressing varying levels of VEGF were established and their response to vandetanib was assessed in tissue culture and as solid tumors.
Results
Vandetanib treatment had no effect on tumor cell clonogenic cell survival in vitro but doses ≥10 nM significantly reduced endothelial cell migration. In vivo, tumors derived from cell clones expressing high levels of VEGF displayed significantly enhanced angiogenesis and more aggressive growth. An intradermal angiogenesis assay was used to demonstrate that a 4-day treatment with vandetanib (50 mg/kg/day) was able to significantly inhibit blood vessel growth induced by both parental and high VEGF-expressing tumor cell clones. In the HT29 tumor model, treatment response to vandetanib (50 mg/kg/day, Monday-Friday for 2 weeks) was greatest in xenografts derived from the highest VEGF-expressing cell clones. A similar trend was noted in the SCCVII tumor model. The present findings indicate that vandetanib therapy effectively counteracted the aggressive feature of tumor growth resulting from VEGF over-expressing tumor cells and suggest that such tumors may be particularly well suited for anti-VEGF interventions.
Keywords: VEGF, tumor vascularity, angiogenesis, anti-angiogenic therapy, vandetanib, ZACTIMA™, ZD6474
It is widely known that angiogenesis plays a key role in the progressive growth of solid tumors and the metastatic cascade of cancer cells (1, 2). Vascular endothelial growth factor (VEGF) is the key mediator of angiogenesis (3). It is the only growth factor shown to be a specific and critical mitogen for endothelial cells while devoid of consistent and significant mitogenic effects in other cell types (4, 5). VEGF induces endothelial cell migration and tube formation (6), functions as a survival factor for endothelial cells (7) and increases vascular permeability which allows leakage of plasma proteins and the development of an extravascular matrix, thus further enhancing the niches for subsequent endothelial cell growth (8). VEGF also induces chemotaxis (9) and the expression of other pro-angiogenic factors (10) such as plasminogen activators (11) and collagenases (5, 12). Its expression is up-regulated in human cancer as a consequence of oncogene activation, loss of tumor suppressor function or hypoxia resulting from suboptimal perfusion (13, 14). Clinically, high levels of plasma VEGF have been associated with poor prognosis in patients with a variety of tumors (15–18).
Recognition of the central role of VEGF in tumor angiogenesis has led to the development of various strategies to inhibit VEGF signaling. Such strategies include antisense oligonucleotides, antibodies or receptor tyrosine kinase inhibitors (19–22). Of these approaches, the small molecule receptor tyrosine kinase inhibitors are particularly attractive. They are orally active and thus easily compatible with long term use. Vandetanib (ZACTIMA™; ZD6474) is a lead compound in this class. It is a once-daily oral anticancer drug that inhibits VEGF, epidermal growth factor (EGF) and RETS (rearranged during transfection) receptor signaling (23). In vivo, vandetanib antitumor effects have been demonstrated in a wide range of preclinical models including lung, prostate, breast, ovarian or vulval tumors (23–26). Moreover, vandetanib also has the potential to inhibit metastasis by prevention of primary tumor dissemination, as well as by inhibiting the growth of metastatic foci (27, 28). Currently, vandetanib is being evaluated in over 50 clinical trials of different tumor types (www.clinicaltrial.gov).
Though evidence exists to suggest that increased VEGF expression levels may be related to more aggressive tumor behavior and poor treatment outcome (15–18), little is known about whether VEGF levels affect VEGF-directed anticancer therapy. Insights into this question are difficult to obtain from clinical trial data due to the significant inter-patient variations, heterogeneous tumor populations and differences in patient treatments associated with such studies. Preclinical investigations employing different tumor cell lines or models with varying levels of endogenous VEGF expression also cannot address this issue adequately because other differences between the models including genetic backgrounds, intrinsic drug sensitivity, tumor-host interactions or tumor immunogenicities, may be involved. To address this topic directly human colorectal tumor (HT29) and murine squamous cell carcinoma (SCCVII) models from clonal cell lines with altered VEGF expression levels were established and characterized (29). The clones were established by clonal selection from tumor cells infected with a recombinant adeno-associated virus (rAAV) vector containing the VEGF165 gene. Consequently the lines carry the same genetic background in the presence of varying levels of stable VEGF expression. In vitro the clonal cell lines have similar growth characteristics and expression profiles of other key pro-angiogenic factors, but in vivo, tumors from high VEGF expressing clones grow faster, have increased blood vessel densities and are better oxygenated (29). The goal of the present study was to examine the therapeutic impact of targeting VEGF signaling using the small molecule VEGFR2 associated tyrosine kinase inhibitor vandetanib in tumors derived from cancer cell lines of varying VEGF expression.
Materials and Methods
Cell lines
Human colon carcinoma (HT29) and murine squamous cell carcinoma (SCCVII) cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and Stanford University (Palo Alto, CA, USA), respectively. Cells were maintained in Dulbecco’s modified Eagle’s medium (HT29) and alpha-minimal essential medium (SCCVII) supplemented with 10% fetal calf serum (FBS) and 0.01% (v/v) L glutamine and penicillin/streptomycin (Invitrogen, Grand Island, NY, USA). Human microvascular endothelial cells of the lung (HMVEC L) were obtained from Cambrex Corporation (Baltimore, MD, USA) and maintained in EGM 2 medium (Clonetics, Inc., San Diego, CA, USA) supplemented with 2 ng/mL VEGF. All cells were maintained at 37°C in a 5% CO2 incubator.
Generation of stable tumor cell lines by rAAV transfection
HT29 or SCCVII clonal cell lines expressing various levels of VEGF were derived as previously described (29). Briefly, 1×104 HT29 or SCCVII cells were suspended in 50 μL of serum- and antibiotic-free medium. Recombinant adeno-associated virus (rAAV) vectors containing the cassette for human VEGF165 and a neomycin resistance gene were then added at a multiplicity of infection (MOI) of 10,000. The cells were incubated for 3 h before changing to selection media that contained 1 mg/mL geneticin for another 48 h at 37°C. Surviving cells were plated at a low density in 60-mm dishes to obtain clones. Stable cell lines were maintained in the appropriate cell media with the addition of 500 μg/mL geneticin.
Animals and tumor models
Either 1×106 HT29 tumor cells or 1×105 SCCVII tumor cells (in a volume of 0.02 mL phosphate-buffered saline) were injected intramuscularly into a single hind limb of 6- to 8-week-old female NCR nu/nu or C3H/HeJ mice (Frederick Cancer Research Facility, MD, USA), respectively. The mice were maintained under specific pathogen-free conditions (University of Florida Health Science Center Vivarium) with food and water provided ad libitum.
Drug preparation
Vandetanib was a gift from AstraZeneca Pharmaceuticals (Macclesfield, UK). Stock solutions of drug (10 mM) were prepared by suspending the agent in 10% (v/v) Tween 80 and N-2-hydroxyethylpiperazine-N′-2-ethane-sulfonic acid (HEPES). Working dilutions were made by serial dilution of the stock solution in sterile saline. All drug preparations were kept refrigerated in the dark and used within 1 week of preparation.
Clonogenic cell survival
HT29 or SCCVII cells were seeded into 60 mm tissue culture dishes and allowed to attach overnight. Vandetanib was added at doses ranging from 0–100 nM/mL for a period of 24 h. After treatment, the cells were trypsinized, counted and plated at three different concentrations in 60-mm petri dishes. Ten to fourteen days later, plates were stained with crystal violet and colonies of greater than 50 cells were counted with an aid of a dissecting microscope.
Endothelial cell migration
To assess endothelial cell migration in vitro, 1×105 HMVEC-L cells were seeded in modified Boyden chambers and treated with 5 to 500 nM vandetanib or vehicle control. After 48 h of treatment, the number of cells migrating through the 8 μm porous membrane was counted using a microscope at a ×20 magnification in 10 randomly selected fields on each filter.
Intradermal tumor angiogenesis
The extent of induction of angiogenesis by HT29 or SCCVII tumor cells and the degree of inhibition of this process by vandetanib treatment in vivo were determined using an intradermal tumor cell angiogenesis assay as described previously (30, 31). Briefly, 1×105 HT29 or SCCVII cells suspended in 10 μL of phosphate-buffered saline were injected intradermally at four sites on the ventral surface of a female nude mouse. One drop of 0.4% trypan blue solution was added to the injected mixture to aid visualization of the injection sites. Vandetanib was administered daily by oral gavage (50 mg/kg) for 4 days commencing on the day before tumor cell inoculation. Control mice received the blank vandetanib carrier. Mice were euthanized 3 days after tumor cell injection. The skin was carefully separated from the underlying muscle and the number of vessels counted using a dissecting microscope. Scoring of all of the reaction areas was carried out at the same magnification (×5) and only vessels readily detected at this magnification were counted. The sites of injection, recognized by local swelling and blue staining, were exposed by carefully removing fat or other tissue covering the area. All vessels that touched the edge of the tumor inoculates were counted. All the animals in the experiments were pre-coded and vessel counts in each animal were scored twice. The resultant data points for each treatment group were pooled for statistical analysis (Wilcoxon rank sum test).
Assessment of tumor response to vandetanib
When tumors reached a size of ~200 mm3, mice were randomly assigned to control or treatment groups. Vandetanib was administered daily by oral gavage (0.1 mL/10 g body weight) for a period of 2 weeks (Monday to Friday). Control mice received vandetanib blank carrier by oral gavage. Following treatment, tumor size was measured by passing the tumor-bearing leg through a series of increasing diameter holes in an acrylic plate. The diameter of the smallest hole that the tumor bearing leg could pass through was recorded. The diameter was converted to a tumor volume using the following formula: tumor volume = 1/6(πd3) − 100, where d is the hole diameter and 100 represents a volume correction factor determined for a mouse leg without a tumor (32). The time for the tumors in the various treatment groups to grow to 5 times the starting size were recorded and compared (Wilcoxon rank–sum test).
Results
Clonal cell lines expressing various levels of VEGF were established from both HT29 and SCCVII parental lines by viral infection as previously described (29). Three clones were ultimately chosen for each tumor model: the first expressed VEGF at a level comparable to that of the parental cell line, the second expressed VEGF at an intermediate level, and the third expressed VEGF at a high level (29). In the HT29 model, the VEGF expression level of parental cells was 0.25 μg/mL; clones selected for study expressed 0.3, 6.1 and 18 μg/mL VEGF, respectively. These clones were named v1-8, v1-3 and v2-8 on the basis of the numbering system used during the original clone selection process (29). A similar nomenclature also was applied in the SCCVII model. For that cell line, neither the parental cells nor the v1-2 clone expressed detectable levels of human VEGF; clones v1-9 and v2-7 expressed 6.9 and 13.8 μg/mL, respectively. Importantly, manipulation of the VEGF expression levels did not significantly affect the in vitro growth rate of these lines nor the expression levels of other pro-angiogenic growth factors including bFGF, PDGF, Ang 1 and Ang 2 (29). In addition, RT-PCR confirmed the lack of expression of VEGFR1 and VEGFR2 on these tumor cell lines (29).
Initial studies examined the inherent in vitro sensitivities of the parental and clonal cell lines to a range of doses of vandetanib. The results showed that doses as high as 100 nM (HT29) or 100 μM (SCCVII) did not affect tumor cell survival in either the parental lines or any of the clonal cell lines (Figure 1). Endothelial cell (HMVEC-L) survival was also not affected by vandetanib doses up to 100 nM (data not shown). However, unlike tumor cells, vandetanib doses ≥50 nM did inhibit endothelial cell growth (32) and endothelial cell migration through a porous membrane was impaired at doses ≥10 nM (Figure 2).
Figure 1.
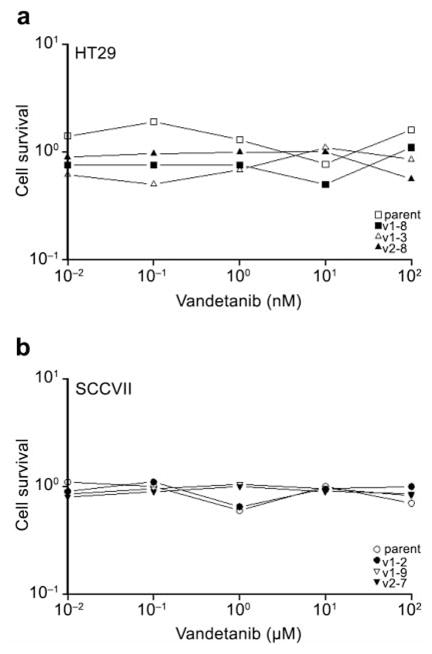
Clonogenic cell survival of HT29 (a) and SCCVII (b) parental and clonal cell lines assessed 24 h after treatment with a range of doses of vandetanib. Data shown are the average of 2 independent experiments.
Figure 2.

The effects of vandetanib treatment on endothelial cell migration through a modified Boyden chamber after 24-hour exposure to 5–500 nM vandetanib. Results for 3 independent experiments are indicated by the different shaded bars. In all cases, data obtained for vandetanib doses ≥10 nM were significantly different (p<0.01) versus controls (Student’s t-test).
To determine the effect of vandetanib treatment on HT29 or SCCVII tumor cell-induced angiogenesis in vivo, an intradermal assay (30, 31) was used. Mice received an intradermal inoculation of parental or high-expressing VEGF165 (v2–8, HT29; v2-7, SCCVII) clonal tumor cells (1×105). Vandetanib (50 mg/kg/day) or its carrier was administered by oral gavage for 4 days commencing on the day prior to tumor cell inoculation. Three days later, all mice were euthanized and the skin flap containing the inoculation site was excised. The number of blood vessels intersecting each inoculate was counted. The results (Figure 3) showed that even this short-term treatment of a few days significantly decreased the numbers of vessels induced by the tumor cell inoculates. Both the parental and high-expressing VEGF variants of the HT29 tumor model showed an approximately 2-fold reduction in the number of blood vessels induced (Figure 3a). A similar reduction in new blood vessel growth also was observed when mice injected with the high VEGF expressing SCCVII clone (v2-7) were treated with vandetanib (Figure 3b). These findings indicate that vandetanib treatment could be equally effective in blocking VEGF signaling in both low and high VEGF-expressing tumors.
Figure 3.

Nude mice (5/group) were injected intradermally with 1×105 HT29 (a) or SCCVII (b) tumor cells at 4 sites per mouse. Three days later, the numbers of blood vessels induced at each inoculate were counted. In groups receiving vandetanib (50 mg/kg), the drug was given daily commencing on the day before tumor cell inoculation. Data shown are the median and 75th and 90th percentiles. Asterisks indicate statistical significance (p<0.05, Wilcoxon rank–sum test) compared with the carrier-treated control groups.
It has previously been shown that vandetanib treatment can increase both the growth delay and the efficacy of ionizing radiation in HT29 xenografts (32). In the present study, the response of tumors established from parental HT29 cells was compared to that of tumors derived from HT29 cell clones with increased VEGF expression. Once established (~200 mm3), tumors were treated with 50 mg/kg oral doses of vandetanib daily for a two week period (days 1–5 and 8–12). In the HT29 tumor model the anti-VEGF therapy resulted in tumor growth delays which increased with increasing VEGF expression (4 days for the parental cell line; 10 days for the high-expressing VEGF clone) (Figure 4a). Mice bearing SCCVII tumors were only treated with vandetanib for a period of 5 days because the rapid growth rate of this tumor model prevented the intended 2-week course of vandetanib from being given. Even so, the 5-day vandetanib treatment did lead to the greatest responses in tumors derived from the higher VEGF expressing SCCVII clones (Figure 4b).
Figure 4.

Response of tumors from HT29 (a) and SCCVII (b) parental and clonal cell lines to vandetanib treatment. Vandetanib (50 mg/kg) was administered on a daily basis (Monday-Friday) for a 2-week period commencing when the tumors reached a size of approximately 200 mm3. Asterisks indicate a p-value of less than 0.05 when the growth delay of each group was compared via the Wilcoxon rank-sum test to that of the parental group.
Discussion
The central role of VEGF in tumor angiogenesis and disease progression has been well established (2, 3). VEGF impacts multiple steps in the angiogenic process including endothelial proliferation, migration, survival, and chemotaxis (3, 33). VEGF mRNA overexpression has been associated with an array of cancer types (34) and evidence exists to implicate VEGF as an indicator of poor prognosis in human cancer (15, 18, 35, 36). To ascertain whether VEGF expression influences the response of tumors to anti-VEGF therapy, clonal cell lines expressing varying VEGF levels (29) were grown as solid tumors which display a difference in tumor vascularity and hypoxia (29). The response of these tumor models to the small molecule VEGFR2 signaling inhibitor vandetanib was then assessed in the absence of confounding interpretational difficulties associated with inter-tumor model comparisons.
In in vitro assays, vandetanib had little effect on HT29 and SCCVII tumor cell survival (Figure 1) but did inhibit endothelial cell function at low doses (Figure 2), supporting the notion that the in vivo effects of this agent reflect its anti-angiogenic properties.
The present results (Figure 3) confirmed previous observations (29) that higher VEGF, expressing tumor cells (both HT29 and SCCVII clones) induced significantly more blood vessels when assessed in an intradermal angiogenesis assay (29). Vandetanib treatment reduced the tumor cell induced angiogenesis in both tumor cell types irrespective of their VEGF expression (Figure 3). For the HT29 model, inhibition of angiogenesis was observed for both high VEGF-expressing cells and parental tumor cells (Figure 3a). Inhibition of angiogenesis by vandetanib treatment was evident for both the high VEGF-expressing SCCVII clone and the parental tumor cells (Figure 3b), although the suppression was significant only for the high VEGF-expressing cells (Figure 3b). These findings suggest vandetanib treatment can efficiently counteract the pro-angiogenic effect of VEGF and impair tumor-induced angiogenesis regardless of the level of VEGF expression.
Xenograft tumors grown from HT29 cells expressing higher levels of VEGF were increasingly more aggressive (grew more rapidly from 200 to 1000 mm3, Figure 4a) than those from parental HT29 cells, as has been reported previously (29). When such tumors were treated with vandetanib, the greatest tumor growth delays were observed in xenograft tumors derived from clonal cells expressing the highest levels of VEGF (Figure 4a). Since vandetanib treatment led to 16–18 day response in all HT29 tumor groups (parent, v1-8, v1-3, v2-8), this implies that at least in this model, this agent effectively counteracted the aggressive feature of tumor growth resulting from VEGF over-expression. Increasing tumor responses (longer growth delays) in tumors derived from the highest VEGF-expressing tumor clone (v2-7) treated with vandetanib were also observed in the SCCVII tumor model (Figure 4b).
The results of this study indicate that vandetanib treatment may overcome the aggressive tumor behavior associated with overexpression of VEGF. In both the human colorectal model HT29 and the rodent carcinoma model SCCVII, greater in vivo response to vandetanib was observed with increasing levels of VEGF expression. Taken together these findings support the use of VEGFR2-associated VEGF signaling inhibition with small molecules such as vandetanib in cancer patients with high levels of VEGF expression.
Acknowledgments
This work was supported by U.S. Public Health Service grant CA089655. The authors thank Chris Pampo and Sharon Lepler for excellent technical assistance.
References
- 1.Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science. 2006;312:1171–1175. doi: 10.1126/science.1125950. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 3.Roskoski R., Jr Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol. 2007;62:179–213. doi: 10.1016/j.critrevonc.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Griffin RJ, Williams BW, Wild R, Cherrington JM, Park H, Song CW. Simultaneous inhibition of the receptor kinase activity of vascular endothelial, fibroblast, and platelet-derived growth factors suppresses tumor growth and enhances tumor radiation response. Cancer Res. 2002;62:1702–1706. [PubMed] [Google Scholar]
- 5.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl 3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 6.Stacker SA, Achen MG, Jussila L, Baldwin ME, Alitalo K. Lymphangiogenesis and cancer metastasis. Nat Rev Cancer. 2002;2:573–583. doi: 10.1038/nrc863. [DOI] [PubMed] [Google Scholar]
- 7.Liu W, Ahmad SA, Reinmuth N, Shaheen RM, Jung YD, Fan F, Ellis LM. Endothelial cell survival and apoptosis in the tumor vasculature. Apoptosis. 2000;5:323–328. doi: 10.1023/a:1009679307513. [DOI] [PubMed] [Google Scholar]
- 8.Thomas KA. Vascular endothelial growth factor, a potent and selective angiogenic agent. J Biol Chem. 1996;271:603–606. doi: 10.1074/jbc.271.2.603. [DOI] [PubMed] [Google Scholar]
- 9.Koch AE, Harlow LA, Haines GK, Amento EP, Unemori EN, Wong WL, Pope RM, Ferrara N. Vascular endothelial growth factor. A cytokine modulating endothelial function in rheumatoid arthritis. J Immunol. 1994;152:4149–4156. [PubMed] [Google Scholar]
- 10.Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21:1104–1117. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- 11.Mandriota SJ, Seghezzi G, Vassalli JD, Ferrara N, Wasi S, Mazzieri R, Mignatti P, Pepper MS. Vascular endothelial growth factor increases urokinase receptor expression in vascular endothelial cells. J Biol Chem. 1995;270:9709–9716. doi: 10.1074/jbc.270.17.9709. [DOI] [PubMed] [Google Scholar]
- 12.Unemori EN, Ferrara N, Bauer EA, Amento EP. Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J Cell Physiol. 1992;153:557–562. doi: 10.1002/jcp.1041530317. [DOI] [PubMed] [Google Scholar]
- 13.Shweiki D, Neeman M, Itin A, Keshet E. Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: implications for tumor angiogenesis. Proc Natl Acad Sci USA. 1995;92:768–772. doi: 10.1073/pnas.92.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rak J, Yu JL, Klement G, Kerbel RS. Oncogenes and angiogenesis: signaling three-dimensional tumor growth. J Investig Dermatol Symp Proc. 2000;5:24–33. doi: 10.1046/j.1087-0024.2000.00012.x. [DOI] [PubMed] [Google Scholar]
- 15.Ai KX, Lu LY, Huang XY, Chen W, Zhang HZ. Prognostic significance of S100A4 and vascular endothelial growth factor expression in pancreatic cancer. World J Gastroenterol. 2008;14:1931–1935. doi: 10.3748/wjg.14.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poon RT, Fan ST, Wong J. Clinical implications of circulating angiogenic factors in cancer patients. J Clin Oncol. 2001;19:1207–1225. doi: 10.1200/JCO.2001.19.4.1207. [DOI] [PubMed] [Google Scholar]
- 17.Jubb AM, Pham TQ, Hanby AM, Frantz GD, Peale FV, Wu TD, Koeppen HW, Hillan KJ. Expression of vascular endothelial growth factor, hypoxia inducible factor 1alpha, and carbonic anhydrase IX in human tumours. J Clin Pathol. 2004;57:504–512. doi: 10.1136/jcp.2003.012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li YH, Hu CF, Shao Q, Huang MY, Hou JH, Xie D, Zeng YX, Shao JY. Elevated expressions of survivin and VEGF protein are strong independent predictors of survival in advanced nasopharyngeal carcinoma. J Transl Med. 2008;6:1. doi: 10.1186/1479-5876-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi W, Siemann DW. Inhibition of renal cell carcinoma angiogenesis and growth by antisense oligonucleotides targeting vascular endothelial growth factor. Br J Cancer. 2002;87:119–126. doi: 10.1038/sj.bjc.6600416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madhusudan S, Harris AL. Drug inhibition of angiogenesis. Curr Opin Pharmacol. 2002;2:403–414. doi: 10.1016/s1471-4892(02)00184-4. [DOI] [PubMed] [Google Scholar]
- 21.Manley PW, Martiny-Baron G, Schlaeppi JM, Wood JM. Therapies directed at vascular endothelial growth factor. Expert Opin Investig Drugs. 2002;11:1715–1736. doi: 10.1517/13543784.11.12.1715. [DOI] [PubMed] [Google Scholar]
- 22.Schenone S, Bondavalli F, Botta M. Antiangiogenic agents: an update on small molecule VEGFR inhibitors. Curr Med Chem. 2007;14:2495–2516. doi: 10.2174/092986707782023622. [DOI] [PubMed] [Google Scholar]
- 23.McCarty MF, Wey J, Stoeltzing O, Liu W, Fan F, Bucana C, Mansfield PF, Ryan AJ, Ellis LM. ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor with additional activity against epidermal growth factor receptor tyrosine kinase, inhibits orthotopic growth and angiogenesis of gastric cancer. Mol Cancer Ther. 2004;3:1041–1048. [PubMed] [Google Scholar]
- 24.Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JA, Boffey SJ, Valentine PJ, Curwen JO, Musgrove HL, Graham GA, Hughes GD, Thomas AP, Stokes ES, Curry B, Richmond GH, Wadsworth PF, Bigley AL, Hennequin LF. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645–4655. [PubMed] [Google Scholar]
- 25.Taguchi F, Koh Y, Koizumi F, Tamura T, Saijo N, Nishio K. Anticancer effects of ZD6474, a VEGF receptor tyrosine kinase inhibitor, in gefitinib (“Iressa™”)-sensitive and resistant xenograft models. Cancer Sci. 2004;95:984–989. doi: 10.1111/j.1349-7006.2004.tb03187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryan AJ, Wedge SR. ZD6474 – a novel inhibitor of VEGFR and EGFR tyrosine kinase activity. Br J Cancer. 2005;92(Suppl 1):S6–13. doi: 10.1038/sj.bjc.6602603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drevs J, Konerding MA, Wolloscheck T, Wedge SR, Ryan AJ, Ogilvie DJ, Esser N. The VEGF receptor tyrosine kinase inhibitor, ZD6474, inhibits angiogenesis and affects microvascular architecture within an orthotopically implanted renal cell carcinoma. Angiogenesis. 2004;7:347–354. doi: 10.1007/s10456-005-1394-3. [DOI] [PubMed] [Google Scholar]
- 28.Matsumori Y, Yano S, Goto H, Nakataki E, Wedge SR, Ryan AJ, Sone S. ZD6474, an inhibitor of vascular endothelial growth factor receptor tyrosine kinase, inhibits growth of experimental lung metastasis and production of malignant pleural effusions in a non-small cell lung cancer model. Oncol Res. 2006;16:15–26. doi: 10.3727/000000006783981260. [DOI] [PubMed] [Google Scholar]
- 29.Norris CM, Shi W, Siemann DW. Impact of VEGF expression on the physiological characteristics of clonal cell lines. In Vivo. 2006;20:815–821. [PubMed] [Google Scholar]
- 30.Auerbach R, Kubai L, Sidky Y. Angiogenesis induction by tumors, embryonic tissues, and lymphocytes. Cancer Res. 1976;36:3435–3440. [PubMed] [Google Scholar]
- 31.Runkel S, Hunter N, Milas L. An intradermal assay for quantification and kinetics studies of tumor angiogenesis in mice. Radiat Res. 1991;126:237–243. [PubMed] [Google Scholar]
- 32.Brazelle WD, Shi W, Siemann DW. VEGF-associated tyrosine kinase inhibition increases the tumor response to single and fractionated dose radiotherapy. Int J Radiat Oncol Biol Phys. 2006;65:836–841. doi: 10.1016/j.ijrobp.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 33.Ho QT, Kuo CJ. Vascular endothelial growth factor: biology and therapeutic applications. Int J Biochem Cell Biol. 2007;39:1349–1357. doi: 10.1016/j.biocel.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berger DP, Herbstritt L, Dengler WA, Marme D, Mertelsmann R, Fiebig HH. Vascular endothelial growth factor (VEGF) mRNA expression in human tumor models of different histologies. Ann Oncol. 1995;6:817–825. doi: 10.1093/oxfordjournals.annonc.a059322. [DOI] [PubMed] [Google Scholar]
- 35.Kamat AA, Merritt WM, Coffey D, Lin YG, Patel PR, Broaddus R, Nugent E, Han LY, Landen CN, Jr, Spannuth WA, Lu C, Coleman RL, Gershenson DM, Sood AK. Clinical and biological significance of vascular endothelial growth factor in endometrial cancer. Clin Cancer Res. 2007;13:7487–7495. doi: 10.1158/1078-0432.CCR-07-1017. [DOI] [PubMed] [Google Scholar]
- 36.Logan-Collins JM, Lowy AM, Robinson-Smith TM, Kumar S, Sussman JJ, James LE, Ahmad SA. VEGF expression predicts survival in patients with peritoneal surface metastases from mucinous adenocarcinoma of the appendix and colon. Ann Surg Oncol. 2008;15:738–744. doi: 10.1245/s10434-007-9699-7. [DOI] [PubMed] [Google Scholar]