

Abstract
Streams are highly heterogeneous ecosystems, in terms of both geomorphology and hydrodynamics. While flow is recognized to shape the physical architecture of benthic biofilms, we do not yet understand what drives community assembly and biodiversity of benthic biofilms in the heterogeneous flow landscapes of streams. Within a metacommunity ecology framework, we experimented with streambed landscapes constructed from bedforms in large-scale flumes to illuminate the role of spatial flow heterogeneity in biofilm community composition and biodiversity in streams. Our results show that the spatial variation of hydrodynamics explained a remarkable percentage (up to 47%) of the variation in community composition along bedforms. This suggests species sorting as a model of metacommunity dynamics in stream biofilms, though natural biofilm communities will clearly not conform to a single model offered by metacommunity ecology. The spatial variation induced by the hydrodynamics along the bedforms resulted in a gradient of bacterial beta diversity, measured by a range of diversity and similarity indices, that increased with bedform height and hence with spatial flow heterogeneity at the flume level. Our results underscore the necessity to maintain small-scale physical heterogeneity for community composition and biodiversity of biofilms in stream ecosystems.
Biofilms (attached and matrix-enclosed microbial communities) are an important form of microbial life in streams and rivers, where they can greatly contribute to ecosystem functions and even large-scale carbon fluxes (1, 3). Streams are inherently heterogeneous and are characterized by a largely unidirectional downstream flow of water that controls the dispersal of suspended microorganisms (21), biofilm community composition (7), architecture (2), and metabolism (13), for instance. However, we do not understand how diverse microorganisms assemble into biofilm communities based on flow heterogeneity and related dispersal in these ecosystems.
Dispersal, as the propagation and immigration of biota, can have important consequences for biodiversity and ecosystem functioning in heterogeneous landscapes (18, 25). Landscape topography and turbulent transport affect dispersal, a relationship that is well studied in the dispersal of plant seeds (31) but not in the microbial world. Only recently have microbial ecologists begun to understand the role of dispersal in large-scale biogeographic patterns (29) and metacommunity ecology (24, 44). This growing body of research on microbial dispersal and its consequences for spatial patterns of community assembly and composition rests entirely on free-living bacteria, while no comparable data exist for microbial biofilms. The confirmation of detachment as an intrinsic behavior in many biofilms has led to the appreciation of dispersal as an insurance policy for these microbial communities to seed new habitats during resource limitation or aging of the parental biofilm (4). However, microbial ecology lacks conceptual models to predict postemigration processes, such as cell propagation, immigration, and community assembly during colonization of new surfaces. The perception of biofilms as microbial landscapes and, at the same time, as integrated parts of the landscape they inhabit offers the possibility to test models for habitat selection by dispersal cells (4). In this study, we focused on the assembly of biofilm communities by dispersal cells in spatially variable-flow environments; we did not measure dispersal as the emigration of cells from established biofilms. We adopted metacommunity ecology as a framework that encapsulates environmental heterogeneity and dispersal (18) to illuminate the mechanisms underlying community assembly.
If the effects of microbial diversity on ecosystem functions are to be understood, we need to address the proper spatial resolution at which microorganisms assemble into communities and at which their functioning becomes manifest. In streams, this is typically at the level of habitats and microhabitats ranging from meters to centimeters, where characteristic geomorphological features (e.g., bedforms) and induced hydrodynamic fields develop and where spatial variations in biofilm metabolism become apparent (13). The ensemble of these small-scale variations translates into the landscape heterogeneity of the streambed.
The aim of this study was to test whether spatial flow heterogeneity generating diverse microhabitats induces spatial species turnover and increases the biodiversity of microbial biofilms. Microbial metacommunity ecology predicts mass effects rather than species sorting to drive community composition in ecosystems with low residence time, such as streams (14, 18, 24). To test this prediction, we constructed six streambed landscapes from bedforms of defined dimensions differing in height; the mean flow (at flume scale) was kept constant, whereas the spatial heterogeneity of flow increased across the gradient of the six landscapes. The inoculum (i.e., the stream water and naturally contained microorganisms) and water chemistry were equal in all flumes. This allowed us to isolate flow heterogeneity as a potential driver of biofilm community composition in a high-energy ecosystem. We used terminal restriction fragment length polymorphism (T-RFLP) analysis of bacterial 16S rRNA gene sequences from winter and summer communities and related bacterial community composition and microbial biomass to the hydrodynamics in representative microhabitats using causal modeling and forward selection of explanatory variables (9, 23).
MATERIALS AND METHODS
Constructed landscapes and sampling.
We studied the effect of spatial flow variation on biofilm community composition and biodiversity in six 40-m-long streamside flumes. To induce landscape heterogeneity, streambed landscapes were constructed from 38 fixed bedforms (1 m long) in streamside flumes (length, 40 m; width, 0.40 m). The triangular bedforms were installed adjacently in a periodic manner (Fig. 1a) to ensure undisturbed upstream flow fields (12). Five of the six flumes contained graded bedforms with given heights of 2, 4, 6, 8, and 10 cm, respectively; one flume without bedforms served as a control. The resulting flow environment resembles that of low-submergence flow as typical for headwater streams. Bedforms were impermeable to avoid vertical hydrodynamic exchange induced by the bedforms (12) and hence confounding effects of a subsurface habitat. A monolayer of clean (thoroughly washed), graded stream gravel (median size, 9.2 mm; lower quartile, 6.3 mm; upper quartile, 14.3 mm) (Oberer Seebach, Austria) covered the flumes and mimicked the benthic layer of a streambed. This experimental design allowed us to isolate the effect of physical heterogeneity on bacterial biodiversity and community composition.
FIG. 1.

Flow fields over the streambed in the control flume and the bedforms, successively increasing in height; these bedforms were used to generate landscapes of increasing flow heterogeneity in the flumes. (a) Bedform (side view); arrows indicate the position of the sampled microhabitat along a bedform. (b) Maps (bird's-eye view) of turbulent kinetic energy (joules) and flow velocity and direction (red arrows) in the control flume (no bedforms) and along the individual bedforms (2, 4, 6, 8, and 10 cm high) as derived from ADV measurements (5 mm above the sediment). The x axis refers to the position along an individual bedform, and the y axis refers to the flume width.
The flume-level flow rate was adjusted to 2.25 ± 0.10 liters s−1. Each flume contained a baffle adjacent to the inflow and a tailgate to achieve uniform flow over the entire flume. Nearly identical mean flume-level flow velocities among flumes were attained by adjusting the slope of each flume individually. Flumes were continuously fed from the same header tank with raw stream water (Oberer Seebach, Austria) in a once-through mode to ensure identical water chemistry and microbial inoculum in the landscapes. Water temperature was monitored daily, and light and concentrations of inorganic nutrients and dissolved organic carbon were measured twice a week. We performed one experiment in winter and one in summer to account for environmental dynamics in addition to hydrodynamics.
Sterile unglazed ceramic coupons (1 by 2 cm), glued on larger tiles (5 by 5 cm) to prevent them from erosion, served as a substratum for biofilm growth (7). Coupons were placed on the gravel to achieve hydrodynamic conditions similar to those in their adjacent environments. Coupons were sampled from four distinct microhabitats (upstream sides, crests, and downstream sides of the bedforms and the trough between two consecutive bedforms [Fig. 1]). Samples were collected from triplicate bedforms from the downstream half of the flumes to account for the possibility of poorly directed water flow and induced gradients of biodiversity in the upstream parts of the flumes. This resulted in 72 samples per sampling date and measured parameter (community composition, bacterial biomass, and chlorophyll a). Coupons from triplicate bedforms (same downstream location in all flumes) were pooled and processed as composite samples for bacterial community composition. Samples were collected 10 times (every fourth day) during winter and 5 times (days 11, 17, 37, 48, and 55) during summer.
Physical heterogeneity.
Three-dimensional flow velocity was measured with an acoustic Doppler velocimeter (ADV) (Vectrino Nortek) equipped with a four-beam side-looking probe; this is a noninvasive technique. Data were collected at a sampling rate of 50 Hz (1 min), yielding a time series (n = 3,000) for each location. In each flume, three-dimensional velocity was mapped over one of the graded bedforms (ca. 5 mm above ground) at regularly distributed nodes of a 5- by 5-cm grid. For each node, we calculated the three-dimensional vector of flow velocity (Rxyz), the turbulent kinetic energy (TKE), and the turbulence intensity (TI) according to the following equations: (11):
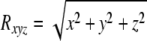 |
(1) |
where x, y, and z are velocity components as Cartesian coordinates,
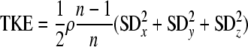 |
(2) |
where SDx, SDy, and SDz are the respective standard deviations (from n = 3,000 measurements) of the velocity components and ρ is the density of water, and
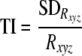 |
(3) |
where SDRxyz is the standard deviation of the three-dimensional velocity. Turbulence intensity is a measure of turbulence that is standardized for mean velocity, while turbulent kinetic energy includes the kinetic energy of mean velocity and turbulence; both describe the fluctuating hydrodynamic environment experienced by benthic biota. Rhodamine injections visualized the flow patterns along the bedforms and the wake-induced turbulence between consecutive bedforms; additionally, average flume-level flow velocity was determined weekly from conservative tracer additions. ADV measurements along four parallel longitudinal lines along one bedform in each flume (n = 80) yielded reliable estimates of the spatial variation of the flow environment.
Analysis of bacterial 16S rRNA gene.
Biofilms were removed from the coupons using ethanol-flamed tweezers and spatulas and homogenized by vortexing. An aliquot of approximately 250 μg homogenate (or the whole sample if less homogenate was obtained) was subjected to DNA extraction and purification using the UltraClean soil DNA isolation kit (MoBio, Carlsbad, CA). Noncolonized ceramic coupons served as negative controls. The fluorescently labeled primers used for PCR were the Bacteria-specific primer 27F (6-carboxyfluorescein labeled) and the universal primer 1492R (JOE [6-carboxy-4′,5′-dichloro-2′,7′-dimethoxyfluorescein] labeled; Thermo Electron, Germany), which give a 1,503-base-pair product of the 16S rRNA gene (20). PCR was performed as described by Moeseneder et al. (30). PCR products were cleaned using gel electrophoresis and the QIAquick gel extraction kit (Qiagen). Restriction digests were done as described earlier (30), using approximately 300 ng DNA and the enzyme HhaI. The products were desalted by gel filtration using MultiScreen-HV 96-well plates (Millipore) loaded with Sephadex G-50 (Sigma). The dried product was resuspended in 10 μl highly deionized formamide and 0.5-μl size marker GS2500 Rox (Applied Biosystems), denatured at 95°C, and immediately placed on ice. Fluorescently labeled DNA fragments were separated in a 3130 Xl capillary sequencer (Applied Biosystems), and electropherograms were analyzed using the GeneMapper software.
Restriction fragments smaller than 30 bp and larger than 900 bp were excluded from further analysis to avoid detection of primers and uncertainties of size determination. Peaks that were >2% of the maximum peak height were clearly distinguishable from background noise. The relative contribution of the respective operational taxonomic units (OTUs) to the total community was estimated as peak height divided by the cumulative peak height of the given sample. T-RFLP analysis can provide reproducible and sufficiently quantitative results (8, 27). Fragments containing the forward and the reverse primers were analyzed separately. T-RFLP patterns produced with the forward primer showed generally more heterogeneity in restriction fragment size than the corresponding patterns containing the reverse primer. All patterns of community composition and diversity were akin. The results presented in this paper therefore refer to the forward fragments because of their higher information content.
Microbial biomass.
Biofilms were detached and disaggregated as described by Besemer et al. (7). Briefly, coupons with biofilm were shaken (1 h) in 0.025-mmol/liter tetrasodium pyrophosphate and subsequently sonicated (180 s, 40 W output; Branson). Microbial cells in the supernatant were stained with the nucleic acid stain Sytox (Invitrogen) and counted and sized using flow cytometry (Cell Lab Quanta; Beckman Coulter). Cell numbers from flow cytometry were compared with epifluorescence microscopy counts; we found very good agreement between the two methods (slope of the regression line, 1.029; n = 62 [T. J. Battin, G. Steniczka, C. Preiler, and P. Paolini, unpublished data]). We used the allometric relationship described by Norland (32) to derive biomass from cell size. Chlorophyll a was extracted with pro analysi-grade acetone (12 h, 4°C) in the dark. Samples were vortexed, and the supernatant was filtered (GF/F Whatman) and assayed fluorometrically (EX435/EM675) using spinach (Sigma) as a standard.
Data analyses.
We used multivariate statistics and various diversity indices to analyze T-RFLP data. The responses of community composition and microbial biomass to the hydrodynamics in the microhabitats along the bedforms were explored using causal modeling on distance matrices (23). Distance matrices for community composition and biomass were calculated for each individual sampling date to remove temporal variation. The distance between T-RFLP samples was calculated using the Bray-Curtis index:
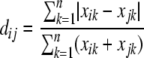 |
(4) |
where xik and xjk are the relative abundances of OTU k in samples i and j. The Bray-Curtis index was chosen as a reliable descriptor of the true resemblance between samples (10, 23). A normalized Euclidean distance matrix was computed from water depth, flow velocity, turbulent kinetic energy, and turbulence intensity in the four microhabitats. A normalized Euclidean distance matrix was calculated from chlorophyll a and bacterial biomass. The hydrodynamic distance matrix was compared to the community composition and biomass distance matrices from each date, using the partial Mantel's matrix randomization test (23, 28) with Spearman's rank correlation and 10,000 permutations. This method calculates the correlation between two matrices while controlling for the effect of a third matrix.
To identify the hydrodynamic parameters that contributed most to the variability of community composition and microbial biomass, we used a forward selection procedure (9). This procedure prevents overestimation of explained variance by using two stopping criteria, the alpha significance level and the coefficient of determination, calculated using a global test. Prior to analysis, the community composition data were Hellinger transformed and the biomass data were z standardized. The proportion of variation explained by the model containing all four explanatory variables (water depth, velocity, turbulent kinetic energy, and turbulence intensity) was calculated as the adjusted coefficient of multiple determination (9, 35). Explanatory variables were checked for multicollinearity using variance inflation factors. The R packages “packfor” (15) and “vegan” (33) were used for these calculations.
The use of a single diversity index is limited for molecular fingerprint data (5, 8), and most direct comparisons of quantitative and qualitative measures of microbial diversity have focused on alpha diversity, an approach that is prone to misinterpretations of diversity over a range of taxa and spatial scales (26). Therefore, we employed various diversity indices of the Hill family (17), namely, richness, the Shannon entropy, and the Gini-Simpson coefficient, which differ in their sensitivities toward rare species. When these diversity indices are expressed in terms of their number equivalents (i.e., the effective number of equally likely elements), they can be expressed in a single equation (17, 19):
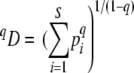 |
(5) |
where pi is the proportion of OTU i in the sample, S is the total number of OTUs, and the exponent q determines sensitivity toward rare or common species. At q = 0 all OTUs are equally weighted (richness), at q = 1 all OTUs are weighted according to their relative frequency (Shannon entropy), and at q = 2 the index is disproportionately sensitive to common species (Gini-Simpson coefficient). Equation 5 is undefined at q = 1 but finds its limit as
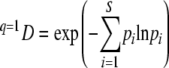 |
(6) |
Expressed as number equivalents, the regional diversity qDγ can be decomposed into two independent orthogonal components (19):
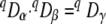 |
(7) |
with qDα being the local diversity (i.e., the effective number of OTUs) and qDβ being a measure for diversity between local communities independent from alpha diversity (i.e., the effective number of distinct communities).
For each flume we calculated qDα as the average local diversity
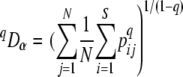 |
(8) |
where pi is the proportion of OTU i in sample j, and we calculated qDγ as the regional diversity
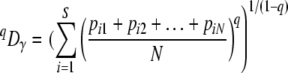 |
(9) |
where N = 4, the number of microhabitats along a bedform. Again, equations 8 and 9 are undefined at q = 1, and limits for qDα and qDγ are calculated from equation 6. qDβ is then computed from equation 7. This analysis of beta diversity has major advantages: it is entirely independent of alpha diversity, it yields a continuum of beta diversity measures differing in sensitivity toward rare or common species, and it integrates a range of popular diversity indices used in general ecology. Furthermore, we used the average Bray-Curtis distance between samples from one flume at a given time as an additional measure for within-flume diversity. The Bray-Curtis distance is not a monotonic transformation of any of the diversity indices used (19).
All diversity measurements were z standardized within each date to remove temporal variation according to
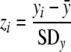 |
(10) |
where yi is any value of the variable y and ȳ and SDy are the mean and standard deviation of the respective variable (23). Standardized data were regressed on the standard deviation of flow velocity along one bedform, which is the major independent measure of flow heterogeneity and a reasonable descriptor of the flow heterogeneity at flume-level; additionally, they were regressed on the standard deviations of water depth, turbulent kinetic energy, and turbulence intensity. PAST (16) and R 2.7.0 (37) were used for statistical analyses.
RESULTS
Flow heterogeneity and environmental conditions.
High-resolution ADV measurements revealed flow fields with distinct spatial patterning of flow velocity and turbulence along the bedforms (Fig. 1; Table 1). Flow velocity accelerated on the upstream side and decelerated on the downstream side of the bedforms. Turbulent kinetic energy increased accordingly and was highest downstream of the bedform crest (Fig. 1), whereas turbulence intensity was highest between consecutive bedforms, where wake-induced eddies formed. Independent spot measurements on multiple graded bedforms confirmed the spatial reproducibility of this high-resolution mapping over one bedform. At the flume level, these local changes generated increasingly heterogeneous landscapes in terms of both roughness and induced flow fields. The spatial heterogeneity of flow, expressed as the standard deviation of the average three-dimensional flow velocity vector (Table 1), increased from 1.6 cm s−1 to 6.3 cm s−1 across all six flumes, while the mean flume-level flow velocity (7.6 ± 0.4 cm s−1) remained nearly identical among flumes.
TABLE 1.
Characterization of the near-bed hydrodynamics in the control flume (without bedforms) and above bedforms successively increasing in height
| Height (cm) of bedform | Mean ± SD (n = 80) |
|||
|---|---|---|---|---|
| Depth (cm) | Rxyz (cm s−1)a | Turbulence intensity (%) | Turbulent kinetic energy (J) | |
| Control (no bedform) | 6.3 ± 0.3 | 7.9 ± 1.6 | 41.9 ± 32.5 | 2.0 ± 0.6 |
| 2 | 6.5 ± 0.8 | 8.0 ± 1.9 | 41.8 ± 38.7 | 2.0 ± 0.9 |
| 4 | 7.5 ± 1.4 | 6.8 ± 2.5 | 48.6 ± 43.7 | 1.7 ± 1.0 |
| 6 | 6.9 ± 1.8 | 8.5 ± 3.5 | 50.0 ± 42.7 | 2.7 ± 1.9 |
| 8 | 6.5 ± 2.1 | 8.6 ± 4.1 | 50.1 ± 44.6 | 2.6 ± 2.0 |
| 10 | 6.7 ± 2.8 | 12.6 ± 6.3 | 50.2 ± 41.1 | 6.6 ± 7.1 |
Rxyz, three-dimensional vector of flow velocity.
The water temperature averaged 5.7 ± 1.5°C and 9.8 ± 2.1°C, and solar radiation (measured at noon) averaged 108 ± 62 μE cm−2 and 375 ± 275 μE cm−2, during winter and summer, respectively. Concentrations of inorganic nutrients and dissolved organic carbon exhibited no significant differences between flumes.
Variability of community composition and biofilm biomass.
T-RFLP analyses of biofilms identified totals of 129 OTUs in winter and 76 OTUs in summer. Pooling all sampling dates, the average OTU numbers (i.e., alpha richness) were 35 and 19 in winter and summer, respectively; no significant trend occurred among flumes (flume-level averages were 32 to 38 OTUs in winter and 16 to 23 OTUs in summer). The bacterial biomass (average ± standard deviation for all samples from one sampling date) ranged from 2.2 ± 0.9 μg cm−2 to 215.6 ± 87.6 μg cm−2 in the winter experiment and from 37.0 ± 8.0 μg cm−2 to 169.2 ± 60.4 μg cm−2 in the summer experiment. Chlorophyll a values varied from 0.1 ± 0.0 μg cm−2 to 5.6 ± 0.4 μg cm−2 in winter and from 0.3 ± 0.1 μg cm−2 to 18.6 ± 5.7 μg cm−2 in summer.
Partial Mantel statistics on distance matrices representing community composition, microbial biomass, and hydrodynamics from each individual sampling date were performed to explore spatial patterns likely induced by the physical environment. We used aggregated microbial biomass combined from bacterial biomass and chlorophyll a to account for possible relationships of bulk biofilm properties, potentially translating into thickness and architecture, with bacterial community composition. We found that the matrix describing hydrodynamics correlated significantly with bacterial community composition matrices in 10 cases (67%) and with the biofilm biomass matrices in 11 cases (73%) (Table 2). Bacterial community composition and biomass matrices correlated in only two cases (13%).
TABLE 2.
Comparison of distance matrices of bacterial community composition, microbial biomass, and hydrodynamics during the winter and summer experiments
| Season and day of growth | Partial Spearman's correlation coefficient betweena: |
||
|---|---|---|---|
| Hydrodynamics and community composition | Hydrodynamics and biomass | Biomass and community composition | |
| Winter | |||
| 4 | 0.303* | 0.274** | 0.044 |
| 8 | 0.073 | 0.230* | 0.145 |
| 12 | 0.272* | 0.222* | −0.133 |
| 16 | 0.470** | 0.313** | −0.019 |
| 20 | 0.378** | 0.094 | −0.012 |
| 24 | 0.196* | 0.261* | 0.127 |
| 28 | 0.107 | 0.212 | 0.361** |
| 32 | 0.133 | 0.415** | −0.132 |
| 36 | 0.430** | 0.263** | 0.053 |
| 40 | 0.202 | 0.299** | 0.250** |
| Summer | |||
| 11 | 0.2381* | 0.3183** | −0.020 |
| 17 | 0.4119** | 0.3439** | 0.129 |
| 37 | 0.3319** | −0.150 | −0.041 |
| 48 | 0.2139* | 0.5829** | −0.076 |
| 55 | −0.011 | 0.032 | 0.028 |
The partial Mantel's randomization test was used for analyses. Values indicate partial Spearman's correlation coefficients between two matrices while correcting for the effect of the third matrix. Boldface indicates statistical significance at a P value of <0.05 (*) or <0.01 (**).
Finally, we used a forward selection procedure (9) to explore the effects of the individual hydrodynamic parameters (velocity, water depth, turbulent kinetic energy, and turbulence intensity) on bacterial community composition and biofilm biomass. Variance inflation factors of less than 10 indicated an acceptable level of multicollinearity between the explanatory variables. Though the adjusted coefficients of multiple determination indicated a significant impact of the model containing all four explanatory variables on community composition in eight cases (53%), a significant contribution of single hydrodynamic parameters was observed in only 5 cases (33%) (Table 3). Turbulent kinetic energy was the only hydrodynamic parameter, which was selected by the procedure in all five cases. Biomass was significantly predicted in 12 cases (80%) by the model containing all explanatory variables. Individual explanatory parameters were selected in 10 cases (67%); however, no single hydrodynamic variable was revealed to be the best predictor of microbial biomass (Table 3).
TABLE 3.
Individual hydrodynamic variables that explain variation of bacterial community composition and biofilm biomassa
| Parameter, season, and day of growth | Coefficient of determinationb for: |
||||
|---|---|---|---|---|---|
| Global test (adjusted r2) | Depth (cm) | Velocity (cm s−1) | Turbulent kinetic energy (J) | Turbulence intensity (%) | |
| Community composition | |||||
| Winter | |||||
| 4 | 0.094* | ||||
| 8 | 0.137** | 0.079** | 0.050* | ||
| 12 | 0.023 | ||||
| 16 | 0.344** | 0.286** | 0.046* | ||
| 20 | 0.230** | ||||
| 24 | 0.029 | ||||
| 28 | 0.076 | ||||
| 32 | 0.121** | ||||
| 36 | 0.152** | 0.050* | 0.087** | ||
| 40 | 0.100* | 0.052** | |||
| Summer | |||||
| 11 | 0.107 | ||||
| 17 | 0.112 | ||||
| 37 | 0.140** | 0.055** | 0.053* | ||
| 48 | 0.096 | ||||
| 55 | 0.036 | ||||
| Biomass | |||||
| Winter | |||||
| 4 | 0.211* | ||||
| 8 | 0.273* | 0.217** | |||
| 12 | 0.240* | 0.233** | |||
| 16 | 0.386** | 0.376** | |||
| 20 | 0.105 | ||||
| 24 | 0.210* | ||||
| 28 | 0.169 | ||||
| 32 | 0.631** | 0.542** | |||
| 36 | 0.590** | 0.121* | 0.081* | 0.382** | |
| 40 | 0.510** | 0.395** | |||
| Summer | |||||
| 11 | 0.324** | 0.241** | |||
| 17 | 0.508** | 0.467** | |||
| 37 | 0.025 | ||||
| 48 | 0.596** | 0.300** | 0.239** | ||
| 55 | 0.254* | 0.143* | |||
The adjusted coefficient of multiple determination (adjusted r2) of the global test represents an unbiased estimate of the explained variation of the model containing all explanatory variables (35); the most important single variables contributing to the observed variation were identified using the forward selection procedure proposed by Blanchet et al. (9).
Boldface indicates statistical significance at a P value of <0.05 (*) or <0.01 (**).
Bacterial richness and diversity.
Computed as richness, beta diversity significantly increased with the standard deviation of flow velocity along the bedforms, and therefore with landscape heterogeneity at the flume level, in winter but not in summer (Fig. 2a and b). In contrast, the Shannon entropy and the Gini-Simpson coefficient revealed significantly increasing trends of beta diversity in both seasons and consistently steeper slopes of the regression lines compared to the richness data (Fig. 2c to f). Regressions were similar for both the Shannon and Simpson indices, despite their different sensitivities toward rare OTUs.
FIG. 2.

Relationships between bacterial beta diversity and landscape flow heterogeneity (as the standard deviation of the average three-dimensional flow velocity vector) during winter and summer growth. Beta diversity was computed as richness (a and b), Shannon entropy (c and d), Gini-Simpson coefficient (e and f), and Bray-Curtis distance (g and h). All indices were z standardized within each sampling date to remove temporal variation.
As the total variation among samples in a community composition table reflects beta diversity (22), we also used the average Bray-Curtis distance between samples from a given flume for a given time as an additional measure for beta diversity. Similarly to the Shannon entropy and the Gini-Simpson coefficient, the Bray-Curtis index revealed significant relationships between beta diversity and landscape heterogeneity in both seasons (Fig. 2g and h). The slopes of the regression lines of all diversity indices were consistently higher in winter. These patterns were broadly consistent when the standard deviations of water depth, turbulent kinetic energy, and turbulence intensity were used as measures of landscape heterogeneity (see Fig. SA1 in the supplemental material).
DISCUSSION
We found species turnover over 1-m-long bedforms that resulted in a gradient of beta diversity that increased with landscape flow heterogeneity induced by bedforms. This finding supports our initial hypothesis and provides clues to habitat heterogeneity as a factor likely influencing microbial biodiversity in streams, where massive loss and homogenization of habitats increasingly threaten biodiversity (36). Similar trends were reported for the beta diversity of benthic algae in streams with contrasting geomorphic heterogeneity (34). We are aware that there likely exist additional purely spatial components of beta diversity at the flume level, which were not included in the present study. However, we sampled bedforms within a limited reach in the flumes and are therefore confident that we essentially captured the beta diversity generated by fine-scale hydrodynamics.
The trends revealed by our multimetric approach are partially due to the presence and absence of individual OTUs, as indicated by the analysis of richness in the winter experiment. A significant contribution, however, is attributable to shifts in the abundance of certain OTUs. The nearly identical trends of the Shannon and Simpson indices suggest that dominant OTUs were sufficient to explain most of the beta diversity patterns. The consistently lower regression slopes during the summer experiment indicate a reduced response of bacterial beta diversity to landscape heterogeneity. Algae dominating biofilms in summer (8.4 ± 7.5 μg chlorophyll a cm−2 in summer versus 3.1 ± 2.0 μg chlorophyll a cm−2 in winter) may affect bacterial community composition in various ways. For instance, they can influence biofilm dynamics through chemical defense (42), or their exudates or simply their alteration of biofilm architecture may decrease the physical controls on bacterial community composition (7).
We recognize the advantages and possible pitfalls of diversity and similarity metrics applied to data from fingerprinting techniques. These techniques have limited ability to detect numerically underrepresented taxa and thus lead to an underestimation of alpha diversity (5, 8). However, they sufficiently estimate relative abundances of common taxa (27) and were found to discriminate communities in a range of environments (43). Thus, metrics including abundance information and focusing on more abundant taxa (i.e., higher-order diversity measures) can be expected to yield reliable results (5). The use of a continuum of metrics of varying order thus seems an equitable way of assessing diversity patterns.
What are the possible mechanisms underlying the observed beta diversity patterns? Mantel statistics suggested local hydrodynamics as one such mechanism in our experiments. Correlations between bacterial community composition and microbial biomass were few and weak when controlling for the effect of hydrodynamics, indicating independent effects of the physical environment on community composition and microbial biomass. However, it is not trivial to identify the various components of flow (e.g., velocity, turbulent kinetic energy, and turbulence intensity) that likely determine bacterial biodiversity in biofilms. The forward selection procedure did not select a single flow component as the major determinant of community composition or biomass, but given the conservative character of the procedure (9), it confidentially substantiates the observed correlations. For instance, it revealed turbulent kinetic energy as a reasonable predictor of community composition, whereas velocity and turbulence intensity predicted the biomasses of nascent and mature biofilms, respectively. Turbulence and related eddies may enhance the downward transport of dispersal cells through the water column to the benthic zone. The same process may enhance mass transfer of nutrients, carbon, and oxygen to the biofilms. In fact, hydrodynamic theory (12) predicts momentum flux and pressure gradients to change along bedforms as used in our experiment and to generate microhabitats differing in turbulence and shear. It is therefore reasonable to assume that our landscapes sorted OTUs capable of coping with the local hydrodynamic environments.
Our results on hydrodynamics, biofilm community composition, and biomass matrices suggest species sorting as a model of biofilm metacommunity dynamics. This seems interesting given the relatively small spatial grain (<1 m) of our study, combined with continuous mixing of water and contained dispersal cells and the predominantly downstream dispersal among local communities (21). According to metacommunity ecology, species sorting explains community composition from local abiotic factors; dispersal remains restricted to colonization but does not allow species to persist in sink habitats (18). Therefore, species sorting is predicted to occur in low-energy ecosystems with extended residence times (24). In contrast, in strongly connected ecosystems with high dispersal, mass effects shift composition patterns away from strict dependence on local conditions as continuous immigration maintains species even in less favorable habitats (18). Mass effects are thus predicted to dominate in high-energy ecosystems with low residence time and continuous mixing, such as in streams (24). We attribute the apparent deviation of our experimental results from theory and evidence from studies of free-living microbial communities (14, 44) to the fact that biofilms attach to stable surfaces and thereby escape the constraints of high-energy ecosystems. The encapsulation of microbial cells within the biofilm matrix reduces their loss and concomitantly increases their residence time relative to that of the free-living cells in the water column.
Immigration of cells from the water column may be too low for mass effects and to eliminate the influence of the local physical factors driving bacterial community composition. This would be corroborated by generally low abundances of biofilm-forming bacteria in the water (7, 39) and by low cell deposition rates (less than 0.03‰ of the total areal transportation flux of cells through the flumes during initial biofilm growth) as estimated in previous experiments (I. Hödl, J. Hödl, G. Singer, K. Besemer, and Tom J. Battin, unpublished data). This suggests that not all dispersers are of the same quality and “search” for the same microhabitat. It is reasonable to assume that advection transports most suspended microorganisms to the streambed before they must then pass the boundary layer to colonize resident biofilms. Swimming (40) and properties such as the coaggregation of single cells into larger entities (38) possibly help to overcome this barrier. Local hydrodynamics may also select for filamentous or chain-building bacteria that can contribute to the formation of streamers in high-shear microhabitats (6). Collectively, these properties may eventually affect the immigration behavior of dispersers and facilitate direct species sorting by hydrodynamics.
Given the unexplained variation in community composition, we recognize that natural biofilm communities in heterogeneous landscapes will not conform to a single model offered by metacommunity ecology. Though mass effects through passive transportation of cells may contribute to the observed community compositions, we suggest that they remain limited because of the aforementioned reasons. Alternatively, neutral models, which have been shown to successfully reproduce observed bacterial abundance distributions (40), could contribute to unravel biofilm metacommunity dynamics. If dispersal is limited, then birth, death, and immigration processes alone can give rise to complex spatial patterns comparable to those of natural biofilms (4, 18).
Manipulating microbial biodiversity in large and natural experimental setups is not a trivial task (41), yet it is necessary if ecological processes are to be understood. We overcame this obstacle by modulating microbial biodiversity through physical landscape heterogeneity and induced hydrodynamics in large streamside flumes. Continuous mixing and fast turnover of water (the water volume of each flume was exchanged approximately 7.5 times per hour) in all flumes fed from the same reservoir enabled us to isolate physical heterogeneity from other factors. We opted for a regression-type experimental design with a clear gradient of increasing landscape heterogeneity. Given the large scale of our flumes, we could not reproduce each level of landscape heterogeneity. However, two independent experiments (summer and winter) underscored the reproducibility of our systems and the power of physical heterogeneity as a driver of biofilm community composition.
Supplementary Material
Acknowledgments
We thank A.-K. Chlup, G. Hochedlinger, G. Steniczka, C. Preiler, C. Baranyi, M. Roura-Carol, E. Sollböck, and H. Hofreiter for their help in the lab and in the field. We thank the organizers of and participants in the EuroDIVERSITY workshop on metacommunity ecology, Uppsala, Sweden, 2008, for fruitful discussions. Robert Findlay, Luc de Meester, and two anonymous reviewers made helpful comments on an earlier version of the paper.
This research was supported by grants from the Austrian Science Fund (P16935-B03) and the European Science Foundation (COMIX, AI0004321) to T.J.B.
Footnotes
Published ahead of print on 18 September 2009.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Battin, T. J., L. A. Kaplan, S. Findlay, C. S. Hopkinson, E. Marti, A. I. Packman, J. D. Newbold, and F. Sabater. 2008. Biophysical controls on organic carbon fluxes in fluvial networks. Nat. Geosci. 1:95-100. [Google Scholar]
- 2.Battin, T. J., L. A. Kaplan, J. D. Newbold, X. Cheng, and C. Hansen. 2003. Effects of current velocity on the nascent architecture of stream microbial biofilms. Appl. Environ. Microbiol. 69:5443-5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Battin, T. J., L. A. Kaplan, J. D. Newbold, and C. M. E. Hansen. 2003. Contributions of microbial biofilms to ecosystem processes in stream mesocosms. Nature 426:439-442. [DOI] [PubMed] [Google Scholar]
- 4.Battin, T. J., W. T. Sloan, S. Kjelleberg, H. Daims, I. M. Head, T. P. Curtis, and L. Eberl. 2007. Microbial landscapes: new paths to biofilm research. Nat. Rev. Microbiol. 5:76-81. [DOI] [PubMed] [Google Scholar]
- 5.Bent, S. J., and L. J. Forney. 2008. The tragedy of the uncommon: understanding limitations in the analysis of microbial diversity. ISME J. 2:689-695. [DOI] [PubMed] [Google Scholar]
- 6.Besemer, K., I. Hödl, G. Singer, and T. J. Battin. Architectural differentiation reflects bacterial community structure in stream biofilms. ISME J., in press. [DOI] [PubMed]
- 7.Besemer, K., G. Singer, R. Limberger, A. K. Chlup, G. Hochedlinger, I. Hödl, C. Baranyi, and T. J. Battin. 2007. Biophysical controls on community succession in stream biofilms. Appl. Environ. Microbiol. 73:4966-4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackwood, C. B., D. Hudleston, D. R. Zak, and J. S. Buyer. 2007. Interpreting ecological diversity indices applied to terminal restriction fragment length polymorphism data: insights from simulated microbial communities. Appl. Environ. Microbiol. 73:5276-5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanchet, F. G., P. Legendre, and D. Borcard. 2008. Forward selection of explanatory variables. Ecology 89:2623-2632. [DOI] [PubMed] [Google Scholar]
- 10.Bloom, S. A. 1981. Similarity indices in community studies: potential pitfalls. Mar. Ecol. Prog. Ser. 5:125-128. [Google Scholar]
- 11.Bradshaw, P. 1971. An introduction to turbulence and its measurement. Pergamon Press, Oxford, United Kingdom.
- 12.Cardenas, M. B., and J. L. Wilson. 2007. Hydrodynamics of coupled flow above and below a sediment-water interface with triangular bedforms. Adv. Water Resour. 30:301-313. [Google Scholar]
- 13.Cardinale, B. J., M. A. Palmer, C. M. Swan, S. Brooks, and N. L. Poff. 2002. The influence of substrate heterogeneity on biofilm metabolism in a stream ecosystem. Ecology 83:412-422. [Google Scholar]
- 14.Crump, B. C., H. E. Adams, J. E. Hobbie, and G. W. Kling. 2007. Biogeography of bacterioplankton in lakes and streams of an Arctic tundra catchment. Ecology 88:1365-1378. [DOI] [PubMed] [Google Scholar]
- 15.Dray, S. 2007. Packfor: forward selection with permutation, R package version 0.0-7. http://biomserv.univ-lyon1.fr/∼dray/software.php.
- 16.Hammer, Ø., D. A. T. Harper, and P. D. Ryan. 2001. PAST: paleontological statistics software package for education and data analysis. Palaeontol. Electron. 4:4A. [Google Scholar]
- 17.Hill, M. O. 1973. Diversity and evenness: a unifying notation and its consequences. Ecology 54:427-432. [Google Scholar]
- 18.Holyoak, M., M. A. Leibold, N. M. Mouquet, R. D. Holt, and M. F. Hoopes. 2005. Metacommunities: a framework for large-scale community ecology, p. 1-31. In M. Holyoak, M. A. Leibold, and R. D. Holt (ed.), Metacommunities. University of Chicago Press, Chicago, IL.
- 19.Jost, L. 2007. Partitioning diversity into independent alpha and beta components. Ecology 88:2427-2439. [DOI] [PubMed] [Google Scholar]
- 20.Lane, D. J. 1991. 16S/23S rRNA sequencing, p. 115-176. In E. Stackebrandt and M. Goodfellow (ed.), Nucleic acid techniques in bacterial systematics. John Wiley & Sons Inc., New York, NY.
- 21.Leff, L. G., J. V. McArthur, and L. J. Shimkets. 1992. Information spiraling: movement of bacteria and their genes in streams. Microb. Ecol. 24:11-24. [DOI] [PubMed] [Google Scholar]
- 22.Legendre, P. 2008. Studying beta diversity: ecological variation partitioning by multiple regression and canonical analysis. J. Plant Ecol. 1:3-8. [Google Scholar]
- 23.Legendre, P., and L. Legendre. 1998. Numerical ecology. Elsevier Science, Amsterdam, The Netherlands.
- 24.Logue, J. B., and E. S. Lindström. 2008. Biogeography of bacterioplankton in inland waters. Freshw. Rev. 1:99-114. [Google Scholar]
- 25.Loreau, M., N. M. Mouquet, and R. D. Holt. 2005. From metacommunities to metaecosystems, p. 418-438. In M. Holyoak, M. A. Leibold, and R. D. Holt (ed.), Metacommunities. The University of Chicago Press, Chicago, IL.
- 26.Lozupone, C. A., M. Hamady, S. T. Kelley, and R. Knight. 2007. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73:1576-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lueders, T., and M. W. Friedrich. 2003. Evaluation of PCR amplification bias by terminal restriction fragment length polymorphism analysis of small-subunit rRNA and mcrA genes by using defined template mixtures of methanogenic pure cultures and soil DNA extracts. Appl. Environ. Microbiol. 69:320-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mantel, N. 1967. The detection of disease clustering and a generalized regression approach. Cancer Res. 27:209-220. [PubMed] [Google Scholar]
- 29.Martiny, J. B. H., B. J. M. Bohannan, J. H. Brown, R. K. Colwell, J. A. Fuhrman, J. L. Green, M. C. Horner-Devine, M. Kane, J. A. Krumins, C. R. Kuske, P. J. Morin, S. Naeem, L. Øvreås, A.-L. Reysenbach, V. H. Smith, and J. T. Staley. 2006. Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4:102-112. [DOI] [PubMed] [Google Scholar]
- 30.Moeseneder, M. M., C. Winter, and G. J. Herndl. 2001. Horizontal and vertical complexity of attached and free-living bacteria of the eastern Mediterranean Sea, determined by 16S rDNA and 16S rRNA fingerprints. Limnol. Oceanogr. 46:95-107. [Google Scholar]
- 31.Nathan, R., and H. C. Muller-Landau. 2000. Spatial patterns of seed dispersal, their determinants and consequences for recruitment. Trends Ecol. Evol. 15:278-285. [DOI] [PubMed] [Google Scholar]
- 32.Norland, S. 1993. The relationship between biomass and volume of bacteria, p. 303-307. In P. F. Kemp, B. F. Sherr, E. B. Sherr, and J. J. Cole (ed.), Handbook of methods in aquatic microbial ecology. Lewis Publishers, Boca Raton, FL.
- 33.Oksanen, J., R. Kindt, P. Legendre, B. O'Hara, G. L. Simpson, and M. H. H. Stevenson. 2008. vegan: community ecology package, R package version 1.11-4. http://cran.r-project.org/.
- 34.Passy, S. I., and F. G. Blanchet. 2007. Algal communities in human-impacted stream ecosystems suffer beta-diversity decline. Divers. Distrib. 13:670-679. [Google Scholar]
- 35.Peres-Neto, P. R., P. Legendre, S. Dray, and D. Borcard. 2006. Variation partitioning of species data matrices: estimation and comparison of fractions. Ecology 87:2614-2625. [DOI] [PubMed] [Google Scholar]
- 36.Poff, N. L., J. D. Olden, D. M. Merritt, and D. M. Pepin. 2007. Homogenization of regional river dynamics by dams and global biodiversity implications. Proc. Natl. Acad. Sci. USA 104:5732-5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.R Development Core Team. 2008. R: a language and environment for statistical computing, 2.7.0. http://www.R-project.org/.
- 38.Rickard, A. H., A. J. McBain, R. G. Ledder, P. S. Handley, and P. Gilbert. 2003. Coaggregation between freshwater bacteria within biofilm and planktonic communities. FEMS Microbiol. Lett. 220:133-140. [DOI] [PubMed] [Google Scholar]
- 39.Rickard, A. H., A. J. McBain, A. T. Stead, and P. Gilbert. 2004. Shear rate moderates community diversity in freshwater biofilms. Appl. Environ. Microbiol. 70:7426-7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sloan, W. T., M. Lunn, S. Woodcock, I. M. Head, S. Nee, and T. P. Curtis. 2006. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 8:732-740. [DOI] [PubMed] [Google Scholar]
- 41.Srivastava, D. S., J. Kolasa, J. Bengtsson, A. Gonzalez, S. P. Lawler, T. E. Miller, P. Munguia, T. Romanuk, D. C. Schneider, and M. K. Trzcinski. 2004. Are natural microcosms useful model systems for ecology? Trends Ecol. Evol. 19:379-384. [DOI] [PubMed] [Google Scholar]
- 42.Steinberg, P. D., R. De Nys, and S. Kjelleberg. 2002. Chemical cues for surface colonization. J. Chem. Ecol. 28:1935-1951. [DOI] [PubMed] [Google Scholar]
- 43.Tiedje, J. M., S. Asuming-Brempong, K. Nüsslein, T. L. Marsh, and S. J. Flynn. 1999. Opening the black box of soil microbial diversity. Appl. Soil Ecol. 13:109-122. [Google Scholar]
- 44.Van der Gucht, K., K. Cottenie, K. Muylaert, N. Vloemans, S. Cousin, S. Declerck, E. Jeppesen, J. M. Conde-Porcuna, K. Schwenk, G. Zwart, H. Degans, W. Vyverman, and L. DeMeester. 2007. The power of species sorting: local factors drive bacterial community composition over a wide range of spatial scales. Proc. Natl. Acad. Sci. USA 104:20404-20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.